
A Transgenic Model Reveals the Role of Klotho in Pancreatic Cancer Development and Paves the Way for New Klotho-Based Therapy

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA Analysis Using UCSC Xena Browser
2.2. Chemicals, Antibodies, and Constructs
2.3. AAV Vector Production and Purification
2.4. Animal Maintenance
2.5. Pancreas-Specific KLOTHO Knockdown Mouse Models
2.6. Mice Tumor Xenograft Studies
2.7. KPC Mouse Models
2.8. Genotyping
2.9. Immunohistochemistry (IHC) Analysis
2.10. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.11. Statistical Analysis
3. Results
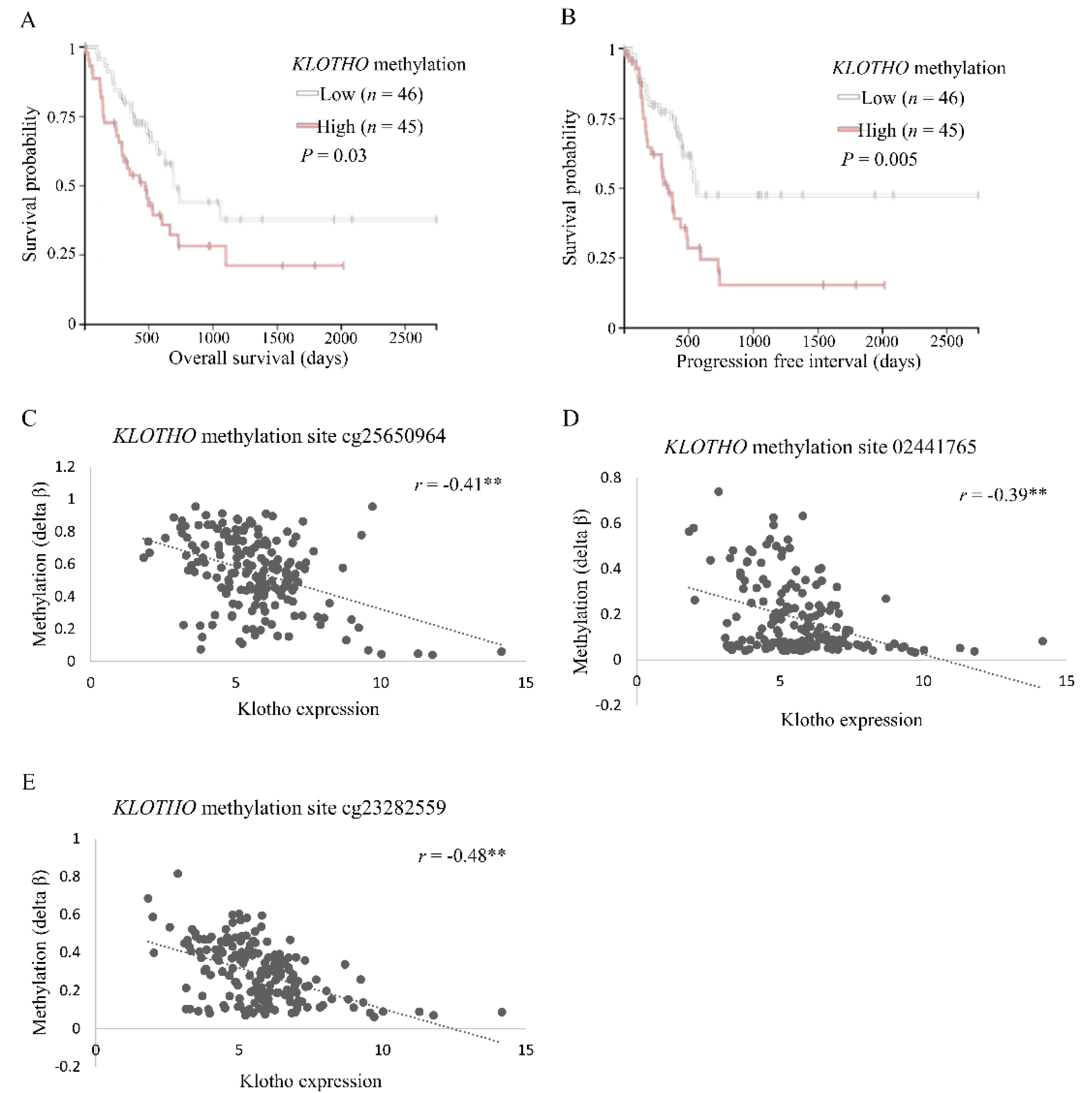
3.1. Levels of Klotho Expression and DNA Methylation in Pancreatic Tumors Correlate with Survival of Cancer Patients
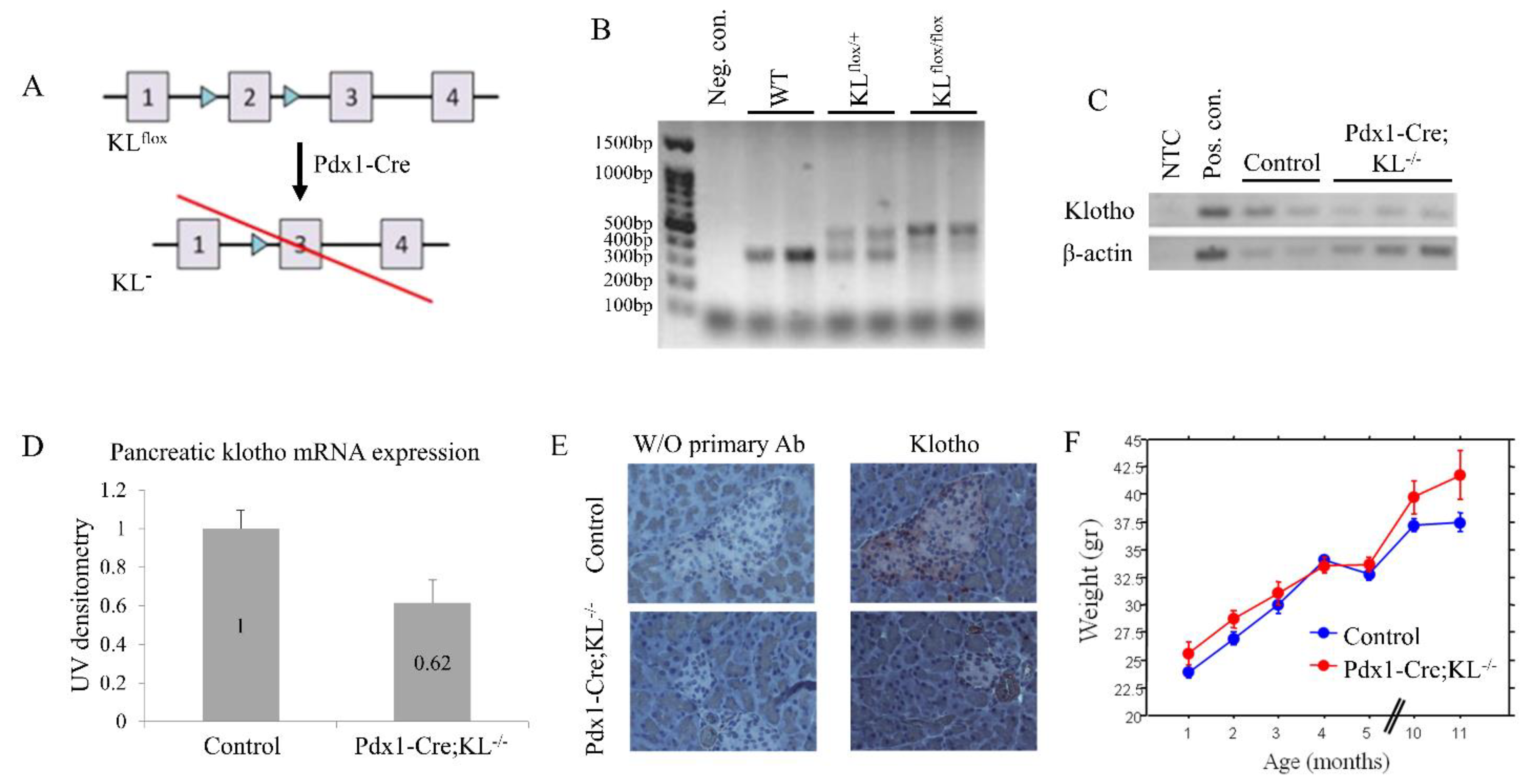
3.2. Generation of Pancreatic KLOTHO Knockdown Mice
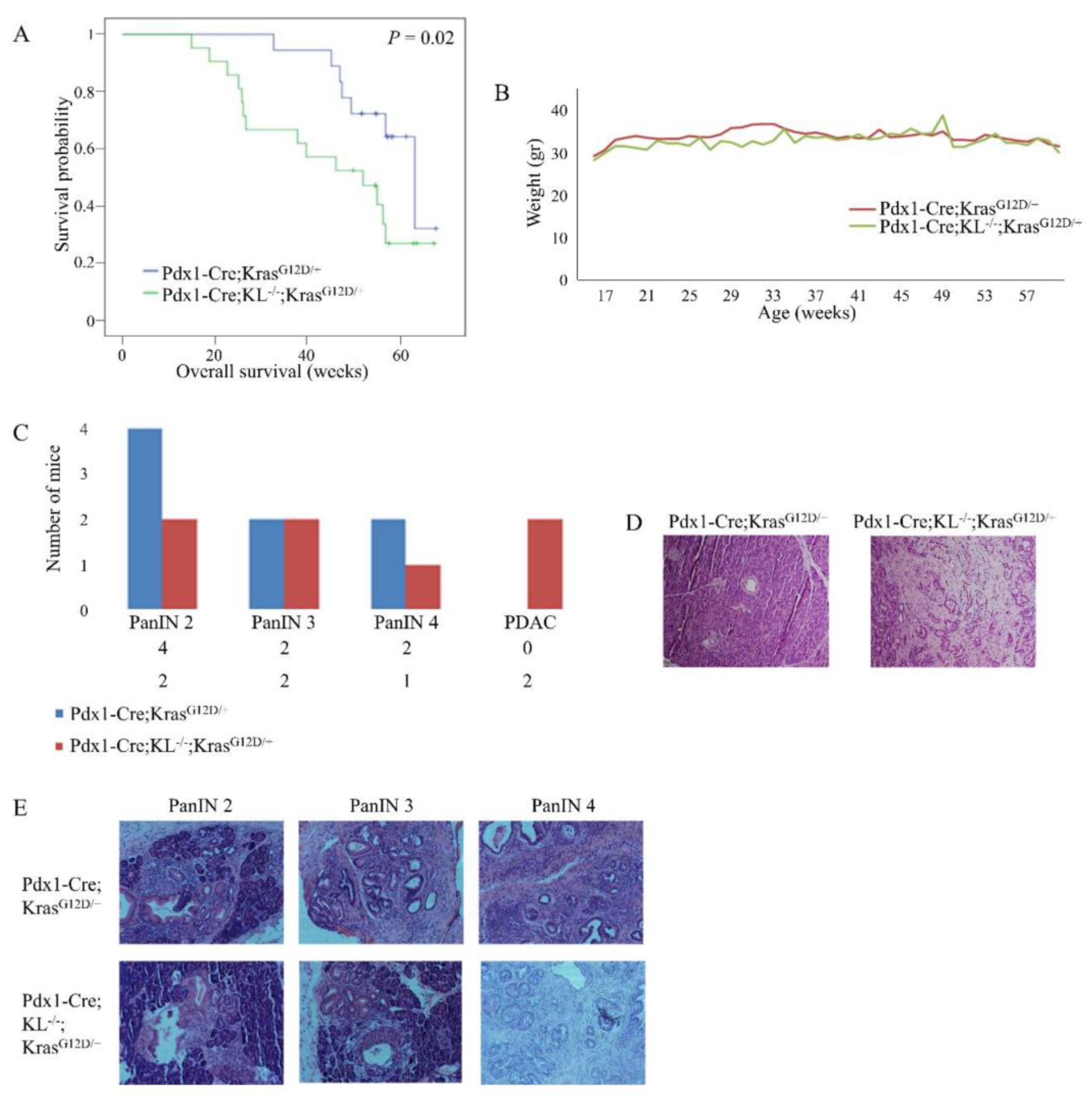
3.3. Loss of Pancreatic Klotho Contributes to Reduced Survival in Mice
3.4. Klotho Cooperates in the Development of PDAC In Vivo
3.5. Treatment with sKL Inhibits Pancreatic Tumors and Prolongs Survival In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and Invasive Ductal Pancreatic Cancer and Its Early Detection in the Mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Siveke, J.T. Genetically Engineered Mouse Models of Pancreatic Cancer: Unravelling Tumour Biology and Progressing Translational Oncology. Gut 2012, 61, 1488–1500. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabeshima, Y. Klotho: A Fundamental Regulator of Aging. Ageing Res. Rev. 2002, 1, 627–638. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the Mouse Klotho Gene Leads to a Syndrome Resembling Ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of Aging in Mice by the Hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the Human Klotho Gene and Its Two Transcripts Encoding Membrane and Secreted Klotho Protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-D.; Podvin, S.; Gillespie, E.; Leeman, S.E.; Abraham, C.R. Insulin Stimulates the Cleavage and Release of the Extracellular Domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. USA 2007, 104, 19796–19801. [Google Scholar] [CrossRef] [Green Version]
- Ligumsky, H.; Rubinek, T.; Merenbakh-Lamin, K.; Yeheskel, A.; Sertchook, R.; Shahmoon, S.; Aviel-Ronen, S.; Wolf, I. Tumor Suppressor Activity of Klotho in Breast Cancer Is Revealed by Structure-Function Analysis. Mol. Cancer Res. 2015, 13, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho Converts Canonical FGF Receptor into a Specific Receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.-C.; Moe, O.W.; et al. Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Hoefs, S.; van der Kemp, A.W.; Topala, C.N.; Bindels, R.J.; Hoenderop, J.G. The Beta-Glucuronidase Klotho Hydrolyzes and Activates the TRPV5 Channel. Science 2005, 310, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-K.; Ortega, B.; Kurosu, H.; Rosenblatt, K.P.; Kuro-O, M.; Huang, C.-L. Removal of Sialic Acid Involving Klotho Causes Cell-Surface Retention of TRPV5 Channel via Binding to Galectin-1. Proc. Natl. Acad. Sci. USA 2008, 105, 9805–9810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-o, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A Tumor Suppressor and a Modulator of the IGF-1 and FGF Pathways in Human Breast Cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef]
- Abramovitz, L.; Rubinek, T.; Ligumsky, H.; Bose, S.; Barshack, I.; Avivi, C.; Kaufman, B.; Wolf, I. KL1 Internal Repeat Mediates Klotho Tumor Suppressor Activities and Inhibits BFGF and IGF-I Signaling in Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 4254–4266. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Groen, A.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.-L.; et al. α-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Sun, Z. In Vivo Pancreatic β-Cell—Specific Expression of Antiaging Gene Klotho: A Novel Approach for Preserving β-Cells in Type 2 Diabetes. Diabetes 2015, 64, 1444–1458. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Sun, Z. Antiaging Gene Klotho Enhances Glucose-Induced Insulin Secretion by up-Regulating Plasma Membrane Levels of TRPV2 in MIN6 β-Cells. Endocrinology 2012, 153, 3029–3039. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Sun, Z. Antiaging Gene Klotho Attenuates Pancreatic β-Cell Apoptosis in Type 1 Diabetes. Diabetes 2015, 64, 4298–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsugi, T.; Ohno, T.; Ohyama, Y.; Uchiyama, T.; Saito, Y.; Matsumura, Y.; Aizawa, H.; Itoh, H.; Kurabayashi, M.; Kawazu, S.; et al. Decreased Insulin Production and Increased Insulin Sensitivity in the Klotho Mutant Mouse, a Novel Animal Model for Human Aging. Metabolism 2000, 49, 1118–1123. [Google Scholar] [CrossRef]
- Lojkin, I.; Rubinek, T.; Orsulic, S.; Schwarzmann, O.; Karlan, B.Y.; Bose, S.; Wolf, I. Reduced Expression and Growth Inhibitory Activity of the Aging Suppressor Klotho in Epithelial Ovarian Cancer. Cancer Lett. 2015, 362, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Zhou, J.; Shu, G.; Liu, D.-C.; Zhou, J.; Chen, J.; Yuan, L. Restoration of Klotho Gene Expression Induces Apoptosis and Autophagy in Gastric Cancer Cells: Tumor Suppressive Role of Klotho in Gastric Cancer. Cancer Cell Int. 2013, 13, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbel Rubinstein, T.; Shahmoon, S.; Zigmond, E.; Etan, T.; Merenbakh-Lamin, K.; Pasmanik-Chor, M.; Har-Zahav, G.; Barshack, I.; Vainer, G.W.; Skalka, N.; et al. Klotho Suppresses Colorectal Cancer through Modulation of the Unfolded Protein Response. Oncogene 2019, 38, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho Inhibits Transforming Growth Factor-Beta1 (TGF-Beta1) Signaling and Suppresses Renal Fibrosis and Cancer Metastasis in Mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Wang, X.; Zhao, W.; Wu, J. Klotho Inhibits Growth and Promotes Apoptosis in Human Lung Cancer Cell Line A549. J. Exp. Clin. Cancer Res. 2010, 29, 99. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jeong, D.-J.; Kim, J.; Lee, S.; Park, J.-H.; Chang, B.; Jung, S.-I.; Yi, L.; Han, Y.; Yang, Y.; et al. The Anti-Aging Gene KLOTHO Is a Novel Target for Epigenetic Silencing in Human Cervical Carcinoma. Mol. Cancer 2010, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Chen, J.; Liu, B.; Zhan, J. Klotho Acts as a Tumor Suppressor in Cancers. Pathol. Oncol. Res. 2013, 19, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Wolf, I. The Role of Alpha-Klotho as a Universal Tumor Suppressor. Vitam. Horm. 2016, 101, 197–214. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Wang, X.; Jie, P.; Lu, H.; Zhang, S.; Lin, X.; Lam, E.K.; Cui, Y.; Yu, J.; et al. Klotho Is Silenced through Promoter Hypermethylation in Gastric Cancer. Am. J. Cancer Res. 2011, 1, 111–119. [Google Scholar] [PubMed]
- Rubinek, T.; Shulman, M.; Israeli, S.; Bose, S.; Avraham, A.; Zundelevich, A.; Evron, E.; Gal-Yam, E.N.; Kaufman, B.; Wolf, I. Epigenetic Silencing of the Tumor Suppressor Klotho in Human Breast Cancer. Breast Cancer Res. Treat. 2012, 133, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Camilli, T.C.; Xu, M.; O’Connell, M.P.; Chien, B.; Frank, B.P.; Subaran, S.; Indig, F.E.; Morin, P.J.; Hewitt, S.M.; Weeraratna, A.T. Loss of Klotho during Melanoma Progression Leads to Increased Filamin Cleavage, Increased Wnt5A Expression, and Enhanced Melanoma Cell Motility. Pigment. Cell Melanoma Res. 2011, 24, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Gu, Y.; Chen, Y. Identification of Novel Predictive Markers for the Prognosis of Pancreatic Ductal Adenocarcinoma. Cancer Investig. 2014, 32, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers , D.; Brooks, A.N.; et al. The UCSC Xena Platform for Cancer Genomics Data Visualization and Interpretation. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm-Benartzi, C.S.; Koestler, D.C.; Karagas, M.R.; Flanagan, J.M.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; Houseman, E.A.; Brown, R. Review of Processing and Analysis Methods for DNA Methylation Array Data. Br. J. Cancer 2013, 109, 1394–1402. [Google Scholar] [CrossRef] [Green Version]
- Piedra, J.; Ontiveros, M.; Miravet, S.; Penalva, C.; Monfar, M.; Chillon, M. Development of a Rapid, Robust, and Universal Picogreen-Based Method to Titer Adeno-Associated Vectors. Hum. Gene Ther. Methods 2015, 26, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Olauson, H.; Lindberg, K.; Amin, R.; Jia, T.; Wernerson, A.; Andersson, G.; Larsson, T.E. Targeted Deletion of Klotho in Kidney Distal Tubule Disrupts Mineral Metabolism. J. Am. Soc. Nephrol. 2012, 23, 1641–1651. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-X.; Huang, L.-Y.; Peng, J.-J.; Liang, L.; Shi, D.-B.; Zheng, H.-T.; Cai, S.-J. Klotho Suppresses Growth and Invasion of Colon Cancer Cells through Inhibition of IGF1R-Mediated PI3K/AKT Pathway. Int. J. Oncol. 2014, 45, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Zhong, J.; Gan, L.H.; Chen, S.J.; Jin, H.C.; Wang, X.; Wang, L.J. Klotho, an Anti-Senescence Related Gene, Is Frequently Inactivated through Promoter Hypermethylation in Colorectal Cancer. Tumour Biol. 2011, 32, 729–735. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbel Rubinstein, T.; Reuveni, I.; Hesin, A.; Klein-Goldberg, A.; Olauson, H.; Larsson, T.E.; Abraham, C.R.; Zeldich, E.; Bosch, A.; Chillón, M.; et al. A Transgenic Model Reveals the Role of Klotho in Pancreatic Cancer Development and Paves the Way for New Klotho-Based Therapy. Cancers 2021, 13, 6297. https://doi.org/10.3390/cancers13246297
Arbel Rubinstein T, Reuveni I, Hesin A, Klein-Goldberg A, Olauson H, Larsson TE, Abraham CR, Zeldich E, Bosch A, Chillón M, et al. A Transgenic Model Reveals the Role of Klotho in Pancreatic Cancer Development and Paves the Way for New Klotho-Based Therapy. Cancers. 2021; 13(24):6297. https://doi.org/10.3390/cancers13246297
Chicago/Turabian StyleArbel Rubinstein, Tammi, Inbal Reuveni, Arkadi Hesin, Anat Klein-Goldberg, Hannes Olauson, Tobias E. Larsson, Carmela R. Abraham, Ella Zeldich, Assumpció Bosch, Miguel Chillón, and et al. 2021. "A Transgenic Model Reveals the Role of Klotho in Pancreatic Cancer Development and Paves the Way for New Klotho-Based Therapy" Cancers 13, no. 24: 6297. https://doi.org/10.3390/cancers13246297
APA StyleArbel Rubinstein, T., Reuveni, I., Hesin, A., Klein-Goldberg, A., Olauson, H., Larsson, T. E., Abraham, C. R., Zeldich, E., Bosch, A., Chillón, M., Hollander, K. S., Shabtay-Orbach, A., Vainer, G. W., Wolf, I., & Rubinek, T. (2021). A Transgenic Model Reveals the Role of Klotho in Pancreatic Cancer Development and Paves the Way for New Klotho-Based Therapy. Cancers, 13(24), 6297. https://doi.org/10.3390/cancers13246297