
The Autophagy Inhibitor Chloroquine, Alone or in Combination with mTOR Inhibitors, Displays Anti-Tumor Effects in In Vitro and In Vivo Lung Carcinoid Models



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability
2.4. Flow Cytometry
2.5. Drug Cytotoxicity Assay
2.6. Western Blotting
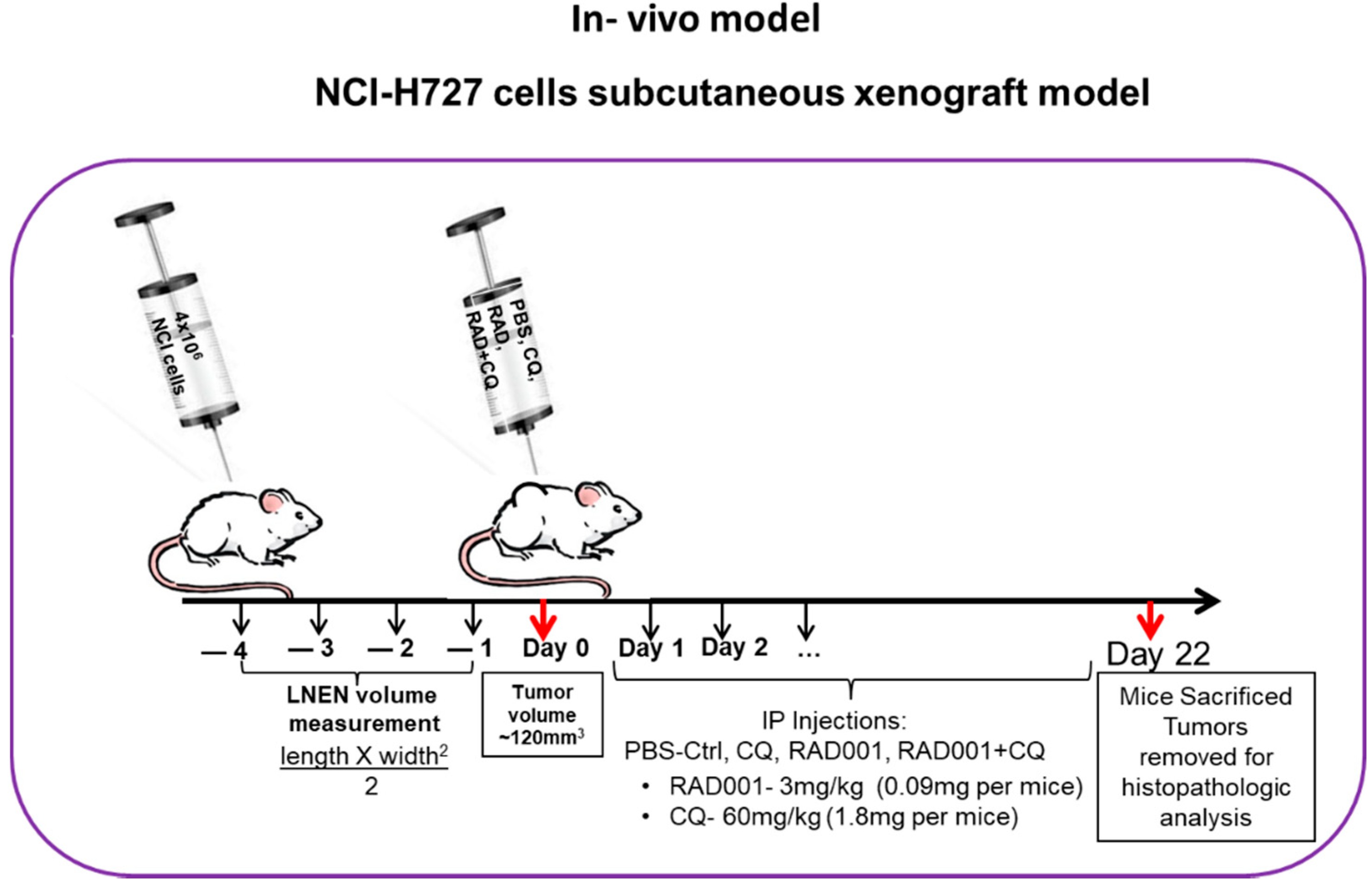
2.7. Subcutaneous Murine Xenograft Model
2.8. Statistical Analyses
3. Results
3.1. CQ and the mTORi RAD001 and Torin1 Inhibit LNEN Cell Viability
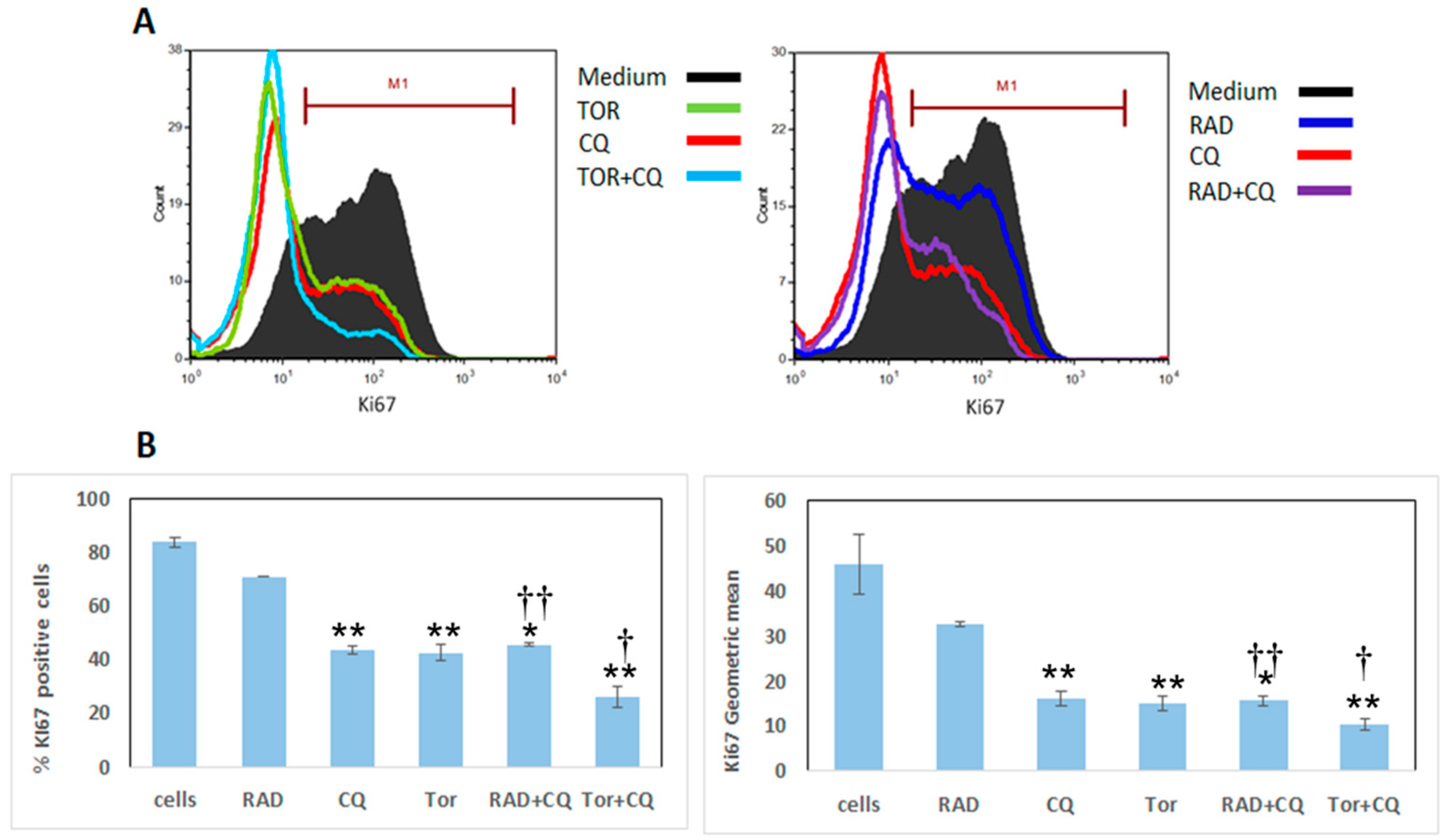
3.2. CQ, RAD001, and Torin1 Inhibit LNEN Cell Proliferation
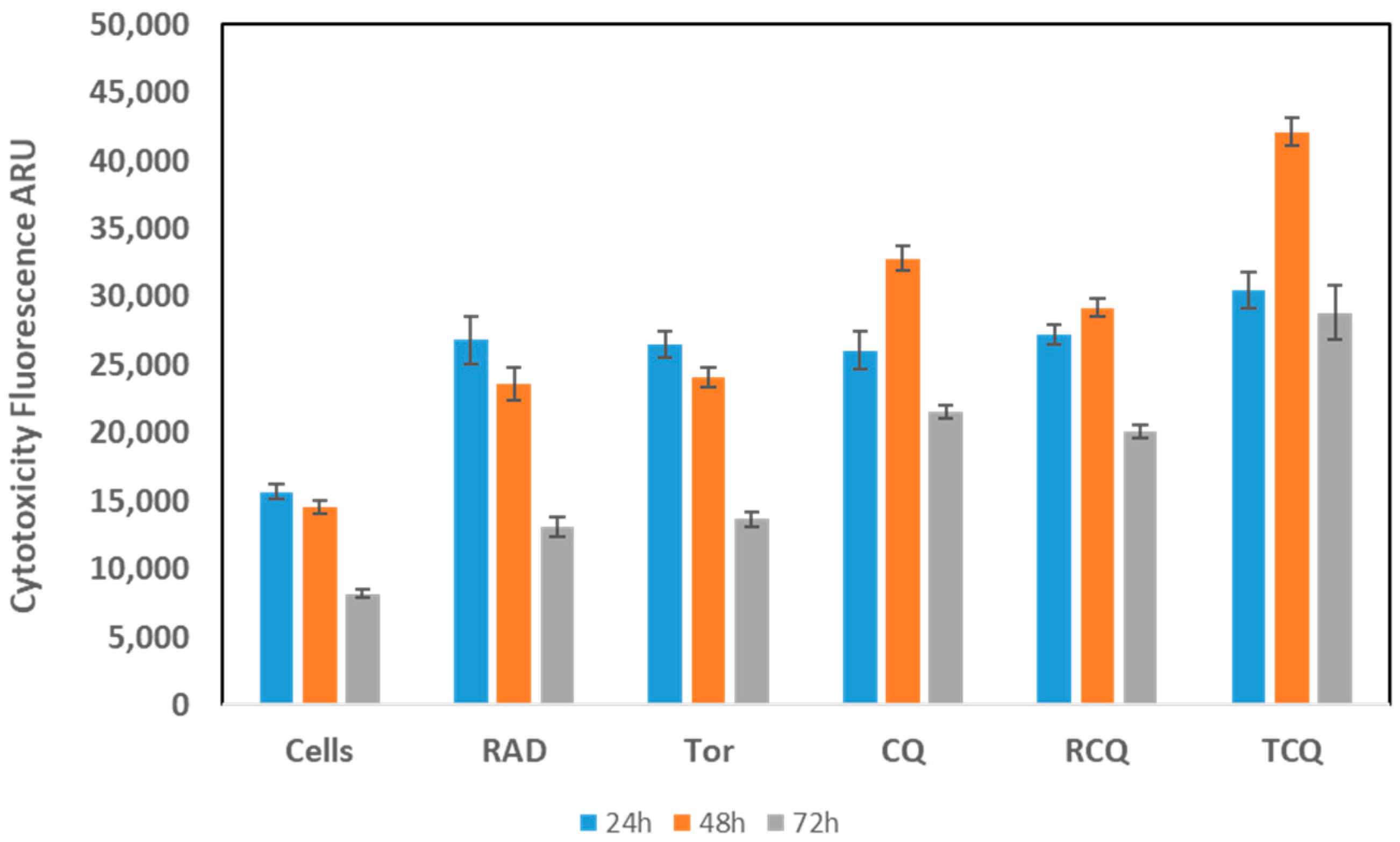
3.3. CQ, Alone or in Combination with mTORi, Induces NCI-H727 Cell Death
3.4. mTORi and CQ Have Synergistic Effects, and Their Combination Suppresses mTOR Activation
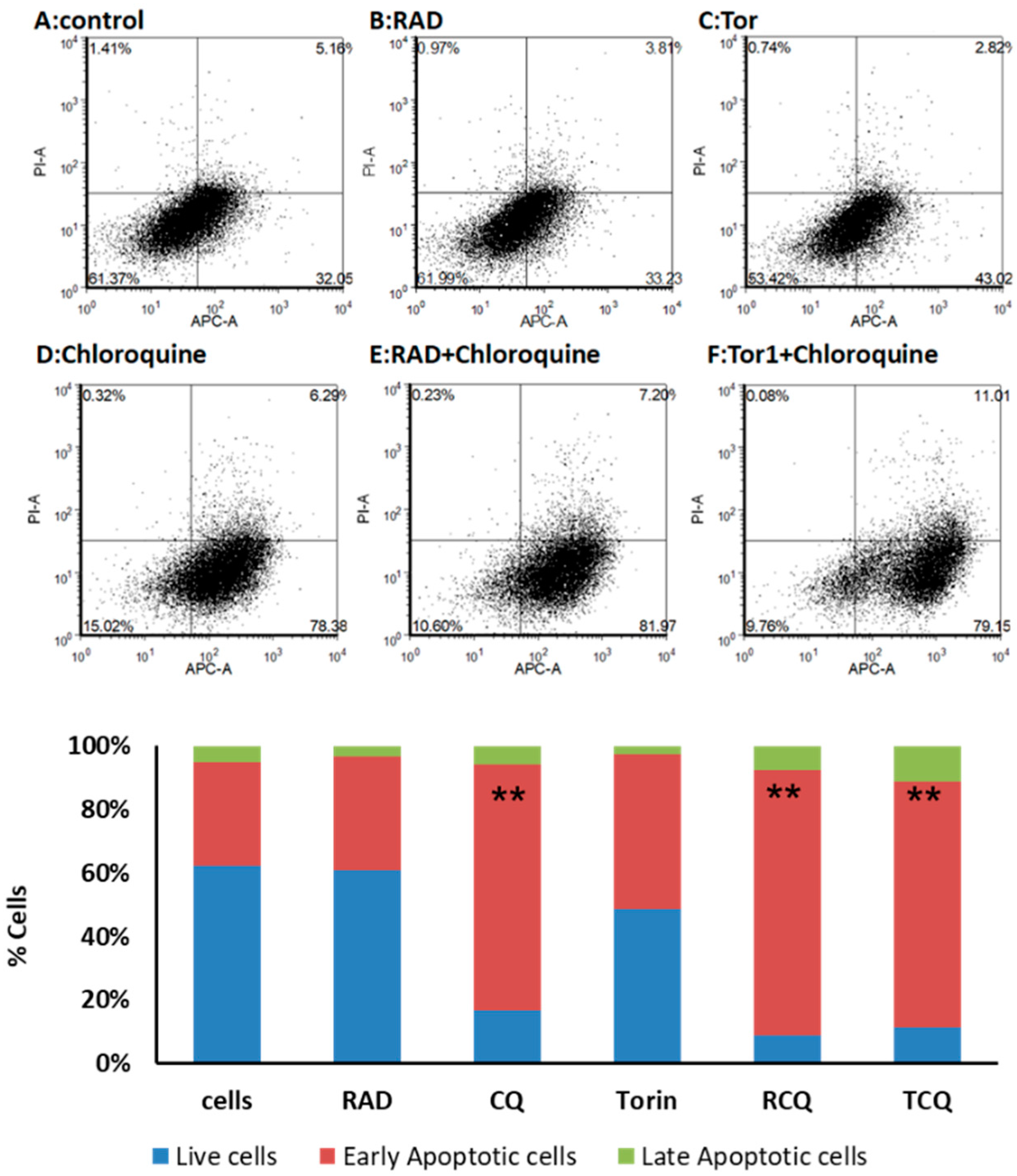
3.5. CQ and mTORi Induce Apoptosis in NCI-H727 Cells
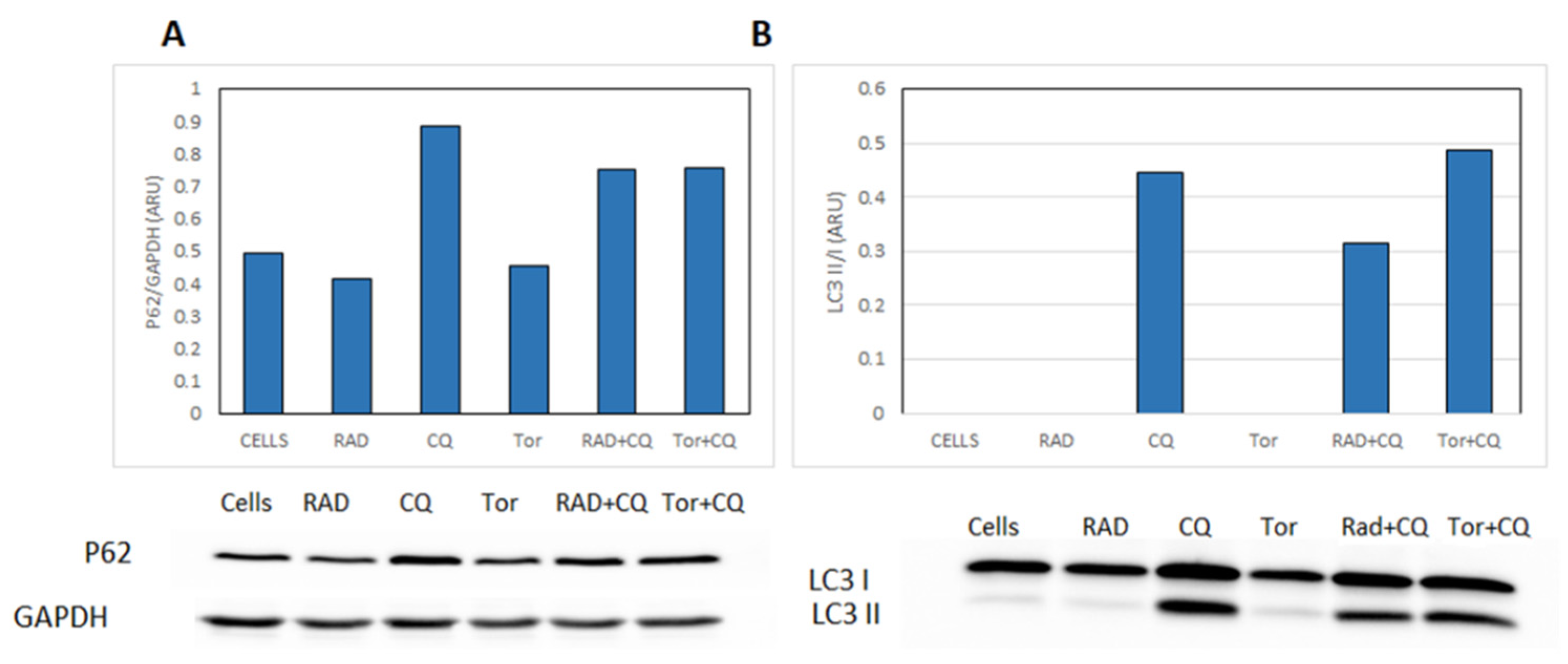
3.6. CQ Inhibits Autophagy and Diminishes mTORi Induction of Autophagy in NCI-H727 Cells
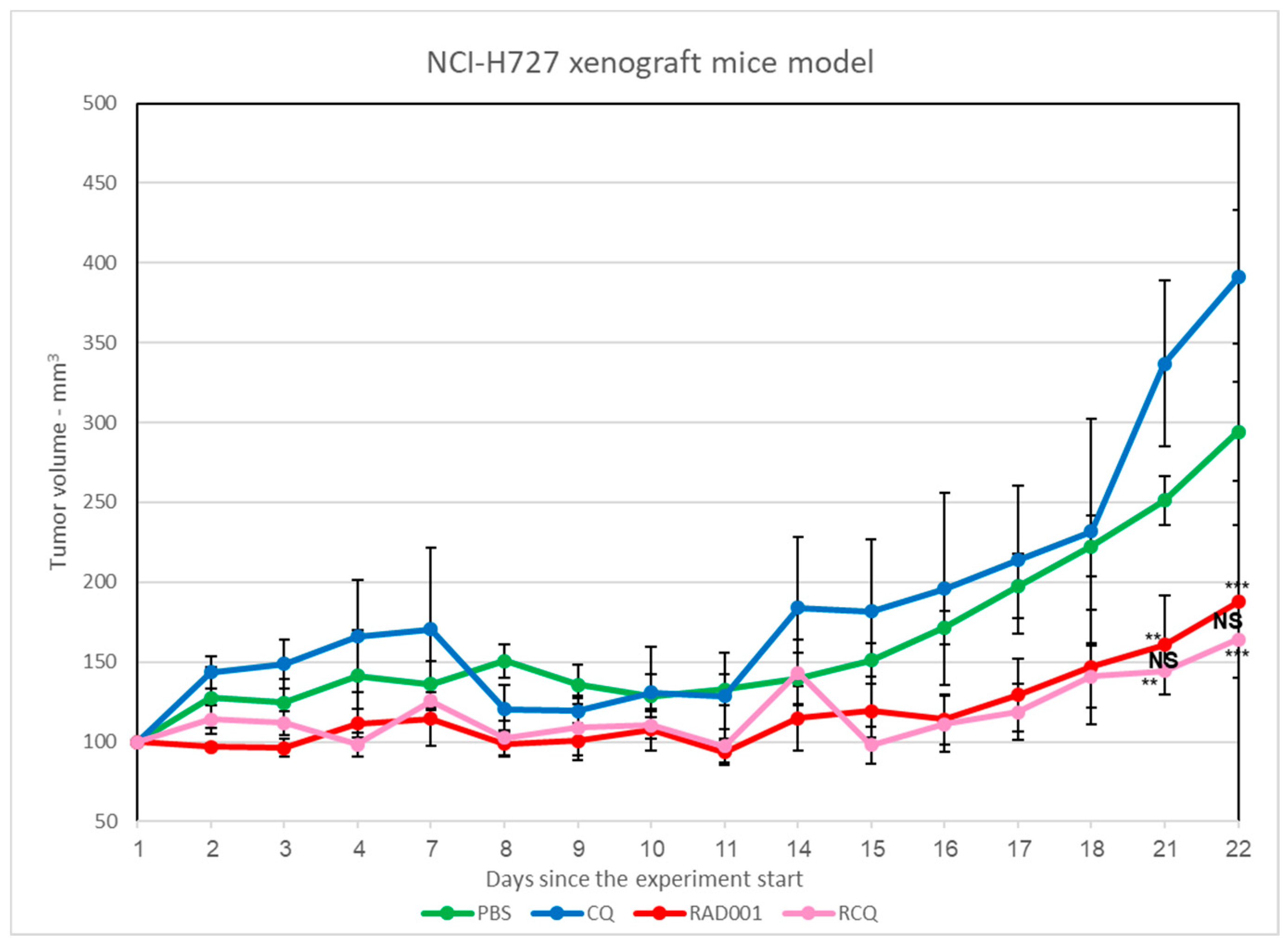
3.7. The Addition of CQ to mTORi Decreases Tumor Volume Compared to mTORi Alone in the NCI-H727 Subcutaneous Murine Xenograft Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Torniai, M.; Scortichini, L.; Tronconi, F.; Rubini, C.; Morgese, F.; Rinaldi, S.; Mazzanti, P.; Berardi, R. Systemic treatment for lung carcinoids: From bench to bedside. Clin. Transl. Med. 2019, 8, 22. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.I.; Krenning, E.P.; Teunissen, J.J.; Bergsma, H.; Kam, B.L.; Kwekkeboom, D.J. Comparison of response evaluation in patients with gastroenteropancreatic and thoracic neuroendocrine tumors after treatment with [177Lu-DOTA0,Tyr3] octreotate. J. Nucl. Med. 2013, 54, 1689–1696. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef]
- Righi, L.; Volante, M.; Rapa, I.; Tavaglione, V.; Inzani, F.; Pelosi, G.; Papotti, M. Mammalian target of rapamycin signaling activation patterns in neuroendocrine tumors of the lung. Endocr. Relat. Cancer 2010, 17, 977–987. [Google Scholar] [CrossRef][Green Version]
- Efeyan, A.; Sabatini, D.M. mTOR and cancer: Many loops in one pathway. Curr. Opin. Cell Biol. 2010, 22, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Wolin, E.M. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013, 335, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Avniel-Polak, S.; Leibowitz, G.; Riahi, Y.; Glaser, B.; Gross, D.J.; Grozinsky-Glasberg, S. Abrogation of Autophagy by Chloroquine Alone or in Combination with mTOR Inhibitors Induces Apoptosis in Neuroendocrine Tumor Cells. Neuroendocrinology 2016, 103, 724–737. [Google Scholar] [CrossRef]
- O’Reilly, K.A.; Rojo, F.; She, Q.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy 2013, 9, 626–628. [Google Scholar] [CrossRef]
- Avniel-Polak, S.; Leibowitz, G.; Doviner, V.; Gross, D.J.; Grozinsky-Glasberg, S. Combining chloroquine with RAD001 inhibits tumor growth in a NEN mouse model. Endocr. Relat. Cancer 2018, 25, 677–686. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- He, H.; Dang, Y.; Dai, F.; Guo, Z.; Wu, J.; She, X.; Pei, Y.; Chen, Y.; Ling, W.; Wu, C.; et al. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J. Biol. Chem. 2003, 278, 29278–29287. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 2004, 279, 47704–47710. [Google Scholar] [CrossRef]
- Wu, H.; Dang, Y.; Su, W.; Liu, C.; Ma, H.; Shan, Y.; Pei, Y.; Wan, B.; Guo, J.; Yu, L. Molecular cloning and characterization of rat LC3A and LC3B--two novel markers of autophagosome. Biochem. Biophys. Res. Commun. 2006, 339, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.K.; Kim, J.H.; Starenki, D.; Park, J.I. Autophagy sensitivity of neuroendocrine lung tumor cells. Int. J. Oncol. 2013, 43, 2031–2038. [Google Scholar] [CrossRef]
- Grimaldi, A.; Balestrieri, M.L.; D’Onofrio, N.; Di Domenico, G.; Nocera, C.; Lamberti, M.; Tonini, G.; Zoccoli, A.; Santini, D.; Caraglia, M.; et al. The synergistic effect of everolimus and chloroquine on endothelial cell number reduction is paralleled by increased apoptosis and reduced autophagy occurrence. PLoS ONE. 2013, 8, e79658. [Google Scholar] [CrossRef]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz Jr, B.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.; Kim, T.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.; Lee, H.J.; Lee, K.; Bang, Y. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Xue, J.; Moyer, A.; Peng, B.; Wu, J.; Hannafon, B.N.; Ding, W. Chloroquine is a zinc ionophore. PLoS ONE 2014, 9, e109180. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knigin, A.; Avniel-Polak, S.; Leibowitz, G.; Oleinikov, K.; Gross, D.J.; Grozinsky-Glasberg, S. The Autophagy Inhibitor Chloroquine, Alone or in Combination with mTOR Inhibitors, Displays Anti-Tumor Effects in In Vitro and In Vivo Lung Carcinoid Models. Cancers 2021, 13, 6327. https://doi.org/10.3390/cancers13246327
Knigin A, Avniel-Polak S, Leibowitz G, Oleinikov K, Gross DJ, Grozinsky-Glasberg S. The Autophagy Inhibitor Chloroquine, Alone or in Combination with mTOR Inhibitors, Displays Anti-Tumor Effects in In Vitro and In Vivo Lung Carcinoid Models. Cancers. 2021; 13(24):6327. https://doi.org/10.3390/cancers13246327
Chicago/Turabian StyleKnigin, Adi, Shani Avniel-Polak, Gil Leibowitz, Kira Oleinikov, David J. Gross, and Simona Grozinsky-Glasberg. 2021. "The Autophagy Inhibitor Chloroquine, Alone or in Combination with mTOR Inhibitors, Displays Anti-Tumor Effects in In Vitro and In Vivo Lung Carcinoid Models" Cancers 13, no. 24: 6327. https://doi.org/10.3390/cancers13246327
APA StyleKnigin, A., Avniel-Polak, S., Leibowitz, G., Oleinikov, K., Gross, D. J., & Grozinsky-Glasberg, S. (2021). The Autophagy Inhibitor Chloroquine, Alone or in Combination with mTOR Inhibitors, Displays Anti-Tumor Effects in In Vitro and In Vivo Lung Carcinoid Models. Cancers, 13(24), 6327. https://doi.org/10.3390/cancers13246327