
PARP1 Inhibitor and Trabectedin Combination Does Not Increase Tumor Mutational Burden in Advanced Sarcomas—A Preclinical and Translational Study


 ,
, 
 , , , , ,
, , , , ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
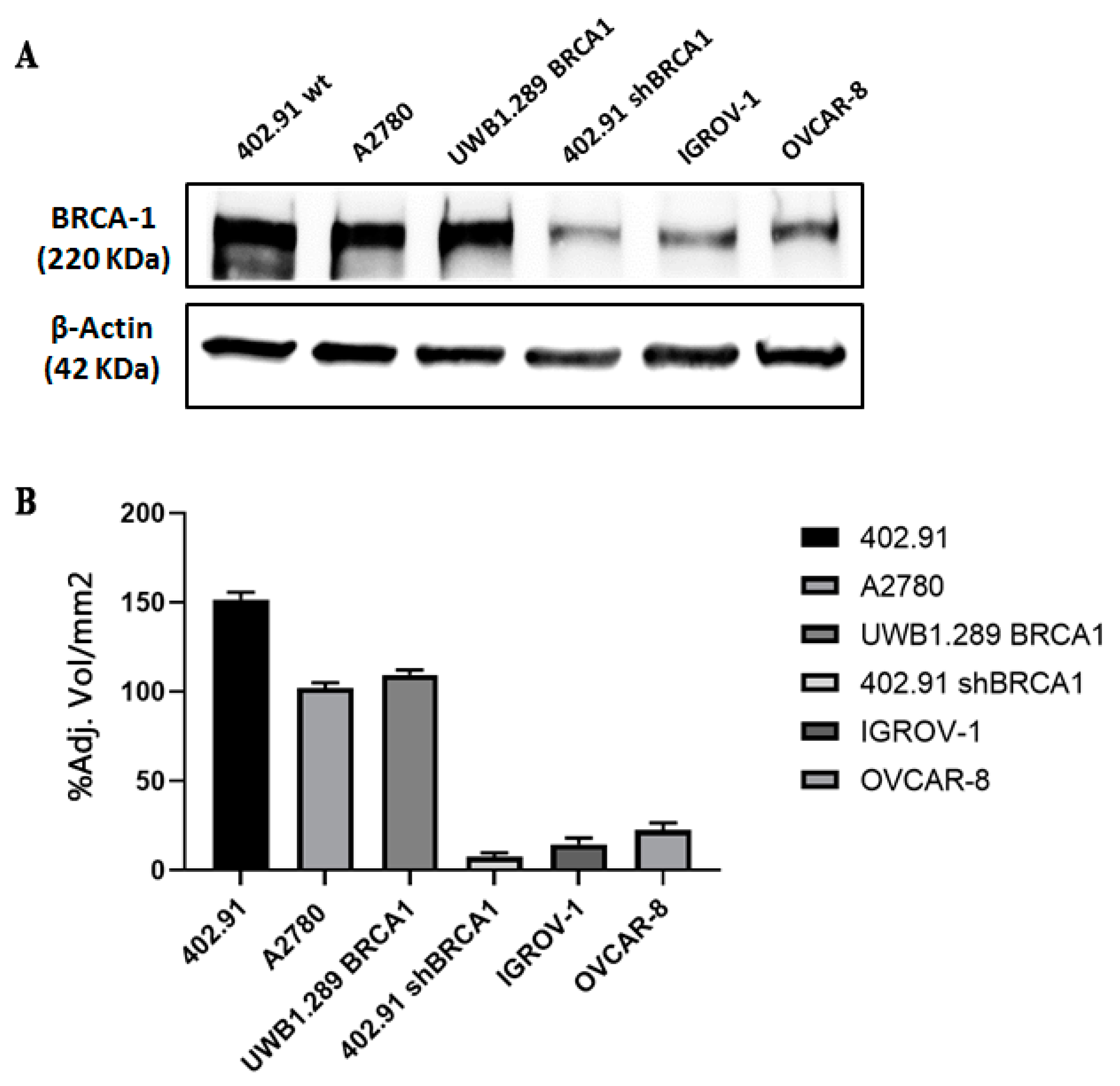
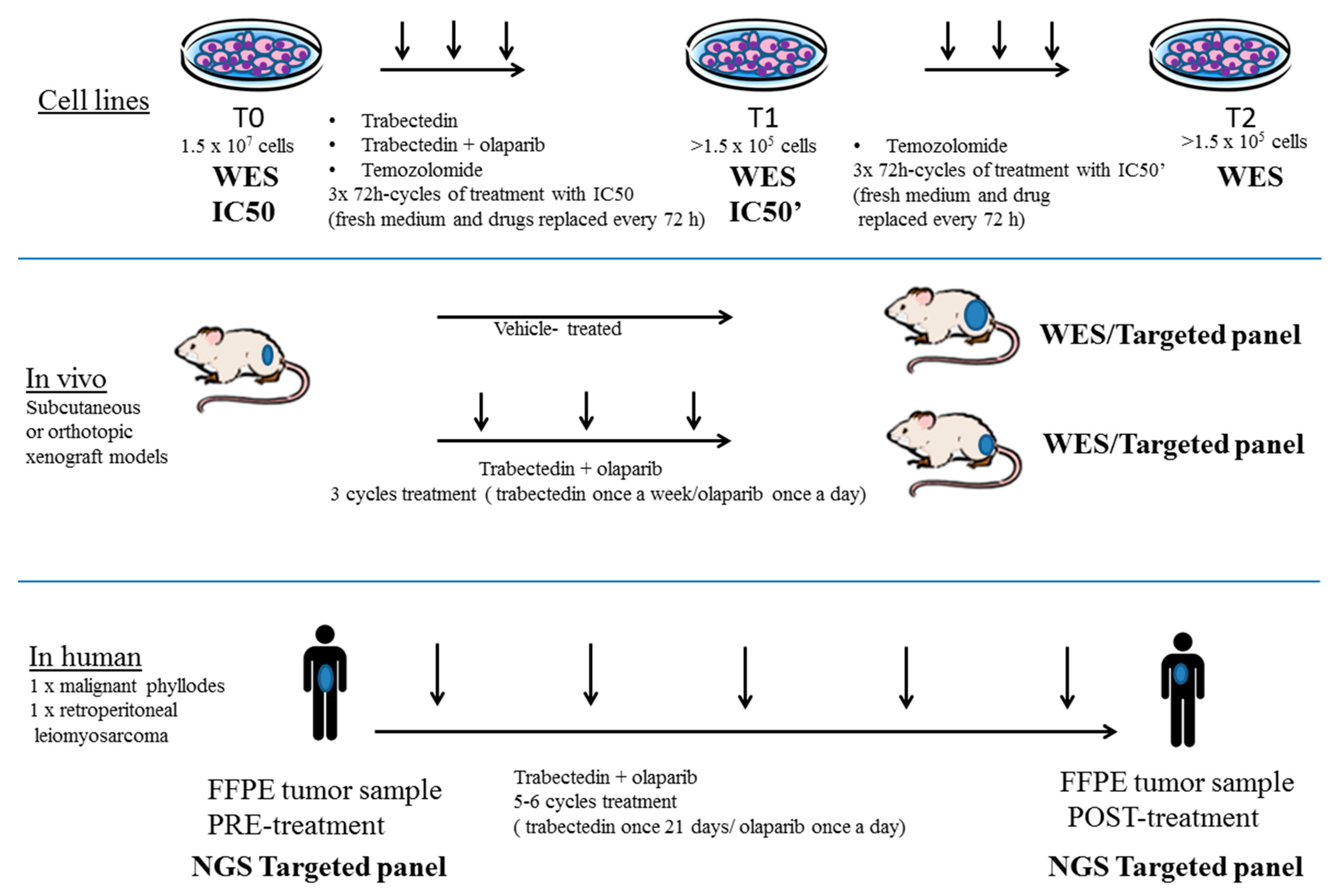
2.1. Cell Cultures, BRCA1 Stable Silencing and Drug Treatment
2.2. Cell Viability Assay and IC50 Determination
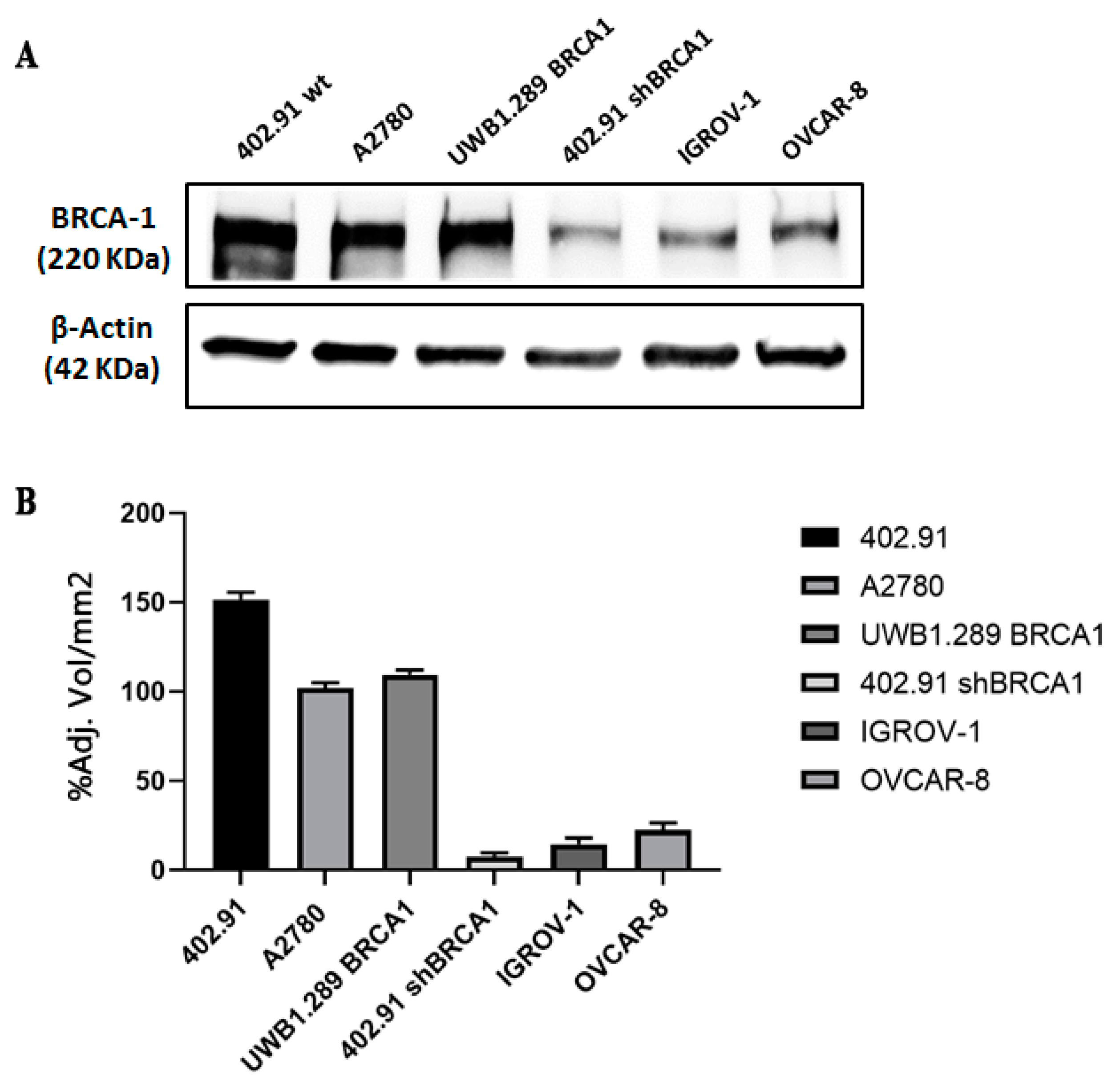
2.3. Western Blot
2.4. Sarcoma Xenograft Models in NOD/SCID Mice
2.5. TOMAS Trial Patients
2.6. DNA Extraction
2.7. Genetic Analyses after Whole Exome Sequencing and NGS Targeted Approach
2.8. Mutational Signature Analyses
3. Results
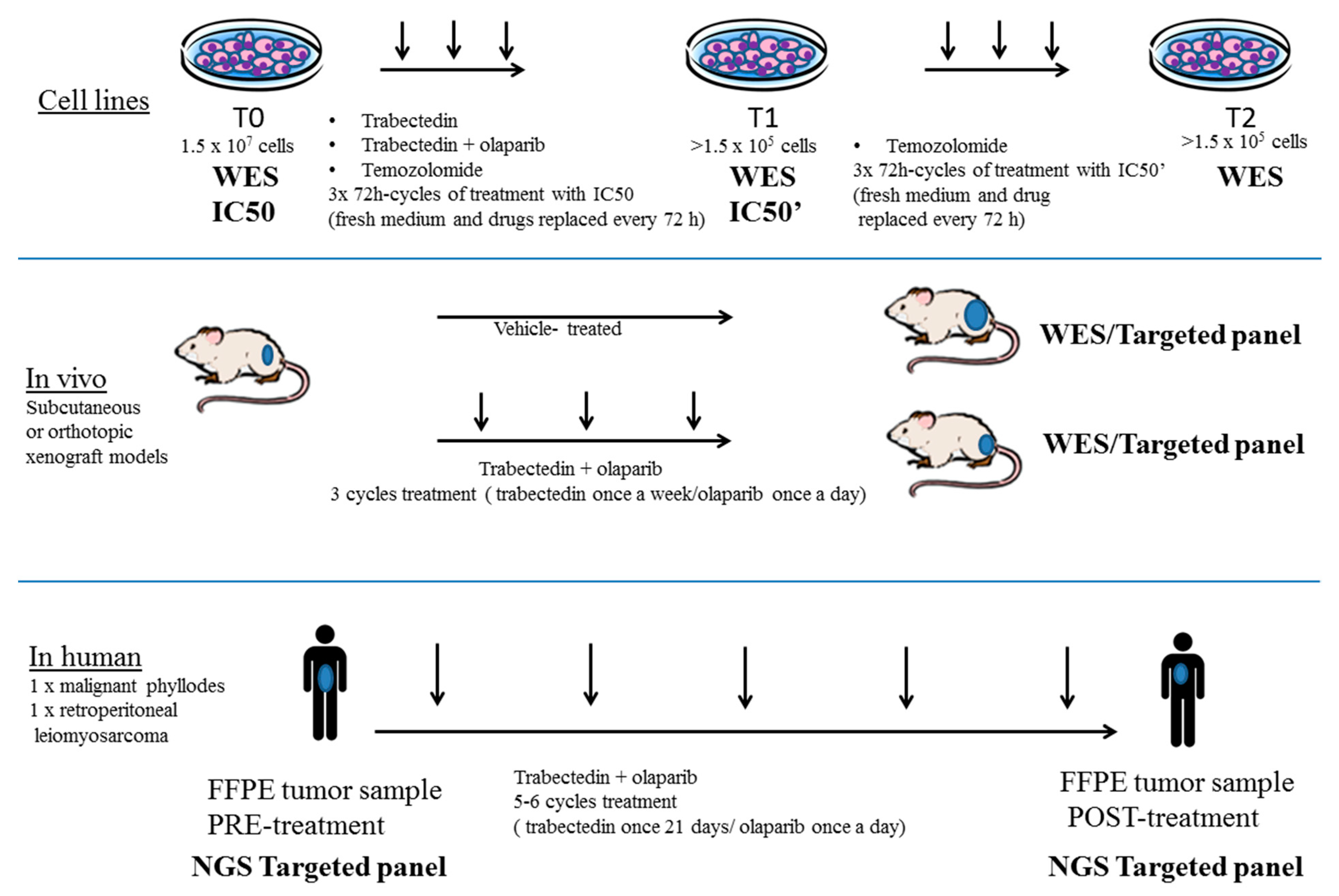
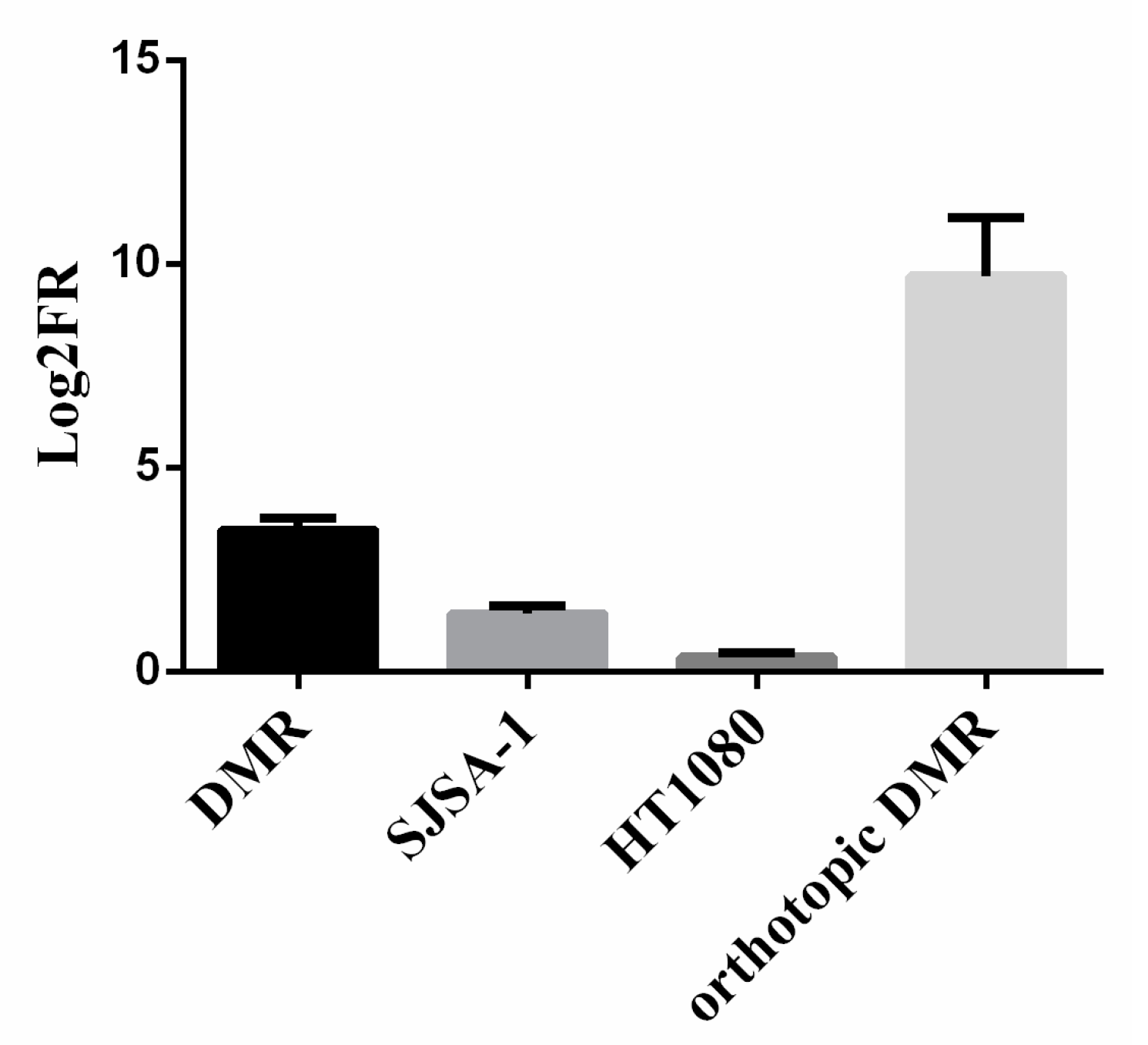
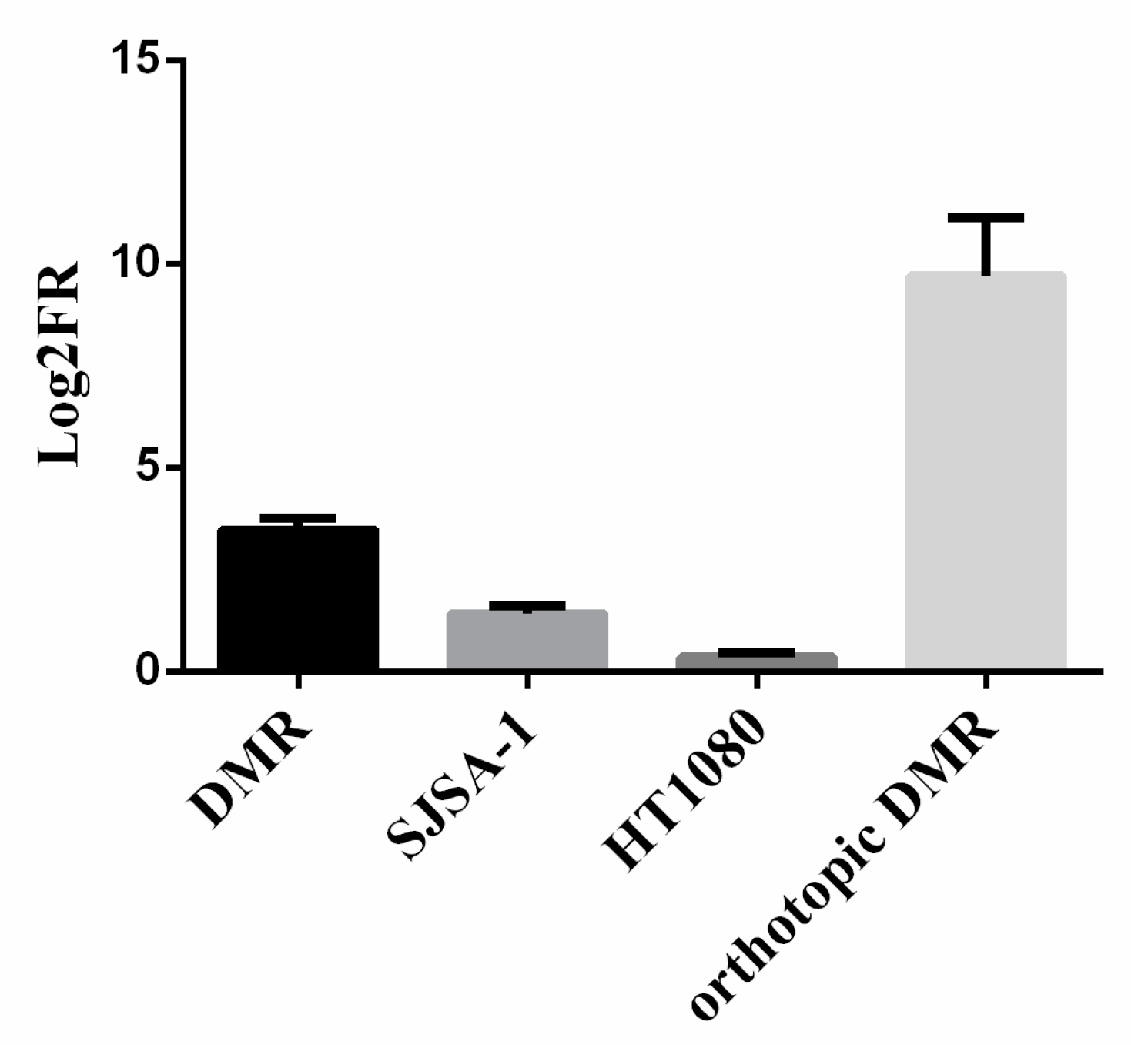
3.1. Generation of a Preclinical and Translational Platform of Bone and Soft Tissue Sarcoma Cell Lines, Xenografts and Tumor Samples from Patients Treated with Trabectedin and Olaparib
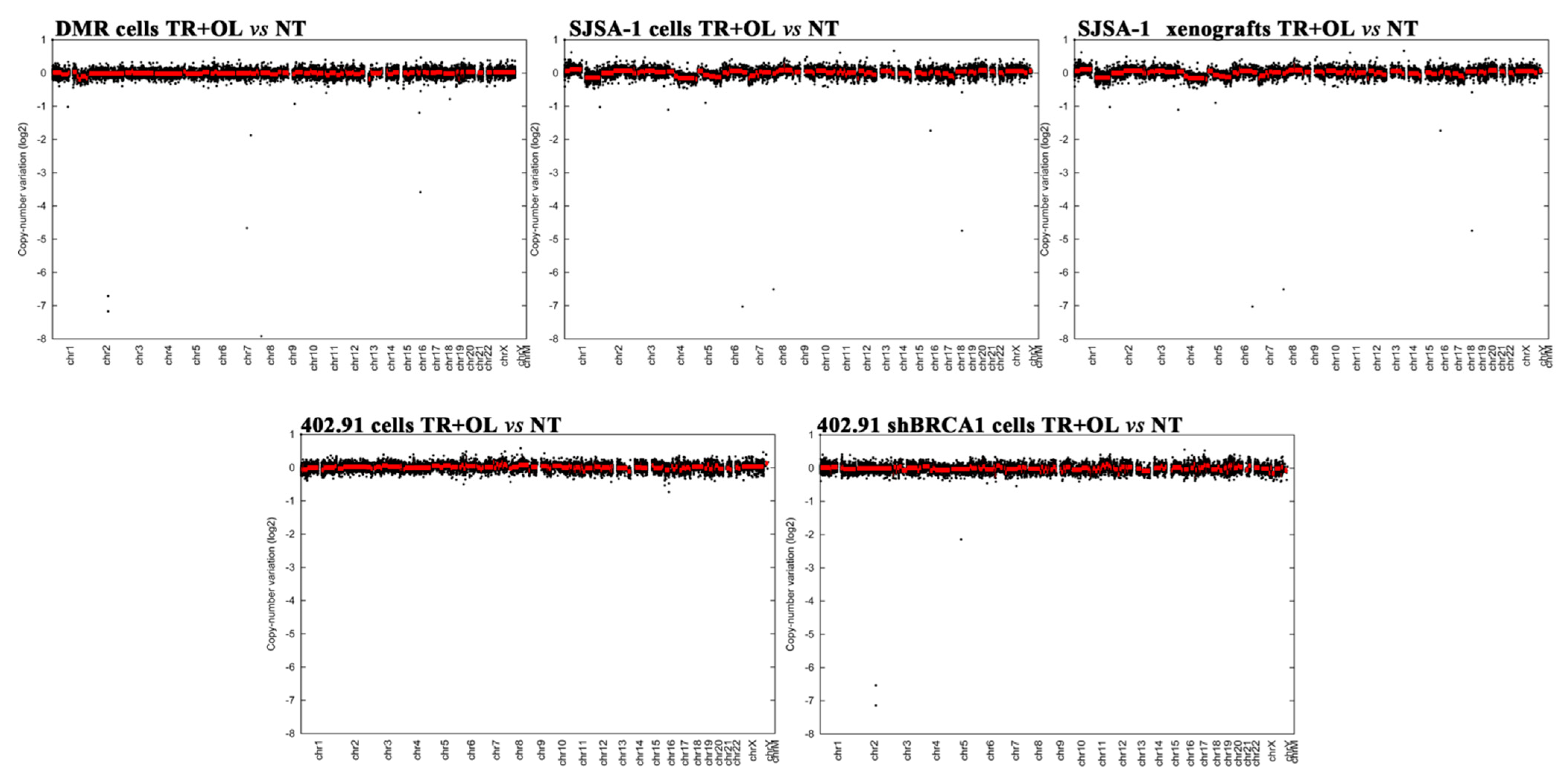
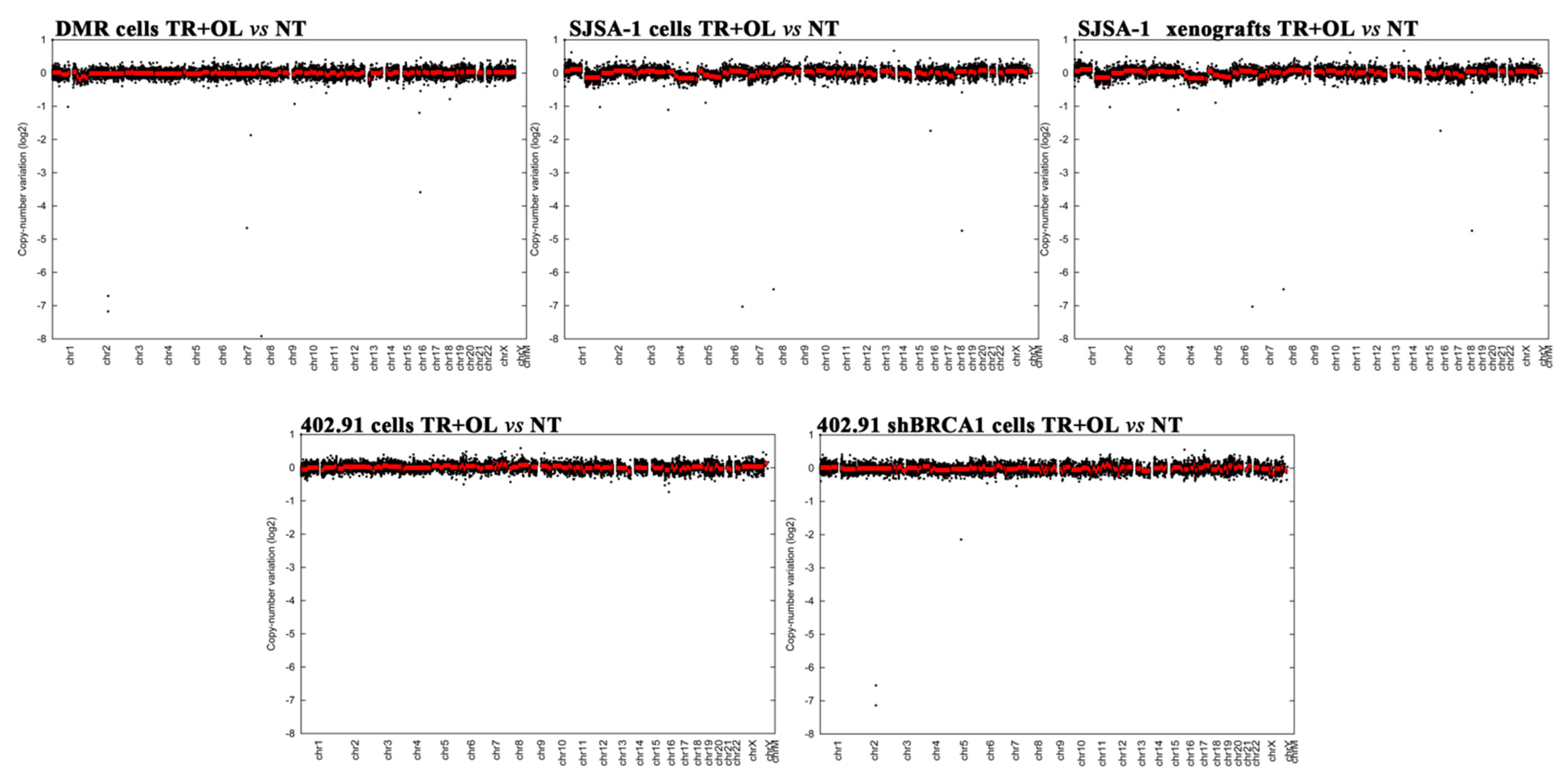
3.2. Genetic Analysis of DNA Damage Response and Repair (DDRR) Genes and Copy Number Alteration in a Preclinical and Translational Platform of Bone and Soft Tissue Sarcomas Treated with Trabectedin and Olaparib
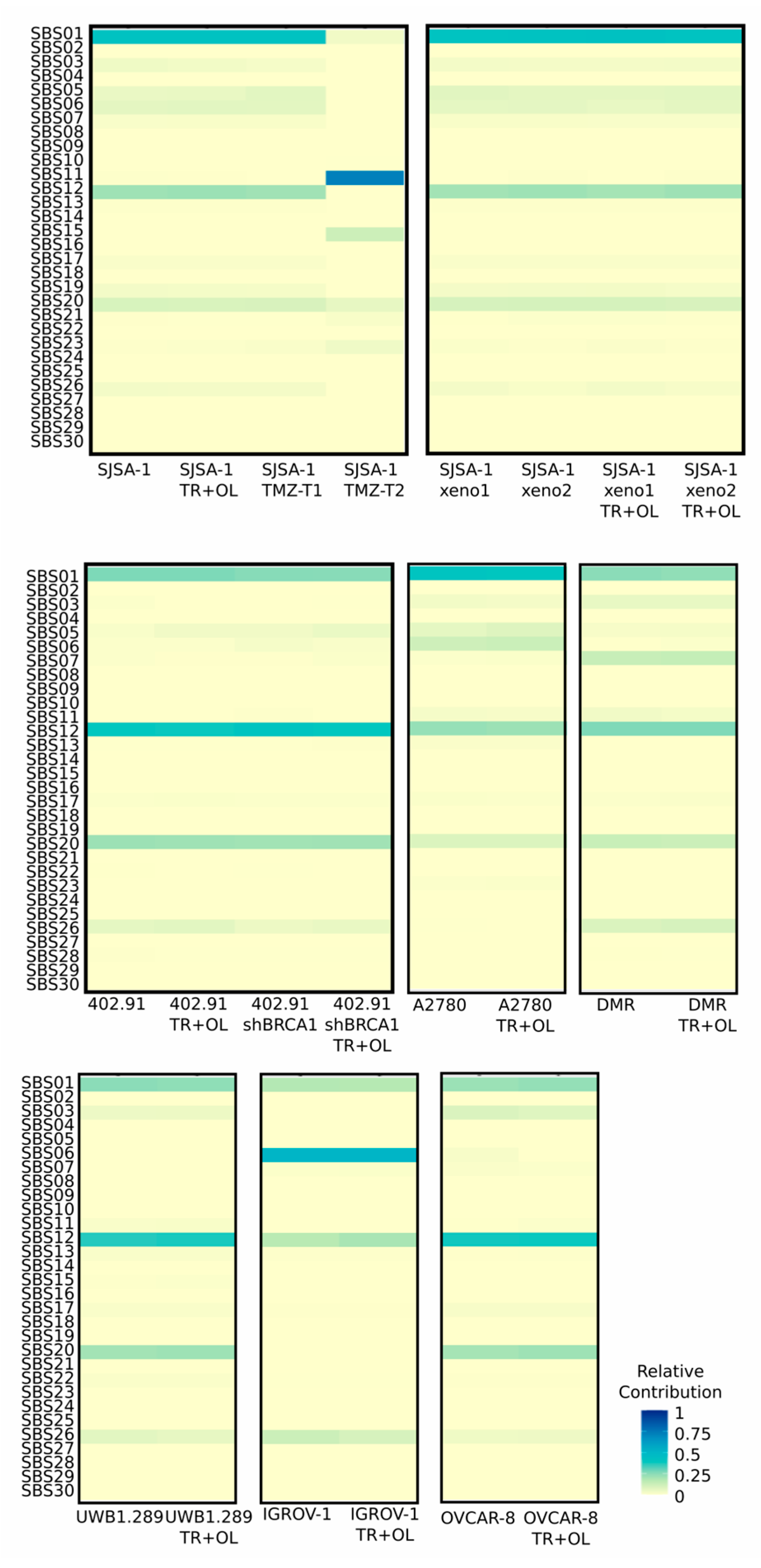
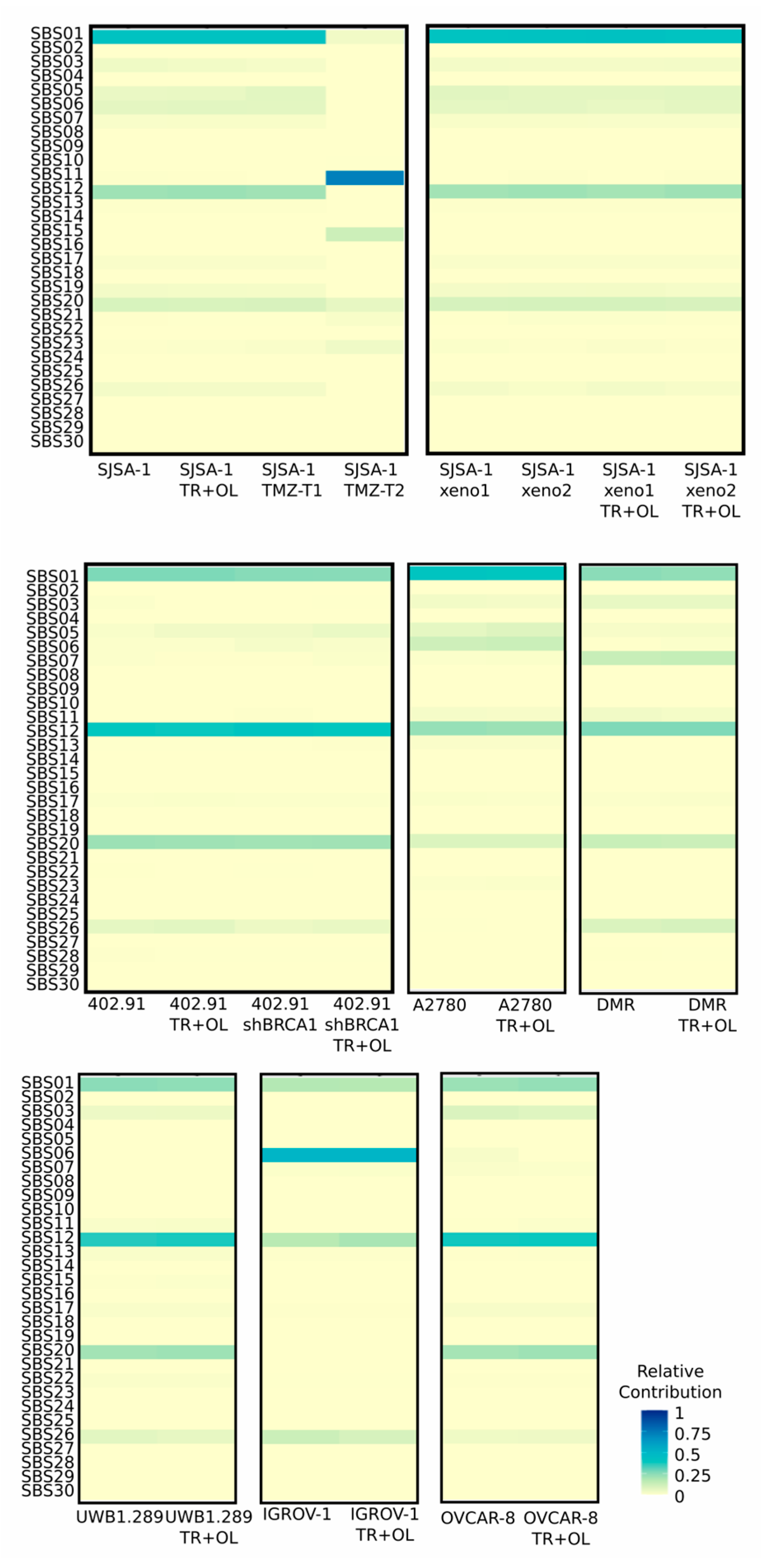
3.3. Treatment with Trabectedin and Olaparib Combination Did Not Influence Single-Base Substitution (SBS) Signatures
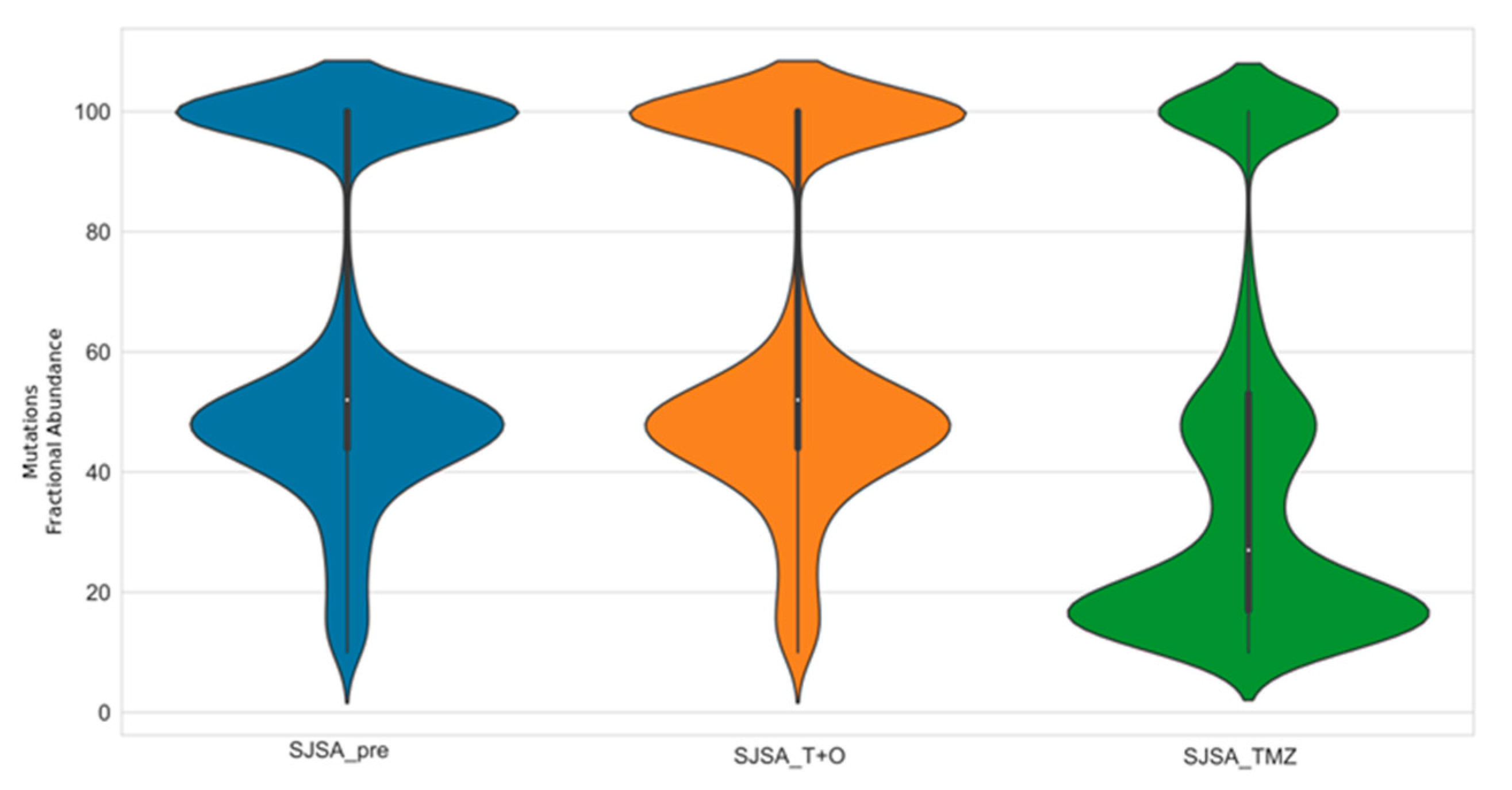
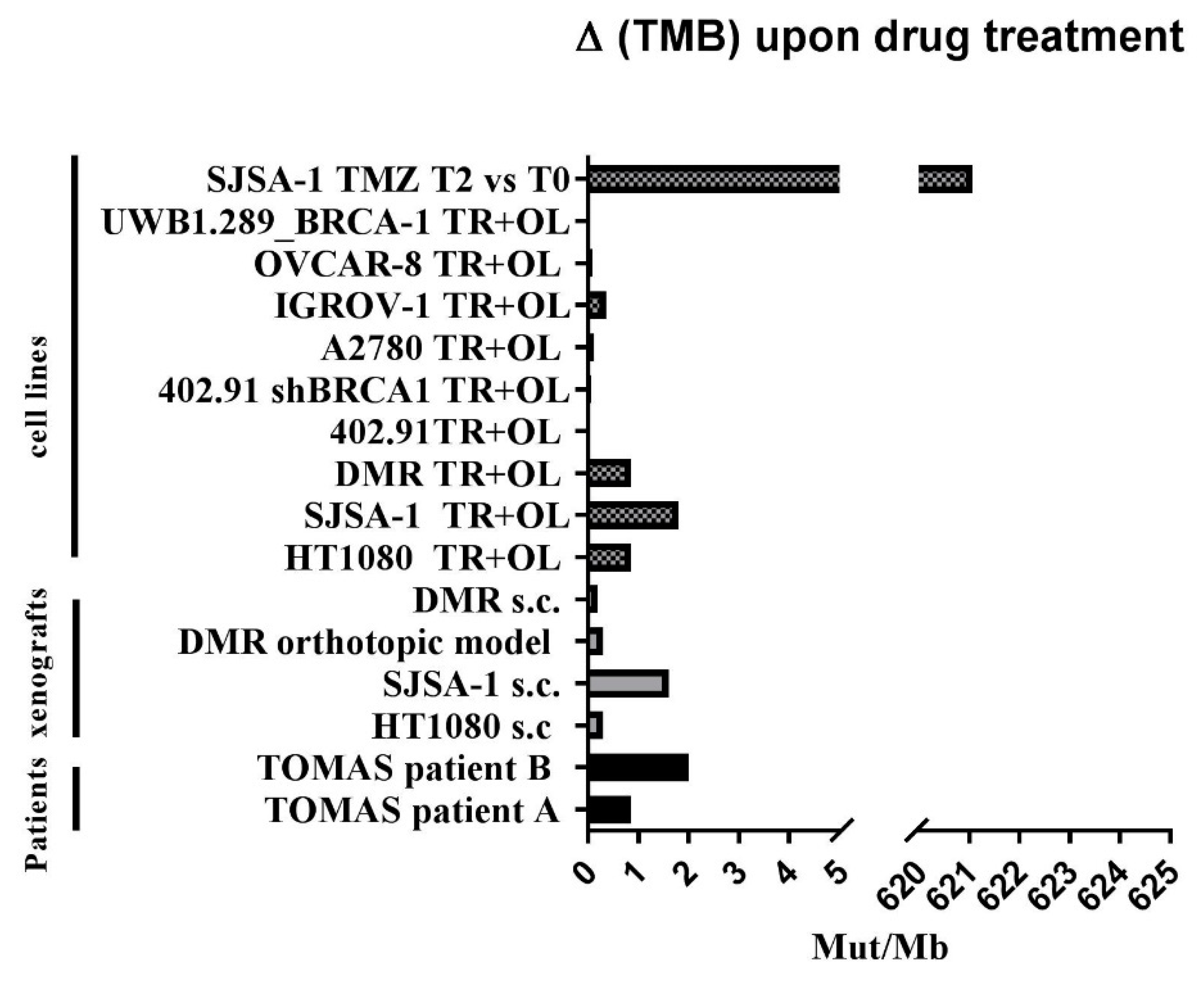
3.4. Trabectedin and Olaparib Treatment Did Not Impinge on Tumor Mutational Burden
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havel, J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Emma, J.; Taylor, M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Sharfman, W.H.; Bhatia, S.; Hwu, W.; Gajewski, T.F.; Craig, L. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D. Tumor Mutational Load Predicts Survival after Immunotherapy across Multiple Cancer Types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The Predictive Value of Tumor Mutation Burden on Efficacy of Immune Checkpoint Inhibitors in Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- VanAllen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes, M.H.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Mcgranahan, N.; Furness, A.J.S.; Rosenthal, R.; Lyngaa, R.; Saini, S.K.; Jamal-hanjani, M.; Gareth, A.; Birkbak, N.J.; Hiley, C.T.; Watkins, T.B.K.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Grignani, G.; Merlini, A.; Sangiolo, D.; Ambrosio, D.; Pignochino, Y. Delving into PARP Inhibition from Bench to Bedside and Back. Pharmacol. Ther. 2020, 206, 107446. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; Mcvey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase E-Mutated and Microsatellite-Instable Endometrial Cancers with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- VanGool, I.C.; Eggink, F.A.; Freeman-mills, L.; Stelloo, E.; De Bruyn, M.; Palles, C.; Nout, R.A.; De Kroon, C.D.; Elisabeth, M.; Nijman, H.W.; et al. POLE Proofreading Mutations Elicit an Anti-Tumor Immune Response in Endometrial Cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martì, J.; Fernandez-Cortés, M.; Serrano-Sáenz, S.; Zamudio-martinez, E.; Delgado-bellido, D.; Garcia-diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Final Analysis of a Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Morice, P.; Leary, A.; Dolladille, C.; Chrétien, B.; Poulain, L.; González-martín, A.; Moore, K.; Reilly, E.M.O.; Ray-coquard, I.; Alexandre, J. Myelodysplastic Syndrome and Acute Myeloid Leukaemia in Patients Treated with PARP Inhibitors: A Safety Meta-Analysis of Randomised Controlled Trials and a Retrospective Study of the WHO Pharmacovigilance Database. Lancet Haematol. 2021, 8, 122–134. [Google Scholar] [CrossRef]
- Ordóñez, J.L.; Amaral, A.T.; Carcaboso, A.M.; Herrero-Martín, D.; Del Carmen García-Macías, M.; Sevillano, V.; Alonso, D.; Pascual-Pasto, G.; San-Segundo, L.; Vila-Ubach, M.; et al. The PARP Inhibitor Olaparib Enhances the Sensitivity of Ewing Sarcoma to Trabectedin. Oncotarget 2015, 6, 18875–18890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laroche, A.; Chaire, V.; Le Loarer, F.; Algéo, M.P.; Rey, C.; Tran, K.; Lucchesi, C.; Italiano, A. Activity of Trabectedin and the PARP Inhibitor Rucaparib in Soft-Tissue Sarcomas. J. Hematol. Oncol. 2017, 10, 84. [Google Scholar] [CrossRef]
- Pignochino, Y.; Capozzi, F.; D’Ambrosio, L.; Dell’Aglio, C.; Basiricò, M.; Canta, M.; Lorenzato, A.; Vignolo Lutati, F.; Aliberti, S.; Palesandro, E.; et al. PARP1 Expression Drives the Synergistic Antitumor Activity of Trabectedin and PARP1 Inhibitors in Sarcoma Preclinical Models. Mol. Cancer 2017, 16, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grignani, G.; Ambrosio, L.D.; Pignochino, Y.; Palmerini, E.; Zucchetti, M.; Boccone, P.; Aliberti, S.; Paolo, A.; Tos, D.; Novara, L.; et al. Trabectedin and Olaparib in Patients with Advanced and Non-Resectable Bone and Soft-Tissue Sarcomas (TOMAS): An Open-Label, Phase 1b Study from the Italian Sarcoma Group. Lancet Oncol. 2018, 19, 1360–1371. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B Cells Are Associated with Survival and Immunotherapy Response in Sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Corti, G.; Bartolini, A.; Crisafulli, G.; Novara, L.; Rospo, G.; Montone, M.; Negrino, C.; Mussolin, B.; Buscarino, M.; Isella, C.; et al. A Genomic Analysis Workflow for Colorectal Cancer Precision Oncology. Clin. Colorectal. Cancer 2019, 18, 91–101. [Google Scholar] [CrossRef]
- Crisafulli, G.; Mussolin, B.; Cassingena, A.; Montone, M.; Bartolini, A.; Barault, L.; Martinetti, A.; Morano, F.; Pietrantonio, F.; Sartore-Bianchi, A.; et al. Whole Exome Sequencing Analysis of Urine Trans-Renal Tumour DNA in Metastatic Colorectal Cancer Patients. ESMO Open 2019, 4, e000572. [Google Scholar] [CrossRef]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Williams, E.D.; Beresford-Smith, B. Xenome—A Tool for Classifying Reads from Xenograft Samples. Bioinformatics 2012, 28, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Svensson, F.; Lang, T.; Johansson, M.E.V.; Hansson, G.C. The Central Exons of the Human MUC2 and MUC6 Mucins Are Highly Repetitive and Variable in Sequence between Individuals. Sci. Rep. 2018, 8, 17503. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; Sarotto, I.; et al. Adaptive Mutability of Colorectal Cancers in Response to Targeted Therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Siravegna, G.; Lazzari, L.; Crisafulli, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Martino, C.; Lanman, R.B.; Nagy, R.J.; Fairclough, S.; et al. Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Article Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell 2018, 34, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A Tool for Visualizing and Exploring Patterns of Small Mutational Events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; We, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Gay, M.; Vila-Casadesús, M.; Franch-Expósito, S.; Hernández-Illán, E.; Lozano, J.J.; Castellví-Bel, S. Mutational Signatures in Cancer (MuSiCa): A Web Application to Implement Mutational Signatures Analysis in Cancer Samples. BMC Bioinform. 2018, 19, 224. [Google Scholar] [CrossRef] [Green Version]
- Blokzijl, F.; Janssen, R.; Van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive Genome-Wide Analysis of Mutational Processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.C.; Renwick, P.J.; Dal Cin, P.; Van den Berghe, H.; Fletcher, C.D. Translocation t(12;16)(q13;p11) in myxoid liposarcoma and round cell liposarcoma: Molecular and cytogenetic analysis. Cancer Res. 1995, 55, 24–27. [Google Scholar] [PubMed]
- Turc-Carel, C.; Aurias, A.; Mugneret, F.; Lizard, S.; Sidaner, I.; Volk, C.; Thiery, J.P.; Olschwang, S.; Philip, I.; Berger, M.P. Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet. Cytogenet. 1988, 32, 229–238. [Google Scholar] [CrossRef]
- Snyder, A.; Chan, T.A. Immunogenic peptide discovery in cancer genomes. Curr. Opin Genet. Dev. 2015, 30, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Bessede, A.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.A.; Guegan, J.; et al. PD1 Inhibition in Soft-Tissue Sarcomas with Tertiary Lymphoid Structures: A Multicenter Phase II Trial. J. Clin. Oncol. 2021, 39 (Suppl. 15), 11507. [Google Scholar] [CrossRef]
- Toulmonde, M.; Brahmi, M.; Giraud, A.; Bessede, A.; Kind, M.; Toulza, E.; Pulido, M.; Cousin, S.; Bourcier, K.; Albert, S.; et al. TRAMUNE, a Phase Ib Study Combining Trabectedin and Durvalumab, Results of the Expansion Cohort in Patients with Advanced Pretreated Soft Tissue Sarcomas. Ann. Oncol. 2020, 31, S1199. [Google Scholar] [CrossRef]
- Blay, J.Y.; Penel, N.; Ray-Coquard, I.L.; Cousin, S.; Bertucci, F.; Bompas, E.; Eymard, J.C.; Saada-Bouzid, E.; Soulie, P.; Boudou-Rouquette, P.; et al. High Clinical Activity of Pembrolizumab in Chordoma, Alveolar Soft Part Sarcoma (ASPS) and Other Rare Sarcoma Histotypes: The French AcSé Pembrolizumab Study from Unicancer. J. Clin. Oncol. 2021, 39, 11520. [Google Scholar] [CrossRef]
- Hindi, N.; Rosenbaum, E.; Jonczak, E.; Hamacher, R.; Rutkowski, P.; Skryd, A.; Connolly, E.; Blay, J.Y.; Gutierrez, A.; Bogefors, C.; et al. Retrospective World-Wide Registry on the Efficacy of Immune Checkpoint Inhibitors in Alveolar Soft Part Sarcoma: Updated Results from Sixty Patients. J. Clin. Oncol. 2021, 39, 11564. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The Role of PD-L1 Expression as a Predictive Biomarker: An Analysis of All US Food and Drug Administration (FDA) Approvals of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and Anti-Inflammatory Effects of Trabectedin on Human Myxoid Liposarcoma Cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11, eaax7918. [Google Scholar] [CrossRef] [PubMed]
- Gejman, R.S.; Chang, A.Y.; Jones, H.F.; DiKun, K.; Hakimi, A.A.; Schietinger, A.; Scheinberg, D.A. Rejection of immunogenic tumor clones is limited by clonal fraction. Elife 2018, 7, e41090. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Lhuillier, C.; Rudqvist, N.P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu Rev. Immunol 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Zapata, I.; Córdoba, S.; Jimeno, J.M.; López-Martín, J.A.; Tercero, J.C.; De La Torre, A.; Vargas, J.A.; Molerón, R.; Sánchez-Prieto, R. In vitro radiosensitisation by trabectedin in human cancer cell lines. Eur. J. Cancer 2008, 44, 1726–1733. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Lopez-Pousa, A.; Peinado-Serrano, J.; Alvarez, R.; Alvarez-Gonzalez, A.; Italiano, A.; Sargos, P.; Cruz-Jurado, J.; Isern-Verdum, J.; et al. Assessment of Safety and Efficacy of Combined Trabectedin and Low-Dose Radiotherapy for Patients with Metastatic Soft-Tissue Sarcomas: A Nonrandomized Phase 1/2 Clinical Trial. JAMA Oncol. 2020, 6, 535–541. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.A.; Veldwijk, M.R.; Herskind, C.; Wenz, F. Radiotherapy, tumor mutational burden, and immune checkpoint inhibitors: Time to do the math. Strahlenther. Onkol. 2018, 194, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Plummer, R.; Jones, C.; Middleton, M.; Wilson, R.; Evans, J.; Olsen, A.; Curtin, N.; Boddy, A.; Mchugh, P.; Newell, D.; et al. Phase I Study of The Poly (ADP-Ribose) Polymerase Inhibitor, AG014699, In Combination with Temozolomide in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2008, 14, 7917–7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
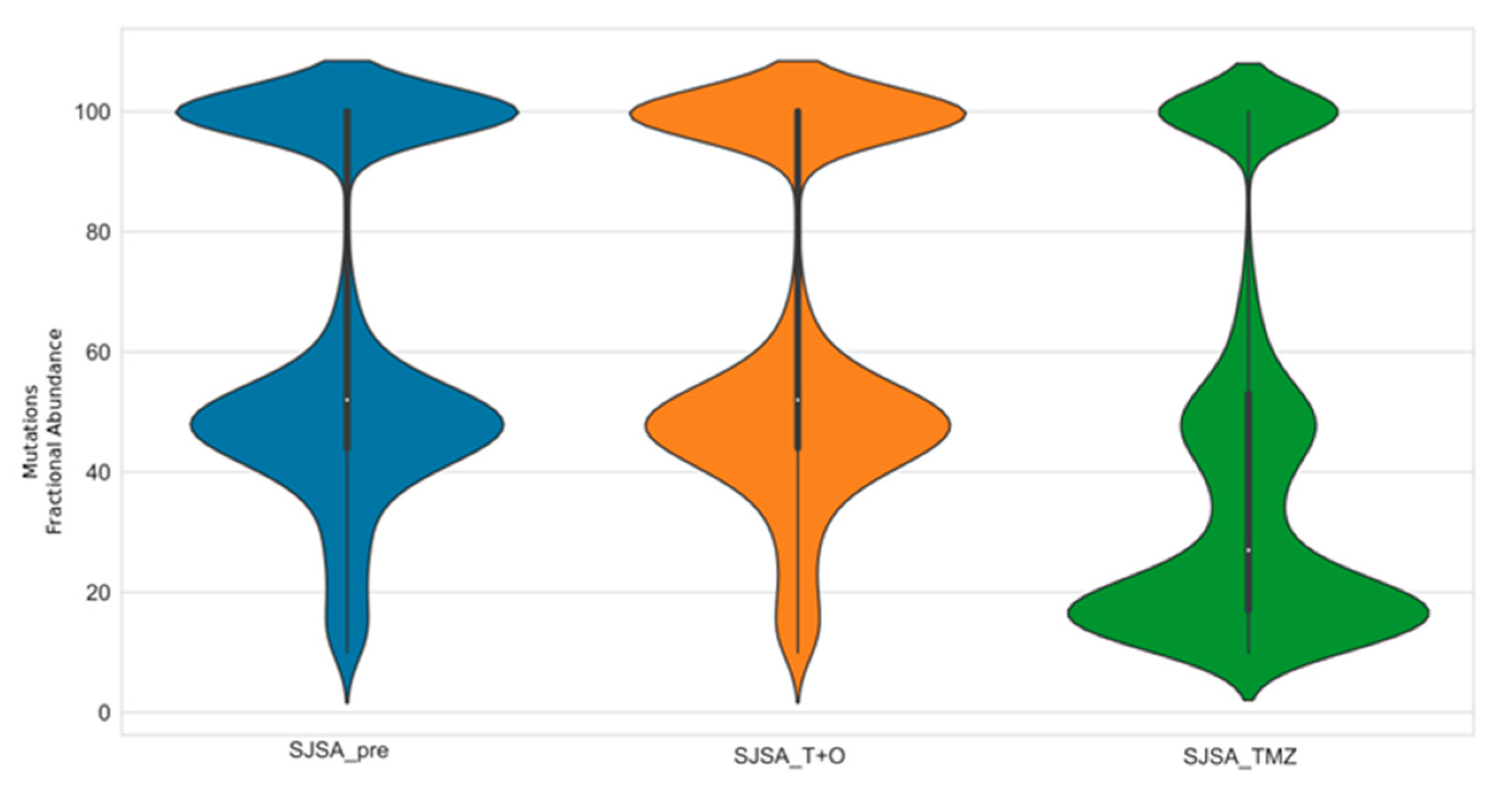{kind=link}
| Cell Line | IC50 Trabectedin (95% CI) nM | IC50 Olaparib (95% CI) µM | Combination Combination Index ± Est. SD | ||
|---|---|---|---|---|---|
| as SINGLE Agent | in Combination with Olaparib | as Single Agent | in Combination with Trabectedin | ||
| DMR | 0.57 (0.45–0.64) | 0.13 (0.12–0.14) | 9.02 (8.11–10.23) | 1.30 (1.18–1.43) | 0.47 ± 0.06 |
| SJSA-1 | 0.25 (0.16–0.39) | 0.18 (0.15–0.19) | 14.55 (6.66–29.64) | 1.80 (1.52–1.93) | 0.86 ± 0.11 |
| HT-1080 | 0.75 (0.68–0.81) | 0.60 (0.59–0.64) | >20 | 6.04 (5.92–6.42) | 0.89 ± 0.15 |
| 402.91 | 0.46 (0.37–0.54) | 0.12 (0.11–0.14) | 7.12 (5.53–7.91) | 1.84 (1.37–2.68) | 0.50 ± 0.12 |
| 402.91 shBRCA1 | 0.19 (0.12–0.28) | 0.14 (0.04–0.53) | 2.92 (2.19–3.89) | 1.40 (0.38–5.27) | 1.24 ± 0.22 |
| A2780 | 0.41 (0.29–0.58) | 0.20 (0.12–0.32) | 8.73 (7.22–10.55) | 1.97 (1.22–3.18) | 0.7 ± 0.11 |
| UWB1.289 BRCA1 | 0.44 (0.30–0.66) | 0.41 (0.37–0.47) | 10.36 (5.37–19.98) | 4.18 (3.67–4.75) | 1.35 ± 0.17 |
| OVCAR-8 | 0.38 (0.12–1.20) | 0.16 (0.04–0.70) | 9.77 (6.06–15.74) | 1.62 (0.37–7.04) | 0.60 ± 0.18 |
| IGROV-1 | 0.62 (0.39–0.99) | 0.326 (0.18–0.58) | 21.04 (13.59–32.57) | 3.26 (1.82–5.85) | 0.68 ± 0.14 |
| Cell Line | IC50 Temozolomide (95% CI) μM |
|---|---|
| DMR | 594 (499–706) |
| HT-1080 | 489 (416–575) |
| 402.91 | 151 (133–172) |
| 402.91 shBRCA1 | 242 (208–281) |
| SJSA-1 | 677 (486–943) |
| SJSA-1 T1 | 1499 (1051–2137) |
| Pathway | Gene | Base Change | Amino Acid Change | Mean Allelic Frequency | ||||
| SJSA-1 Untreated Cells | SJSA-1 TR + OL Treated Cells | SJSA-1 Untreated Xenografts | SJSA-1 TR + OL Treated Xenografts | |||||
| BER | MUTYH | c.T538C | p.Y180H | 48.76 | 50.00 | 39.67 | 39.58 | |
| NEIL1 | c.C826A | p.P276T | 48.28 | 36.73 | 52.94 | 40.60 | ||
| NEIL2 | c.C307T | p.R103W | 65.67 | 63.89 | 66.47 | 67.76 | ||
| DDR | SENP7 | c.G1909T | p.D637Y | 48.60 | 49.21 | 48.34 | 46.57 | |
| INO80D | c.C2876G | p.S959C | 49.05 | 44.97 | 45.58 | 44.19 | ||
| UBA7 | c.A2450G | p.H817R | 44.05 | 50.00 | 48.55 | 57.70 | ||
| UBR5 | c.G3850A | p.A1284T | 46.02 | 43.45 | 49.16 | 53.05 | ||
| YY1AP1 | c.C385T | p.L129F | 40.85 | 42.10 | 48.72 | 41.65 | ||
| HRFA | CNTROB | c.G605T | p.R202L | 45.34 | 55.05 | 44.74 | 44.67 | |
| CNTROB | c.G1316A | p.R439Q | 43.91 | 51.23 | 48.23 | 44.21 | ||
| DDX11 | c.C1040A | p.A347D | 37.70 | 42.21 | 38.87 | 34.82 | ||
| FANCD2 | c.T1367G | p.L456R | 26.66 | 25.67 | 25.12 | 24.77 | ||
| FANCD2 | c.A1868C | p.Q623P | 32.46 | 29.70 | 36.02 | 31.80 | ||
| MCMDC2 | c.C1570T | p.P524S | 28.06 | 29.59 | 29.16 | 31.35 | ||
| NSMCE2 | c.T377G | p.F126C | 45.10 | 47.71 | 52.83 | 45.60 | ||
| SLX4 | c.A1637G | p.Y546C | 53.01 | 51.25 | 47.00 | 52.70 | ||
| SLX4IP | c.G779A | p.G260E | 46.67 | 38.86 | 41.86 | 48.35 | ||
| MMR | RFC1 | c.G2076C | p.E692D | 100.00 | 98.00 | 100.00 | 98.72 | |
| NER | XPC | c.T860G | p.F287C | 53.38 | 46.67 | 50.49 | 49.13 | |
| BIVM-ERCC5 | c.G2003A | p.R668H | 17.62 | 15.28 | 13.29 | 13.71 | ||
| Pathway | Gene | Base Change | Amino Acid Change | Mean Allelic Frequency | ||||
| DMR Untreated Cells | DMR TR + OL Treated Cells | |||||||
| DDR | UPF1 | c.G2204A | p.G735D | 100.00 | 98.56 | |||
| TRRAP | c.C2165T | p.S722F | 17.62 | 15.85 | ||||
| NER | DCAF4L2 | c.G250T | p.D84Y | 53.38 | 47.24 | |||
| Pathway | Gene | Base Change | Amino Acid Change | Mean Allelic Frequency | ||||
| TOMAS Untreated Patient B | TOMAS TR + OL Treated Patient B | |||||||
| DDR | TP53 | c.G248A | p.S215N | 42.44 | 47.76 | |||
| HRFA | WRN | c.G250T | p.D84Y | <10% | 15.38 | |||
| Pathway | Gene | Base Change | Amino Acid Change | Mean Allelic Frequency | ||||
| 402.91 Untreated Cells | 402.91 TR + OL Treated Cells | 402.91 shBRCA1 Untreated Cells | 402.91 shBRCA1 TR + OL Treated Cells | |||||
| DDR | CINP | c.C159G | p.N53K | 49.26 | 52.54 | 46.71 | 51.45 | |
| HERC2 | c.A32G | p.Q11R | 0 | 27.78 | 11.76 | 16.67 | ||
| HUS1B | c.C390A | p.H130Q | 51.36 | 45.11 | 55.67 | 54.41 | ||
| SENP7 | c.A1836T | p.Q612H | 53.73 | 54.55 | 56.67 | 52.54 | ||
| HRFA | DDX11 | c.G1819C | p.A607P | 9.38 | 5.66 | 14.15 | 10.16 | |
| DDX11 | c.C556T | p.R186W | 16.39 | 19.12 | 16.05 | 13.68 | ||
| NBN | c.A511G | p.I171V | 50.43 | 56.96 | 48.81 | 46 | ||
| PARP4 | c.G2695A | p.A899T | 48.89 | 33.66 | 39.71 | 48.91 | ||
| SLX4 | c.C3812T | p.S1271F | 49.80 | 45.28 | 52.89 | 51.01 | ||
| MMR | MSH4 | c.A1766G | p.Y589C | 52.75 | 53.42 | 39.33 | 35.19 | |
| NER | DCAF4 | c.C1034G | p.S345C | 45.88 | 45.38 | 40.56 | 41.95 | |
| MC1R | c.C478T | p.r160W | 99.22 | 85.29 | 58.24 | 59.83 | ||
| c.G178T | p.V60L | 0 | 9.80 | 40.56 | 38.36 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignochino, Y.; Crisafulli, G.; Giordano, G.; Merlini, A.; Berrino, E.; Centomo, M.L.; Chiabotto, G.; Brusco, S.; Basiricò, M.; Maldi, E.; et al. PARP1 Inhibitor and Trabectedin Combination Does Not Increase Tumor Mutational Burden in Advanced Sarcomas—A Preclinical and Translational Study. Cancers 2021, 13, 6295. https://doi.org/10.3390/cancers13246295
Pignochino Y, Crisafulli G, Giordano G, Merlini A, Berrino E, Centomo ML, Chiabotto G, Brusco S, Basiricò M, Maldi E, et al. PARP1 Inhibitor and Trabectedin Combination Does Not Increase Tumor Mutational Burden in Advanced Sarcomas—A Preclinical and Translational Study. Cancers. 2021; 13(24):6295. https://doi.org/10.3390/cancers13246295
Chicago/Turabian StylePignochino, Ymera, Giovanni Crisafulli, Giorgia Giordano, Alessandra Merlini, Enrico Berrino, Maria Laura Centomo, Giulia Chiabotto, Silvia Brusco, Marco Basiricò, Elena Maldi, and et al. 2021. "PARP1 Inhibitor and Trabectedin Combination Does Not Increase Tumor Mutational Burden in Advanced Sarcomas—A Preclinical and Translational Study" Cancers 13, no. 24: 6295. https://doi.org/10.3390/cancers13246295
APA StylePignochino, Y., Crisafulli, G., Giordano, G., Merlini, A., Berrino, E., Centomo, M. L., Chiabotto, G., Brusco, S., Basiricò, M., Maldi, E., Pisacane, A., Leuci, V., Sangiolo, D., D’Ambrosio, L., Aglietta, M., Kasper, B., Bardelli, A., & Grignani, G. (2021). PARP1 Inhibitor and Trabectedin Combination Does Not Increase Tumor Mutational Burden in Advanced Sarcomas—A Preclinical and Translational Study. Cancers, 13(24), 6295. https://doi.org/10.3390/cancers13246295