
RKIP Pleiotropic Activities in Cancer and Inflammatory Diseases: Role in Immunity




Abstract
Simple Summary
Abstract
1. Introduction
2. RKIP Structure
3. RKIP Functions on Cell Signaling
3.1. RKIP-Mediated Inhibition of the Raf-1/MEK/ERK Pathway
3.2. Inhibition of the NF-kB Pathway
3.3. RKIP-Mediated Inhibition of GRK2
3.4. RKIP-Mediated Inhibition of STAT3 Activation
3.5. Regulation of GSK3β Signaling
3.6. Regulation of the Spindle Checkpoint by RKIP
3.7. Clinical Significance of pRKIP in Various Cancers
3.7.1. pRKIP Expression Correlates with Good Prognosis
3.7.2. pRKIP Expression Correlates with Poor Prognosis
4. Regulation of RKIP Expression
4.1. SNAIL
4.2. BACH1
4.3. SP1, CREB, p300, AR
4.4. c-MET
4.5. MMPs
4.6. EZH2
4.7. Methylation
4.8. miRNAs
4.9. PKC
4.10. XIST
5. Effects of RKIP Expression Levels in Various Cancers
5.1. Adenocarcinomas
5.2. Colon Cancer
5.3. Prostate Cancer
5.4. Pancreatic Cancer
5.5. Gliomas
5.6. Renal Cell Carcinoma
5.7. Gastric Cancer
5.8. Lung Cancer
5.9. Leukemia
5.10. Multiple Myeloma
5.11. Other Cancers
6. RKIP–Immune System Cross-Talks
6.1. RKIP and T Cell Function
6.2. RKIP and Cytokine/Interferon Secretion Patterns
6.3. RKIP and Apoptosis
6.4. RKIP and Inhibitory T-Cell Receptors (Immune Checkpoints)
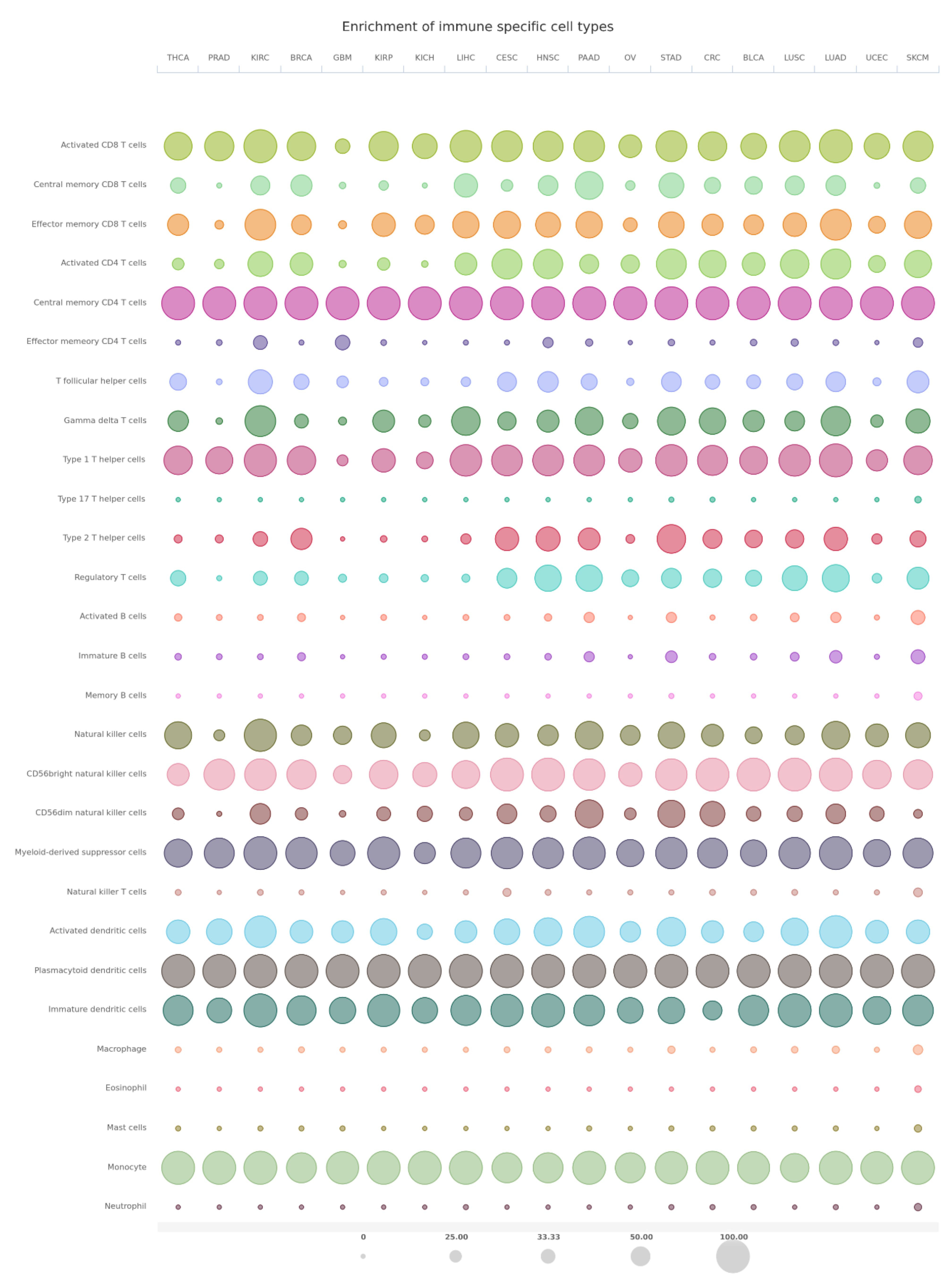
7. Bioinformatics Analyses
8. RKIP in Inflammatory Diseases
8.1. SIRS
8.2. AITP
9. Discussion and Perspectives
10. Induction of RKIP
10.1. Targeting SNAIL
10.2. Targeting BACH1
10.3. Targeting EZH2
- (1)
- Inhibitors of EZH-2 methyltransferase activity.
- (2)
- Inhibitors that break PCR2 structure
- (3)
- Triggering EZH2 degradation
- (4)
- EZH2 inhibitors combined with other therapies
11. Inhibition of RKIP
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | androgen deprivation therapy |
| APCs | antigen-presenting cells |
| AR | androgen receptor |
| ARE | androgen responsive element |
| B-AR | beta-adrenergic receptor |
| BACH1 | BTB and CNC homology 1 |
| Bcl-2 | B-cell lymphoma 2 |
| Bp | base pairs |
| c-FLIP | FLICE-inhibitory protein |
| cAMP | cyclic adenosine monophosphate |
| CD | cluster of differentiation |
| CDK5 | cyclin dependent kinase 5 |
| ceRNA | competing endogenous RNA |
| CREB | cyclic adenosine monophosphate response element-binding protein |
| DC | dendritic cell |
| Dlg | discs-large tumor suppressor |
| DR5 | death receptor 5 |
| E-cad | E-cadherin |
| EGCG | epigallocatechin 3-gallate |
| EMT | epithelial-to-mesenchymal transition |
| EZH2 | enhancer of Zeste homolog 2 |
| FasL | Fas ligand |
| FLICE | FADD-like IL-1β-converting enzyme |
| GPCRs | G-protein-coupled receptors |
| GRK2 | G protein-coupled receptor kinase 2 |
| GRK2 | GPCR kinase 2 |
| GSK3β | Glycogen synthase kinase 3 |
| HDAC | histone deacetylase |
| HHC | human hepatoma |
| HSC | hepatic stellate cells |
| IKK | IkB kinase |
| ITP | immune thrombocytopenic purpura |
| JAK 1/2 | Janus kinase 1/2 |
| JNK | Jun N-terminal kinase |
| lncRNA | long non-coding RNA |
| LPS | lipopolysaccharide |
| MAPK | MAP kinase |
| MDA-9 | melanoma differentiation associated gene 9 |
| mFas | membrane Fas |
| mFasL | membrane FasL |
| MREs | miRNA recognition elements |
| NIK | NF-kB-inducing kinase |
| NPC | nasopharyngeal carcinoma |
| NSCLC | non-small-cell lung cancer |
| PCa | prostate cancer-associated |
| PcG | polycomb group |
| PEBP | phosphatidylethanolamine-binding protein |
| PKC | protein kinase C |
| PRC2 | polycomb repressive complex 2 |
| PSD-95 | postsynaptic density protein |
| RKIP | Raf kinase inhibitory protein |
| RTKs | receptor tyrosine kinases |
| S153 | Serine 153 |
| SEA | staphylococcal enterotoxin A |
| SLE | systemic lupus erythematosus |
| Sp1 | specificity protein 1 |
| STAT | signal transducer and activator of transcription |
| TAK-1 | transforming growth factor B-activated kinase-1 |
| TCR | T cell antigen receptor |
| TILs | Tumor-infiltrating lymphocytes |
| TNBC | triple-negative breast cancer |
| TSA | trichostatin A |
| XIAP | X-linked inhibitor of apoptosis protein |
| XIST | X-inactive specific transcript |
| YY1 | Yin Yang 1 |
| ZO-1 | tight junction protein-1 |
References
- Escara-Wilke, J.; Yeung, K.; Keller, E.T. Raf kinase inhibitor protein (RKIP) in cancer. Cancer Metastasis Rev. 2012, 31, 615–620. [Google Scholar] [CrossRef]
- Bernier, I.; Jolles, P. Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim. Biophys. Acta. 1984, 790, 174–181. [Google Scholar] [CrossRef]
- Perry, A.C.; Hall, L.; Bell, A.E.; Jones, R. Sequence analysis of a mammalian phospholipid-binding protein from testis and epididymis and its distribution between spermatozoa and extracellular secretions. Biochem. J. 1994, 301 Pt 1, 235–242. [Google Scholar] [CrossRef]
- Grandy, D.K.; Hanneman, E.; Bunzow, J.; Shih, M.; Machida, C.A.; Bidlack, J.M.; Civelli, O. Purification, cloning, and tissue distribution of a 23-kDa rat protein isolated by morphine affinity chromatography. Mol. Endocrinol. 1990, 4, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Bollengier, F.; Mahler, A. Localization of the novel neuropolypeptide h3 in subsets of tissues from different species. J. Neurochem. 1988, 50, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Hori, N.; Chae, K.S.; Murakawa, K.; Matoba, R.; Fukushima, A.; Okubo, K.; Matsubara, K. A human cDNA sequence homologue of bovine phosphatidylethanolamine-binding protein. Gene 1994, 140, 293–294. [Google Scholar] [PubMed]
- Seddiqi, N.; Bollengier, F.; Alliel, P.M.; Perin, J.P.; Bonnet, F.; Bucquoy, S.; Jolles, P.; Schoentgen, F. Amino acid sequence of the Homo sapiens brain 21–23-kDa protein (neuropolypeptide h3), comparison with its counterparts from Rattus norvegicusand Bos taurus species, and expression of its mRNA in different tissues. J. Mol. Evol. 1994, 39, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Skinner, J.J.; Rosner, M.R. RKIP structure drives its function: A three-state model for regulation of RKIP. Crit. Rev. Oncog. 2014, 19, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, A.E.; Clark, M.C.; McElheny, D.; Heil, G.; Hong, J.; Liu, X.; Kim, Y.; Joachimiak, G.; Joachimiak, A.; Koide, S.; et al. Raf kinase inhibitory protein function is regulated via a flexible pocket and novel phosphorylation-dependent mechanism. Mol. Cell Biol. 2009, 29, 1306–1320. [Google Scholar] [CrossRef]
- Qin, Q.; Liu, H.; Shou, J.; Jiang, Y.; Yu, H.; Wang, X. The inhibitor effect of RKIP on inflammasome activation and inflammasome-dependent diseases. Cell Mol. Immunol. 2021, 18, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Siyun, W.; Ma, H.; Yan, Y.; Chen, Y.; Fu, S.; Wang, J.; Wang, Y.; Chen, H.; Liu, J. cMET promotes metastasis and epithelial-mesenchymal transition in colorectal carcinoma by repressing RKIP. J. Cell. Physiol. 2021, 5, 3963–3978. [Google Scholar]
- Zaravinos, A.; Bonavida, B.; Chatzaki, E.; Baritaki, S. RKIP: A Key Regulator in Tumor Metastasis Initiation and Resistance to Apoptosis: Therapeutic Targeting and Impact. Cancers 2018, 10, 287. [Google Scholar] [CrossRef]
- Yeung, K.; Janosch, P.; McFerran, B.; Rose, D.W.; Mischak, H.; Sedivy, J.M.; Kolch, W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the Raf kinase inhibitor protein. Mol. Cell Biol. 2000, 20, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.C.; Rose, D.W.; Dhillon, A.S.; Yaros, D.; Gustafsson, M.; Chatterjee, D.; McFerran, B.; Wyche, J.; Kolch, W.; Sedivy, J.M. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol. Cell Biol. 2001, 21, 7207–7217. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Bitar, M.S.; Al-Maghrebi, M.; Behbehani, A.I.; Al-Ali, W.; Rath, O.; Doyle, B.; Tan, K.Y.; Pitt, A.; Kolch, W. Raf kinase inhibitor protein RKIP enhances signaling by glycogen synthase kinase-3β. Cancer Res. 2011, 71, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Al-Mulla, F.; Bitar, M.S.; Feng, J.; Park, S.; Yeung, K.C. A new model for raf kinase inhibitory protein induced chemotherapeutic resistance. PLoS ONE 2012, 7, e29532. [Google Scholar]
- Chatterjee, D.; Sabo, E.; Tavares, R.; Resnick, M.B. Inverse association between Raf kinase inhibitory protein and signal transducers and activators of transcription 3 expression in gastric adenocarcinoma patients: Implications for clinical outcome. Clin. Cancer Res. 2008, 14, 2994–3001. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Al-Mulla, F.; Bitar, M.S.; Taqi, Z.; Yeung, K.C. RKIP: Much more than Raf Kinase inhibitory protein. J. Cell. Physiol. 2013, 8, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Gabriela-Freitas, M.; Pinheiro, J.; Raquel-Cunha, A.; Cardoso-Carneiro, D.; Martinho, O. RKIP as an Inflammatory and Immune System Modulator: Implications in Cancer. Biomolecules 2019, 9, 769. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Baritaki, S. Prognostic and Therapeutic Applications of RKIP in Cancer; Academic Press: Cambridge, MA, USA, 2020; pp. 323–333. [Google Scholar]
- Banfield, M.J.; Barker, J.J.; Perry, A.C.; Brady, R.L. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure 1998, 6, 1245–1254. [Google Scholar] [CrossRef]
- Fu, Z.; Smith, P.C.; Zhang, L.; Rubin, M.A.; Dunn, R.L.; Yao, Z.; Keller, E.T. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J. Natl. Cancer Inst. 2003, 95, 878–889. [Google Scholar] [CrossRef]
- Simister, P.C.; Banfield, M.J.; Brady, R.L. The crystal structure of PEBP-2, a homologue of the PEBP/RKIP family. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58 (Pt 6 Pt 2), 1077–1080. [Google Scholar] [CrossRef]
- Shemon, A.N.; Heil, G.L.; Granovsky, A.E.; Clark, M.M.; McElheny, D.; Chimon, A.; Rosner, M.R.; Koide, S. Characterization of the Raf kinase inhibitory protein (RKIP) binding pocket: NMR-based screening identifies small-molecule ligands. PLoS ONE 2010, 5, e10479. [Google Scholar] [CrossRef]
- Tavel, L.; Jaquillard, L.; Karsisiotis, A.I.; Saab, F.; Jouvensal, L.; Brans, A.; Delmas, A.F.; Schoentgen, F.; Cadene, M.; Damblon, C. Ligand binding study of human PEBP1/RKIP: Interaction with nucleotides and Raf-1 peptides evidenced by NMR and mass spectrometry. PLoS ONE 2012, 7, e36187. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.S.; Tauc, P.; Brochon, J.C.; Maget-Dana, R.; Lelievre, D.; Metz-Boutigue, M.H.; Bureaud, N.; Schoentgen, F. Behaviour of bovine phosphatidylethanolamine-binding protein with model membranes. Evidence of affinity for negatively charged membranes. Eur. J. Biochem. 2001, 268, 5831–5841. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Mc Henry, K.T.; Lane, W.S.; Fenteany, G. A chemical inhibitor reveals the role of Raf kinase inhibitor protein in cell migration. Chem. Biol. 2005, 12, 981–991. [Google Scholar] [CrossRef]
- Shemon, A.N.; Eves, E.M.; Clark, M.C.; Heil, G.; Granovsky, A.; Zeng, L.; Imamoto, A.; Koide, S.; Rosner, M.R. Raf Kinase Inhibitory Protein protects cells against locostatin-mediated inhibition of migration. PLoS ONE 2009, 4, e6028. [Google Scholar] [CrossRef] [PubMed]
- Serre, L.; Vallee, B.; Bureaud, N.; Schoentgen, F.; Zelwer, C. Crystal structure of the phosphatidylethanolamine-binding protein from bovine brain: A novel structural class of phospholipid-binding proteins. Structure 1998, 6, 1255–1265. [Google Scholar] [CrossRef]
- Skinner, J.J.; Wang, S.; Lee, J.; Ong, C.; Sommese, R.; Sivaramakrishnan, S.; Koelmel, W.; Hirschbeck, M.; Schindelin, H.; Kisker, C.; et al. Conserved salt-bridge competition triggered by phosphorylation regulates the protein interactome. Proc. Nat. Acad. Sci. USA 2017, 114, 13453–13458. [Google Scholar] [CrossRef] [PubMed]
- Mandal, J.P.; Shiue, C.N.; Chen, Y.C.; Lee, M.C.; Yang, H.H.; Chang, H.H.; Hu, C.T.; Liao, P.C.; Hui, L.C.; You, R.I.; et al. PKCδ mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free Radic. Biol. Med. 2021, 163, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Galal, Y.; Zaravinos, A.; Bonavida, B. Chapter 13- Regulation of NKG2D by RKIP: Implications on NK-mediated cytotoxicity and cytokine production. In Successes and Challenges of NK Immunotherapy: Breaking Toerance to Cancer Resistance; Academic Press: Cambridge, MA, USA, 2021; pp. 233–265. [Google Scholar]
- Ferrell, J.E. MAP kinases in mitogenesis and development. Curr. Top. Dev. Biol. 1996, 33, 1–60. [Google Scholar] [PubMed]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras/GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Marais, R.; Marshall, C.J. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv. 1996, 27, 101–125. [Google Scholar] [PubMed]
- Morrison, D.K.; Cutler, R.E. The complexity of Raf-1 regulation. Curr. Opin. Cell Biol. 1997, 9, 174–179. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Elion, E.A. Routing MAP kinase cascades. Science 1998, 281, 1625–1626. [Google Scholar] [CrossRef]
- Denouel-Galy, A.; Douville, E.M.; Warne, P.H.; Papin, C.; Laugier, D.; Calothy, G.; Downward, J.; Eychene, A. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr. Biol. 1998, 8, 46–55. [Google Scholar] [CrossRef]
- Joneson, T.; Fulton, J.A.; Volle, D.J.; Chaika, O.V.; Bar-Sagi, D.; Lewis, R.E. Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J. Biol. Chem. 1998, 273, 7743–7748. [Google Scholar] [CrossRef]
- Michaud, N.R.; Therrien, M.; Cacace, A.; Edsall, L.C.; Spiegel, S.; Rubin, G.M.; Morrison, D.K. KSR stimulates Raf-1 activity in a kinase- independent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 12792–12796. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fantl, W.J.; Harrowe, G.; Williams, L.T. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr. Biol. 1998, 8, 56–64. [Google Scholar] [CrossRef]
- Therrien, M.; Michaud, N.R.; Rubin, G.M.; Morrison, D.K. KSR modulates signal propagation within the MAPK cascade. Genes Dev. 1996, 10, 2684–2695. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Kornfeld, K.; Muslin, A.J. The protein kinase KSR interacts with 14-3-3 protein and Raf. Curr. Biol. 1997, 7, 294–300. [Google Scholar] [CrossRef]
- Downward, J. KSR: A novel player in the RAS pathway. Cell 1995, 83, 831–834. [Google Scholar] [CrossRef][Green Version]
- Whitmarsh, A.J.; Cavanagh, J.; Tournier, C.; Yasuda, J.; Davis, R.J. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 1998, 281, 1671–1674. [Google Scholar] [CrossRef]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Rath, O.; Beach, S.; Xiang, X.; Kelly, S.M.; Luo, Z.; Kolch, W.; Yeung, K.C. Regulation of RKIP binding to the N-region of the Raf-1 kinase. FEBS Lett. 2006, 580, 6405–6412. [Google Scholar] [CrossRef]
- Rath, O.; Park, S.; Tang, H.H.; Banfield, M.J.; Brady, R.L.; Lee, Y.C.; Dignam, J.D.; Sedivy, J.M.; Kolch, W.; Yeung, K.C. The RKIP (Raf-1 Kinase Inhibitor Protein) conserved pocket binds to the phosphorylated N-region of Raf-1 and inhibits the Raf-1-mediated activated phosphorylation of MEK. Cell. Signal. 2008, 20, 935–941. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Bitar, M.S.; Thiery, J.P.; Zea, T.T.; Chatterjee, D.; Bennett, L.; Park, S.; Edwards, J.; Yeung, K.C. Clinical implications for loss or diminution of expression of Raf-1 kinase inhibitory protein and its phosphorylated form in ductal breast cancer. Am. J. Cancer Res. 2013, 3, 446–464. [Google Scholar] [PubMed]
- Lee, H.C.; Tian, B.; Sedivy, J.M.; Wands, J.R.; Kim, M. Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology 2006, 131, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Schuierer, M.M.; Bataille, F.; Hagan, S.; Kolch, W.; Bosserhoff, A.K. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004, 64, 5186–5192. [Google Scholar] [CrossRef]
- Das, S.K.; Bhutia, S.K.; Sokhi, U.K.; Azab, B.; Su, Z.; Boukerche, H.; Anwar, T.; Moen, E.L.; Chatterjee, D.; Pellecchia, M.; et al. Raf kinase inhibitor RKIP inhibits MDA-9/syntenin-mediated metastasis in melanoma. Cancer Res. 2012, 72, 6217–6226. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Bai, Y.; Wang, Z.; Beach, S.; Mott, S.; Roy, R.; Braastad, C.; Sun, Y.; Mukhopadhyay, A.; Aggarwal, B.B.; et al. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J. Biol. Chem. 2004, 279, 17515–17523. [Google Scholar] [CrossRef] [PubMed]
- Wottrich, S.; Kaufhold, S.; Chrysos, E.; Zoras, O.; Baritaki, S.; Bonavida, B. Inverse correlation between metastasis suppressor RKIP and the metastasis inducer YY1: Contrasting roles in the regulation of chemo/immune-resistance in cancer. Drug Resist. Updates 2017, 30, 28–38. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- Deiss, K.; Kisker, C.; Lohse, M.J.; Lorenz, K. Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from raf1 to G protein-coupled receptor kinase (GRK) 2. J. Biol. Chem. 2012, 287, 23407–23417. [Google Scholar] [CrossRef]
- Cross-Knorr, S.; Lu, S.; Perez, K.; Guevara, S.; Brilliant, K.; Pisano, C.; Quesenberry, P.J.; Resnick, M.B.; Chatterjee, D. RKIP phosphorylation and STAT3 activation is inhibited by oxaliplatin and camptothecin and are associated with poor prognosis in stage II colon cancer patients. BMC Cancer 2013, 13, 463. [Google Scholar] [CrossRef]
- Nisimova, L.; Wen, S.; Cross-Knorr, S.; Rogers, A.B.; Moss, S.F.; Chatterjee, D. Role of raf kinase inhibitor protein in helicobacter pylori-mediated signaling in gastric cancer. Crit. Rev. Oncog. 2014, 19, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Yesilkanal, A.E.; Rosner, M.R. Targeting Raf Kinase Inhibitory Protein Regulation and Function. Cancers 2018, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Moen, E.L.; Wen, S.; Anwar, T.; Cross-Knorr, S.; Brilliant, K.; Birnbaum, F.; Rahaman, S.; Sedivy, J.M.; Moss, S.F.; Chatterjee, D. Regulation of RKIP function by Helicobacter pylori in gastric cancer. PLoS ONE 2012, 7, e37819. [Google Scholar] [CrossRef]
- Li, S.; Liu, T.; Mo, W.; Hou, Q.; Zhou, Y.; Liu, M.; He, Z.; Liu, Z.; Chen, Q.; Wang, H.; et al. Prognostic value of phosphorylated Raf kinase inhibitory protein at serine 153 and its predictive effect on the clinical response to radiotherapy in nasopharyngeal carcinoma. Radiat. Oncol. 2016, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Yoon, N.K.; Hernandez-Cueto, A.; Mah, V.; Rivera-Pazos, C.M.; Chatterjee, D.; Vega, M.I.; Maresh, E.L.; Horvath, S.; Chia, D.; et al. Expression of phosphorylated raf kinase inhibitor protein (pRKIP) is a predictor of lung cancer survival. BMC Cancer 2011, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; Cabrava-Haimandez, M.D.; Sensi, M.; Canevari, S.; Libra, M.; Penichet, M.; Chen, H.; Berenson, J.R.; Bonavida, B. Unique pattern of overexpression of Raf-1 kinase inhibitory protein in its inactivated phosphorylated form in human multiple myeloma. For. Immunopathol. Dis. Ther. 2011, 2, 2. [Google Scholar] [CrossRef]
- Germain, D.; Frank, D.A. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin. Cancer Res. 2007, 13, 5665–5669. [Google Scholar] [CrossRef]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Bitar, M.S.; Taqi, Z.; Rath, O.; Kolch, W. RAF kinase inhibitory protein (RKIP) modulates cell cycle kinetics and motility. Mol. Biosyst. 2011, 7, 928–941. [Google Scholar] [CrossRef]
- Eves, E.M.; Shapiro, P.; Naik, K.; Klein, U.R.; Trakul, N.; Rosner, M.R. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol. Cell. 2006, 23, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Eves, E.M.; Rosner, M.R. MAP kinase regulation of the mitotic spindle checkpoint. Methods Mol. Biol. 2010, 661, 497–505. [Google Scholar] [PubMed]
- Adams, R.R.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001, 11, 49–54. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Hagan, S.; Al-Ali, W.; Jacob, S.P.; Behbehani, A.I.; Bitar, M.S.; Dallol, A.; Kolch, W. Raf kinase inhibitor protein: Mechanism of loss of expression and association with genomic instability. J. Clin. Pathol. 2008, 61, 524–529. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar] [PubMed]
- Rivat, C.; Rodrigues, S.; Bruyneel, E.; Pietu, G.; Robert, A.; Redeuilh, G.; Bracke, M.; Gespach, C.; Attoub, S. Implication of STAT3 signaling in human colonic cancer cells during intestinal trefoil factor 3 (TFF3)—And vascular endothelial growth factor-mediated cellular invasion and tumor growth. Cancer Res. 2005, 65, 195–202. [Google Scholar] [PubMed]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Paule, B.; Clerc, D.; Rudant, C.; Coulombel, C.; Bonhomme-Faivre, L.; Quillard, J.; Bisson, M. Enhanced expression of interleukin-6 in bone and serum of metastatic renal cell carcinoma. Hum. Pathol. 1998, 29, 421–424. [Google Scholar] [CrossRef]
- Suematsu, S.; Matsusaka, T.; Matsuda, T.; Ohno, S.; Miyazaki, J.; Yamamura, K.; Hirano, T.; Kishimoto, T. Generation of plasmacytomas with the chromosomal translocation t(12;15) in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA 1992, 89, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Esfandi, F.; Mohammadzadeh Ghobadloo, S.; Basati, G. Interleukin-6 level in patients with colorectal cancer. Cancer Lett. 2006, 244, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Wirtz, S.; Nikolaev, A.; Lehr, H.A.; Galle, P.R.; Rose-John, S.; Neurath, M.F. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle 2005, 4, 217–220. [Google Scholar] [CrossRef]
- Atreya, R.; Neurath, M.F. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin. Rev. Allergy Immunol. 2005, 28, 187–196. [Google Scholar] [CrossRef]
- Huerta-Yepez, S.; Ekmekcioglu, S.; Rivera-Pazos, C.; Antonio-Andres, G.; Vega, M.; Baay-Guzman, G.; Grimm, E. Braf Mutations Are Associated with High Levels of Phosphorylated RKIP in Melanoma Cell Lines: Potential Prognostic Significance. Forum Immunopathol. Dis. Ther. 2011, 2, 189–194. [Google Scholar] [CrossRef]
- Beach, S.; Tang, H.; Park, S.; Dhillon, A.S.; Keller, E.T.; Kolch, W.; Yeung, K.C. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene 2008, 27, 2243–2248. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: Downregulation of the NF-κB/Snail/YY1/RKIP circuitry. Nitric Oxide Biol. Chem. 2011, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Tsao, D.A.; Yu, H.S.; Chang, H.R. Nitric oxide enhances expression of raf kinase inhibitor protein in keratinocytes. Exp. Dermatol. 2009, 18, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Shvartsur, A.; Givechian, K.B.; Garban, H.; Bonavida, B. Overexpression of RKIP and its cross-talk with several regulatory gene products in multiple myeloma. J. Exp. Clin. Cancer Res. 2017, 36, 62. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Yun, J.; Eves, E.M.; Newman, M.; Erkeland, S.J.; Hammond, S.M.; Minn, A.J.; Rosner, M.R. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009, 28, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, J.; Farquhar, K.S.; Yun, J.; Frankenberger, C.A.; Bevilacqua, E.; Yeung, K.; Kim, E.J.; Balázsi, G.; Rosner, M.R. Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc. Natl. Acad. Sci. USA 2014, 111, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, O.; Qin, J.; Liu, S.; Sun, S.; Liu, H.; Kuang, J.; Jiang, G.; Zhang, W. Cis-acting elements and trans-acting factors in the transcriptional regulation of raf kinase inhibitory protein expression. PLoS ONE 2013, 8, e83097. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Keller, J.M.; Yeung, K.; Keller, E.T.; Fu, Z. Transcriptional regulation of RKIP expression by androgen in prostate cells. Cell. Physiol. Biochem. 2012, 30, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Sells, M.A.; Boyd, J.T.; Chernoff, J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 1999, 145, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Beshir, A.B.; Ren, G.; Magpusao, A.N.; Barone, L.M.; Yeung, K.C.; Fenteany, G. Raf kinase inhibitor protein suppresses nuclear factor-kappaB-dependent cancer cell invasion through negative regulation of matrix metalloproteinase expression. Cancer Lett. 2010, 299, 137–149. [Google Scholar] [CrossRef]
- Ren, G.; Baritaki, S.; Marathe, H.; Feng, J.; Park, S.; Beach, S.; Bazeley, P.S.; Beshir, A.B.; Fenteany, G.; Mehra, R.; et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012, 72, 3091–3104. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Soengas, M.S.; Capodieci, P.; Polsky, D.; Mora, J.; Esteller, M.; Opitz-Araya, X.; McCombie, R.; Herman, J.G.; Gerald, W.L.; Lazebnik, Y.A. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001, 409, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Dong, Z.; Guo, Y.; Lin, X.; Chen, Z.; Kuang, G.; Yang, Z. Aberrant methylation and loss expression of RKIP is associated with tumor progression and poor prognosis in gastric cardia adenocarcinoma. Clin. Exp. Metastasis 2013, 30, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Song, Y.; Fu, Z.; Yu, W. miR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol. Cancer 2014, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Dong, Z.; Lin, X.; Zhang, M.; Kuang, G.; Zhu, T. Decreased expression and aberrant methylation of Raf kinase inhibitory protein gene in esophageal squamous cell carcinoma. Cancer Investig. 2012, 30, 703–711. [Google Scholar] [CrossRef]
- Wei, H.; Liu, Z.; She, H.; Liu, B.; Gu, J.; Wei, D.; Zhang, X.; Wang, J.; Qi, S.; Ping, F. Promoter methylation and expression of Raf kinase inhibitory protein in esophageal squamous cell carcinoma. Oncol. Lett. 2017, 13, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Minoo, P.; Baker, K.; Goswami, R.; Chong, G.; Foulkes, W.D.; Ruszkiewicz, A.R.; Barker, M.; Buchanan, D.; Young, J.; Jass, J.R. Extensive DNA methylation in normal colorectal mucosa in hyperplastic polyposis. Gut 2006, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.E.; Kim, N.I.; Lee, J.S.; Park, M.H.; Yoon, J.H. Reduced RKIP expression is associated with breast neoplastic progression and is correlated with poor outcomes and aberrant methylation in breast carcinoma. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 467–474. [Google Scholar] [CrossRef]
- Labbozzetta, M.; Poma, P.; Vivona, N.; Gulino, A.; D’Alessandro, N.; Notarbartolo, M. Epigenetic changes and nuclear factor-κB activation, but not microRNA-224, downregulate Raf-1 kinase inhibitor protein in triple-negative breast cancer SUM 159 cells. Oncol. Lett. 2015, 10, 3807–3815. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Weng, X.D.; Wang, L.; Liu, X.H.; Zhu, H.C.; Guo, J.; Ning, J.Z.; Xiao, C.C. LncRNA XIST acts as a tumor suppressor in prostate cancer through sponging miR-23a to modulate RKIP expression. Oncotarget 2017, 8, 94358–94370. [Google Scholar] [CrossRef]
- Cai, S.; Chen, R.; Li, X.; Cai, Y.; Ye, Z.; Li, S.; Li, J.; Huang, H.; Peng, S.; Wang, J.; et al. Downregulation of Micro0RNA-23a suppresses prostate cancer metastasis by targeting the PAK6-LIMK1 signlaing pathway. Oncotarget 2015, 6, 3904–3917. [Google Scholar] [CrossRef] [PubMed]
- Hatzl, S.; Geiger, O.; Kuepper, M.K.; Caraffini, V.; Seime, T.; Furlan, T.; Nussbaumer, E.; Wieser, R.; Pichler, M.; Scheideler, M.; et al. Increased expression of miR-23a mediates a loss of expression in the RAF kinase inhibitor protein RKIP. Cancer Res. 2016, 76, 3644–3654. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Dai, T.; Lin, X.; Zhao, X.; Chen, X.; Wang, C.; Li, X.; Shen, H.; Wang, X. MicroRNA-224 targets RKIP to control cell invasion and expression of metastasis genes in human breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 425, 127–133. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Gossen, M.; Dittmar, G.; Selbach, M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics 2009, 9, 205–209. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- De Castro, J.; Odeh, H.N.; Figy, C.; Yeung, M.L.; Trumbly, R.; Yeung, K.C. Chapter 9- Regulation of RKIP expression in breast cancer cells by miRNAs. In Prognostic and Therapeutic Applications of RKIP in Cancer; Academic Press: Cambridge, MA, USA, 2020; pp. 139–146. [Google Scholar]
- Hagan, S.; Al-Mulla, F.; Mallon, E.; Oien, K.; Ferrier, R.; Gusterson, B.; Curto Garcia, J.J.; Kolch, W. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin. Cancer Res. 2005, 11, 7392–7397. [Google Scholar] [CrossRef]
- Du, Y.; Liu, X.H.; Zhu, H.C.; Wang, L.; Ning, J.Z.; Xiao, C.C. MiR-543 promotes proliferation and epithelial-Mesenchymal transition in prostate cancer via targeting RKIP. Cell Physiol. Biochem. 2017, 41, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, P.; Li, B.; Sun, P.; Zhang, J.; Wang, B.; Jia, B. RKIP suppresses gastric cancer cell proliferation and invasion and enhances apoptosis regulated by microRNA-224. Tumour Biol. 2014, 35, 10095–10103. [Google Scholar] [CrossRef]
- Kim, S.W.; Ramasamy, K.; Bouamar, H.; Lin, A.P.; Jiang, D.; Aguiar, R.C. MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc. Natl. Acad. Sci. USA 2012, 109, 7865–7870. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Vivian, C.J.; Brinker, A.E.; Hampton, K.R.; Lianidou, E.; Welch, D.R. Microenvironmental influences on metastasis suppressor expression and function during a metastatic cell’s journey. Cancer Microenviron. 2014, 7, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Poma, P.; Labbozzetta, M.; Vivona, N.; Porcasi, R.; D’Alessandro, N.; Notarbartolo, M. Analysis of possible mechanisms accounting for raf-1 kinase inhibitor protein downregulation in hepatocellular carcinoma. Omics 2012, 16, 579–588. [Google Scholar] [CrossRef]
- Arthur, S.; Sundaram, U. Protein kinase C-mediated phosphorylation of RKIP regulates inhibition of Na-alanine cotransport by leukotriene D(4) in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2014, 307, C1010–C1016. [Google Scholar] [CrossRef]
- Wu, C.; Liu, F.; Zhou, X.; Cheng, Z.; Yang, X.; Xiao, H.; Chen, Q.; Cai, K. Effect of Protein Kinase C on Proliferation and Apoptosis of T lymphocytes in Idiopathic Thrombocytopenic Purpura Children. Cell Mol. Immunol. 2005, 3, 197–202. [Google Scholar]
- Xiong, W.N.; Xu, Y.J.; Zhang, Z.X.; Sheng, G.X.; Xiong, S.D. A study on the effect of PKC on the T cell proliferation and apoptosis in asthma patient. Zhonghua J. Tuberc. Respir. 2001, 24, 629–630. [Google Scholar]
- Corbit, K.C.; Trakul, N.; Eves, E.M.; Diaz, B.; Marshall, M.; Rosner, M.R. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J. Biol. Chem. 2003, 278, 13061–13068. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Gao, H.Q.; Li, H.B.; Qi, S.J.; Liu, W.L.; Xu, L.; Li, H.; Liu, J.X.; Dong, Z.M. Correlation among RKIP expression, NF-kB p65 levels, and T-lymphocyte subsets in gastric cardia adenocarcinoma. Genet. Mol. Res. 2015, 14, 16491–16496. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking in ammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Zeng, H.; Joo, M.; Everhart, M.B.; Sherrill, T.P.; Li, B.; Cheng, D.-s.; Yull, F.E.; Christman, J.W.; Blackwell, T.S. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J. Immunol. 2006, 176, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvare, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45 beta mediates the NF-kappaB suppression of JNK signaling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bonavida, B. Chapter 3—Pleiotropic activities of RKIP in cancer: Role in survival, EMT, chemo-immunoresistance, and autophagy. In Prognostic and Therapeutic Applications of RKIP in Cancer; Academic Press: Cambridge, MA, USA, 2020; pp. 47–75. [Google Scholar]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wei, J.; Wei, L.; Zhang, X.; Bai, F.; Wen, S.; Wie, Y.; Tan, S.; Lu, Z.; Lin, X. Role of RKIP in human hepatic stellate cell proliferation, invasion, and metastasis. J. Cell Biochem. 2019, 120, 6168–6177. [Google Scholar] [CrossRef]
- Fujimori, Y.; Inkouchi, M.; Takagi, Y.; Kato, K.; Kojima, K.; Sugihara, K. Prognostic value of RKIP and p-ERK in gastric cancer. J. Exp. Clin. Cancer Res. 2012, 31, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, D.; Fan, Q. RKIP suppresses the proliferation and invasion of choriocarcinoma cells through inhibiting the MAPK signaling pathway. Int. J. Clin. Exp. Med. 2015, 8, 22183–22190. [Google Scholar] [PubMed]
- Yang, K.; Li, Y.; Lian, G.; Lin, H.; Shang, C.; Zeng, L.; Chen, S.; Li, J.; Huang, C.; Huang, K.; et al. KRAS promotes tumor metastasis and chemoresistance by repressing RKIP via the MAPK-ERK pathway in pancreatic cancer. Int. J. Cancer 2018, 142, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Jazirehi, A.; Vega, M.I.; Huerta-Yepez, S.; Baritaki, S. Roles Each of Snail, Yin Yang 1 and RKIP in the Regulation of Tumor Cells Chemo-immuno-resistance to Apoptosis. For. Immunopathol. Dis. Therap. 2013, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Militello, L.; Malaponte, G.; Spandidos, D.A.; Salcedo, M.; Bonavida, B. The anti-CD20 mAb LFB-R603 interrupts the dysregulated NF-κB/Snail/RKIP/PTEN resistance loop in B-NHL cells: Role in sensitization to TRAIL apoptosis. Int. J. Oncol. 2011, 38, 1683–1694. [Google Scholar]
- Baritaki, S.; Katsman, A.; Chatterjee, D.; Yeung, K.C.; Spandidos, D.A.; Bonavida, B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J. Immunol. 2007, 179, 5441–5453. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B. RKIP-mediated chemo-immunosensitization of resistant cancer cells via disruption of the NF-κB/Snail/YY1/RKIP resistance-driver loop. Crit. Rev. Oncog. 2014, 19, 431–445. [Google Scholar] [CrossRef]
- Yousuf, S.; Duan, M.; Moen, E.L.; Cross-Knorr, S.; Brilliant, K.; Bonavida, B.; LaValle, T.; Yeung, K.C.; Al-Mulla, F.; Chin, E.; et al. Raf kinase inhibitor protein (RKIP) blocks signal transducer and activator of transcription 3 (STAT3) activation in breast and prostate cancer. PLoS ONE 2014, 9, e92478. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Liu, H.; Baritaki, S.; Del Lourdes Cebrera-Muñoz, M.; Rivera-Pazos, C.; Maldonado-Valenzuela, A.; Valencia-Hipolito, A.; Vega, M.I.; Chen, H.; Berenson, J.R.; et al. Overexpression of Yin Yang 1 in bone marrow-derived human multiple myeloma and its clinical significance. Int. J. Oncol. 2014, 45, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Baritaki, S.; Baay-Guzman, G.; Hernandez-Luna, M.A.; Hernandez-Cueto, A.; Vega, M.I.; Bonavida, B. Contribution of either YY1 or BclXL-induced inhibition by the NO-donor DETANONOate in the reversal of drug resistance, both in vitro and in vivo. YY1 and BclXL are overexpressed in prostate cancer. Nitric Oxide 2013, 29, 17–24. [Google Scholar] [CrossRef]
- Castellano, G.; Torrisi, E.; Ligresti, G.; Malaponte, G.; Militello, L.; Russo, A.E.; McCubrey, J.A.; Canevari, S.; Libra, M. The involvement of the transcription factor Yin Yang 1 in cancer development and progression. Cell Cycle 2009, 8, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hertlein, E.; Bakkar, N.; Sun, H.; Acharyya, S.; Wang, J.; Carathers, M.; Davuluri, R.; Guttridge, D.C. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell Biol. 2007, 27, 4374–4387. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.B.; Majumder, P.; Cooper, J.C.; Yoon, H.; Wade, P.A.; Boss, J.M. Yin yang 1 regulates the expression of snail through a distal enhancer. Mol. Cancer Res. 2009, 7, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, L.; Zhu, J.; Sun, A.; Yu, G.; Chen, M.; Huang, P.; Liu, H.; Shao, G.; Yang, W.; et al. Hippo signaling effector WWTR1 is a metastatic biomarker of gastric cardia adenocarcinoma. Cancer Cell Int. 2019, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Liu, C.L.; Chang, Y.C.; Nieh, S.; Lin, Y.S.; Jao, S.W.; Chen, S.F.; Liu, T.Y. Increased chemoresistance via Snail-Raf kinase inhibitor protein signaling in colorectal cancer in response to a nicotine derivative. Oncotarget 2016, 7, 23512–23520. [Google Scholar] [CrossRef]
- Bai, X.Y.; Zhang, X.C.; Yang, S.Q.; An, S.J.; Chen, Z.H.; Su, J.; Xie, Z.; Gou, L.Y.; Wu, Y.L. Blockade of Hedgehog Signaling Synergistically Increases Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer Cell Lines. PLoS ONE 2016, 11, e0149370. [Google Scholar]
- Heiden, K.B.; Williamson, A.J.; Doscas, M.E.; Ye, J.; Wang, Y.; Liu, D.; Xing, M.; Prinz, R.A.; Xu, X. The sonic hedgehog signaling pathway maintains the cancer stem cell self-renewal of anaplastic thyroid cancer by inducing snail expression. J. Clin. Endocrinol. Metab. 2014, 99, E2178–E2187. [Google Scholar] [CrossRef] [PubMed]
- Al-Mulla, F.; Hagan, S.; Behbehani, A.I.; Bitar, M.S.; George, S.S.; Going, J.J.; García, J.J.; Scott, L.; Fyfe, N.; Murray, G.I.; et al. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J. Clin. Oncol. 2006, 24, 5672–5679. [Google Scholar] [CrossRef] [PubMed]
- Martinho, O.; Granja, S.; Jaraquemada, T.; Caeiro, C.; Miranda-Gonçalves, V.; Honavar, M.; Costa, P.; Damasceno, M.; Rosner, M.R.; Lopes, J.M.; et al. Downregulation of RKIP is associated with poor outcome and malignant progression in gliomas. PLoS ONE 2012, 7, e30769. [Google Scholar] [CrossRef] [PubMed]
- Xinzhou, H.; Ning, Y.; Ou, W.; Xiaodan, L.; Fumin, Y.; Huitu, L.; Wei, Z. RKIp inhibits the migration and invasion of human prostate cancer PC-3 M cells through regulation of extracellular matrix [Research Support, non-U.S.-Gov’t]. Mol. Biol. 2011, 45, 1004–1011. [Google Scholar] [CrossRef]
- Schuierer, M.M.; Bataille, F.; Weiss, T.S.; Hellerbrand, C.; Bosserhoff, A.K. Raf kinase inhibitor protein is downregulated in hepatocellular carcinoma. Oncol. Rep. 2006, 16, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fu, Z.; Binkley, C.; Giordano, T.; Burant, C.F.; Logsdon, C.D.; Simeone, D.M. Raf kinase inhibitory protein inhibits beta-cell proliferation. Surgery 2004, 136, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Kitagawa, Y.; Shen, R.; Shah, R.; Mehra, R.; Rhodes, D.; Keller, P.J.; Mizokami, A.; Dunn, R.; Chinnaiyan, A.M.; et al. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate 2006, 66, 248–256. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Long, B.J.; Grigoryev, D.N.; Nnane, I.P.; Liu, Y.; Ling, Y.Z.; Brodie, A.M. Antiandrogenic effects of novel androgen synthesis inhibitors on hormone-dependent prostate cancer. Cancer Res. 2000, 60, 6630–6640. [Google Scholar]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Vincent, K.; Pichler, M.; Fodde, R.; Berindan-Neagoe, I.; Slack, F.J.; Calin, G.A. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015, 34, 5003–5011. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kraus, W.L. From discovery to function: The expanding roles of long noncoding RNAs in physiology and disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.J.; Yu, J.; Huang, Y.Q.; Yang, J. Circulating Long Noncoding RNA as a Potential Target for Prostate Cancer. Int. J. Mol. Sci. 2015, 16, 13322–13338. [Google Scholar] [CrossRef]
- Tycowski, K.T.; Guo, Y.E.; Lee, N.; Moss, W.N.; Vallery, T.K.; Xie, M.; Steitz, J.A. Viral noncoding RNAs: More surprises. Genes Dev. 2015, 29, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene regulation by non-coding RNAs. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Martens-Uzunova, E.S.; Böttcher, R.; Croce, C.M.; Jenster, G.; Visakorpi, T.; Calin, G.A. Long noncoding RNA in prostate, bladder, and kidney cancer. Eur. Urol. 2014, 65, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Wang, D.; Chen, J.; Tian, Y.; Cai, X.; Peng, H.; Zhu, L.; Huang, A.; Tang, H. LncRNA-AF113014 promotes the expression of Egr2 by interaction with miR-20a to inhibit proliferation of hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0177843. [Google Scholar]
- Li, Q.; Shen, W.; Li, X.; Zhang, L.; Jin, X. The lncRNA n340790 accelerates carcinogenesis of thyroid cancer by regulating miR-1254. Am. J. Transl. Res. 2017, 9, 2181–2194. [Google Scholar] [PubMed]
- Kim, S.O.; Kim, M.R. (-)-Epigallocatechin 3-gallate inhibits invasion by inducing the expression of Raf kinase inhibitor protein in AsPC-1 human pancreatic adenocarcinoma cells through the modulation of histone deacetylase activity. Int. J. Oncol. 2012, 42, 349–358. [Google Scholar] [CrossRef]
- Odabaei, G.; Chatterjee, D.; Jazirehi, A.R.; Goodglick, L.; Yeung, K.; Bonavida, B. Raf-1 kinase inhibitor protein: Structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv. Cancer Res. 2004, 91, 169–200. [Google Scholar]
- Maresch, J.; Birner, P.; Zakharinov, M.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. Additive effect on survival of Raf kinase inhibitor protein and signal transducer and activator of transcription 3 in high-grade glioma. Cancer 2010, 10, 1002. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, M.; Souza, V.C.; Izumi, C.; Barbieri, M.R.; Chammas, R.; Oba-Shinjo, S.M.; Uno, M.; Marie, S.K.N.; Rosa, J.C. Proteomic analysis of low- to high-grade astrocytomas reveals an alteration of the expression level of raf kinase inhibitor protein and nucleophosmin. Proteomics 2010, 10, 2812–2821. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Gao, Y.; Zhao, X.L.; Liu, Y.X.; Sun, B.C.; Yang, J.; Yao, Z. Effects of raf kinase inhibitor protein expression on metastasis and progression of human breast cancer. Mol. Cancer Res. 2009, 7, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Papale, M.; Vocino, G.; Lucarelli, G.; Rutigliano, M.; Gigante, M.; Rocchetti, M.T.; Pesce, F.; Sanguedolce, F.; Bufo, P.; Battaglia, M.; et al. Urinary RKIP/p-RKIP is a potential diagnostic and prognostic marker of clear cell renal cell carcinoma. Oncotarget 2017, 8, 40412–40424. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.; Park, J.Y.; Sung, J.Y.; Park, Y.K.; Kim, Y.W. Reduced expression of Raf-1 kinase inhibitory protein in renal cell carcinoma: A significant prognostic marker. Pathology 2012, 44, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Y.H.; Wang, A.Q.; Yao, B.; Xie, G.; Feng, G.; Zhang, Y.; Cheng, Z.S.; Hui, L.; Dai, T.Z.; et al. Immunohistochemical detection of the Raf kinase inhibitor protein in nonneoplastic gastric tissue and gastric cancer tissue. Med. Oncol. 2010, 27, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Liu, H.; Kong, Q.; Li, B. RKIP expression associated with gastric cancer cell invasion and metastasis. Tumour. Biol. 2012, 33, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers—A different disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Cardile, V.; Malaponte, G.; Loreto, C.; Libra, M.; Caggia, S.; Trovato, F.M.; Musumeci, G. Raf kinase inhibitor protein (RKIP) and phospho-RKIP expression in melanomas. Acta Histochem. 2013, 115, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Zebisch, A.; Caraffini, V.; Sill, H. RAF Kinase Inhibitor Protein in Myeloid Leukemogenesis. Int. J. Mol. Sci. 2019, 20, 5756. [Google Scholar] [CrossRef]
- Crassini, K.; Pyke, T.; Shen, Y.; Stevenson, W.S.; Christopherson, R.I.; Mulligan, S.P.; Best, O.G. Inhibition of the Raf-1 kinase inhibitory protein (RKIP) by locostatin induces cell death and reduces the CXCR4-mediated migration of chronic lymphocytic leukemia cells. Leuk. Lymphoma 2018, 59, 2917–2928. [Google Scholar] [CrossRef] [PubMed]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Buschow, S.I.; Ramazzotti, M.; Reinieren-Beeren, I.M.J.; Heinzerling, L.M.; Westdorp, H.; Stefanini, I.; Beltrame, L.; Hato, S.V.; Ellebaek, E.; Gross, S.; et al. Survival of metastatic melanoma patients after dendritic cell vaccination correlates with expression of leukocyte phosphatidylethanolamine-binding protein 1/Raf kinase inhibitory protein. Oncotarget 2017, 8, 67439–67456. [Google Scholar] [CrossRef]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8⁺ T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef]
- Stairiker, C.J.; Thomas, G.D.; Salek-Ardakani, S. EZH2 as a Regulator of CD8+ T Cell Fate and Function. Front Immunol. 2020, 11, 593203. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Tsung, A.; Irvine, K.R.; Parkhurst, M.R.; Goletz, T.J.; Tsung, K.; Carroll, M.W.; Liu, C.; Moss, B.; Rosenberg, S.A.; et al. gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998, 188, 277–286. [Google Scholar] [CrossRef]
- He, S.; Liu, Y.; Meng, L.; Sun, H.; Wang, Y.; Ji, Y.; Purushe, J.; Chen, P.; Li, C.; Madzo, J.; et al. Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumor immunity. Nat. Commun. 2017, 8, 2125. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Amezquita, R.A.; Guan, T.; Kleinstein, S.H.; Kaech, S.M. Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8+ T Cell Terminal Differentiation and Loss of Multipotency. Immunity 2017, 46, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.T.; Vella, A.T. RKIP contributes to IFN-y synthesis by CD8+ T cells after serial TCR triggering in systemic inflammatory response syndrome. J. Immunol. 2013, 191, 708–716. [Google Scholar] [CrossRef]
- Schuierer, M.M.; Heilmeier, U.; Boettcher, A.; Ugocsai, P.; Bosserhoff, A.K.; Schmitz, G.; Langmann, T. Induction of Raf kinase inhibitor protein contributes to macrophage differentiation. Biochem. Biophys. Res. Commun. 2006, 342, 1083–1087. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, M.C.; Izquierdo, M.; de Murcia, G.; Lopez-Rivas, A. Activation of protein kinase C attenuates early signals in Fas- mediated apoptosis. Eur. J. Immunol. 1997, 27, 1442–1450. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Shetty, S.; Graham, B.A.; Brown, J.G.; Hu, X.; Vegh-Yarema, N.; Harding, G.; Paul, J.T.; Gibson, S.B. Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol. Cell Biol. 2005, 25, 5404–5416. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Yeung, K.; Palladino, M.; Berenson, J.; Bonavida, B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009, 69, 8376–8385. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Baritaki, S.; Huerta-Yepez, S.; Vega, M.I.; Chatterjee, D.; Yeung, K. Novel therapeutic applications of nitric oxide donors in cancer: Roles in chemo- and immunosensitization to apoptosis and inhibition of metastases. Nitric Oxide 2008, 19, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.X.; Li, W.Z.; Guo, Y.L.; Chen, L.; Li, G.H.; Yu, J.J.; Shu, B.; Peng, S. Tumor suppressor RKIP inhibits prostate cancer cell metastasis and sensitizes prostate cancer cells to docetaxel treatment. Neoplasma 2018, 65, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Bonavida, B. Viral infection and cancer: The NF-kappaB/Snail/RKIP loop regulates target cell sensitivity to apoptosis by cytotoxic lymphocytes. Crit. Rev. Immunol. 2010, 30, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Huerta-Yepez, S.; Vega, M.; Baritaki, S.; Spandidos, D.A.; Bonavida, B. The NO TRAIL to YES TRAIL in cancer therapy (review). Int. J. Oncol. 2007, 31, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Ma, X.; Li, X.; Wang, X.; Mei, Q.; Li, X.; Wu, Z.; Han, W. The Accomplices of NF-κB Lead to Radioresistance. Curr. Protein. Pept. Sci. 2015, 16, 279–294. [Google Scholar] [CrossRef]
- Touboul, R.; Bonavida, B. Chapter 17: YY1 expression and PD-1 regulation in CD8 T lymphocytes. In YY1 in the Control of the Pathogenesis and Drug Resistance of Cancer: A Critical Therapeutic Target; Academic Press: Cambridge, MA, USA, 2021; Volume 17, pp. 289–309. [Google Scholar]
- Balkhi, M.Y.; Wittmann, G.; Xiong, F.; Junghans, R.P. YY1 upregulates checkpoint receptors and downregulates type I cytokines in exhausted, chronically stimulated human T cells. iScience 2018, 2, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Scmidt-Supprian, M.; Shi, Y.; Hobeika, E.; Barteneva, N.; Jumaa, H.; Pelanda, R.; Reth, M.; Skok, J.; Rajewsky, K. Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 2007, 21, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lee, J.S.; Galvin, K.M. Everything you have ever wanted to know about Yin Yang 1. Biochim. Biophys. Acta. 1997, 1332, F49–F66. [Google Scholar] [CrossRef]
- Srinivasan, L.; Atchison, M.L. YY1 DNA binding and PcG recruitment requires CtBP. Genes Dev. 2004, 18, 2596–2601. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.J.; Kharchenko, P.V.; Daheron, L.; Park, P.J.; Kingston, R.E. Variable requirements for DNA-binding proteins at polycomb-dependent repressive regions in human HOX clusters. Mol. Cell Biol. 2013, 33, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Atchison, M.L. Function of YY1 in long-distance DNA interactions. Front. Immunol. 2014, 5, 45. [Google Scholar] [CrossRef]
- Baritaki, S.; Huerta-Yepez, S.; Sakai, T.; Spandidos, D.A.; Bonavida, B. Chemotherapeutic drugs sensitize cancer cells to TRAIL-mediated apoptosis: Up-regulation of DR5 and inhibition of Yin Yang 1. Mol. Cancer Ther. 2007, 6, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Garban, H.J.; Bonavida, B. Nitric oxide inhibits the transcription repressor Yin-Yang 1 binding activity at the silencer region of the Fas promoter: A pivotal role for nitric oxide in the up-regulation of Fas gene expression in human tumor cells. J. Immunol. 2001, 167, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Garban, H.J.; Bonavida, B. Nitric oxide sensitizes ovarian tumor cells to Fas-induced apoptosis. Gynecol. Oncol. 1999, 73, 257–264. [Google Scholar] [CrossRef]
- Martinez-Paniagua, M.A.; Baritaki, S.; Huerta-Yepez, S.; Ortiz-Navarrete, V.F.; González-Bonilla, C.; Bonavida, B.; Vega, M.I. Mcl-1 and YY1 inhibition and induction of DR5 by the BH3-mimetic Obatoclax (GX15-070) contribute in the sensitization of B-NHL cells to TRAIL apoptosis. Cell Cycle 2011, 10, 2792–2805. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.I.; Huerta-Yepez, S.; Jazirehi, A.R.; Garban, H.; Bonavida, B. Rituximab (chimeric anti-CD20) sensitizes B-NHL cell lines to Fas-induced apoptosis. Oncogene 2005, 24, 8114–8127. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Vega, M.; Garban, H.; Bonavida, B. Involvement of the TNF-alpha autocrine-paracrine loop, via NF-kappaB and YY1, in the regulation of tumor cell resistance to Fas-induced apoptosis. Clin. Immunol. 2006, 120, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Vega, M.; Escoto-Chavez, S.E.; Murdock, B.; Sakai, T.; Baritaki, S.; Bonavida, B. Nitric oxide sensitizes tumor cells to TRAIL-induced apoptosis via inhibition of the DR5 transcription repressor Yin Yang 1. Nitric Oxide 2009, 20, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B. Rituximab-induced inhibition of antiapoptotic cell survival pathways: Implications in chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene 2007, 26, 3629–3636. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; D’Amore, M.; Lofrumento, D.D. Rituximab-mediated Raf kinase inhibitor protein induction modulates NF-κB in Sjögren syndrome. Immunology 2014, 143, 42–51. [Google Scholar] [CrossRef]
- Martinez-Paniagua, M.A.; Vega, M.I.; Huerta-Yepez, S.; Baritaki, S.; Vega, G.G.; Hariharan, K.; Bonavida, B. Galiximab signals B-NHL cells and inhibits the activities of NF-κB-induced YY1- and snail-resistant factors: Mechanism of sensitization to apoptosis by chemoimmunotherapeutic drugs. Mol. Cancer 2012, 11, 572–581. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Vega, M.I.; Chatterjee, D.; Goodglick, L.; Bonavida, B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of non-Hodgkin’s lymphoma B cells by Rituximab. Cancer Res. 2004, 64, 7117–7126. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.I.; Martínez-Paniagua, M.; Huerta-Yepez, S.; González-Bonilla, C.; Uematsu, N.; Bonavida, B. Dysregulation of the cell survival/anti-apoptotic NF-kappaB pathway by the novel humanized BM-ca anti-CD20 mAb: Implication in chemosensitization. Int. J. Oncol. 2009, 35, 1289–1296. [Google Scholar] [CrossRef]
- Vega, M.I.; Baritaki, S.; Huerta-Yepez, S.; Martinez-Paniagua, M.A.; Bonavida, B. A potential mechanism of rituximab-induced inhibition of tumor growth through its sensitization to tumor necrosis factor-related apoptosis-inducing ligand-expressing host cytotoxic cells. Leuk. Lymphoma 2011, 52, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hays, E.; Bonavida, B. YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist. Updates 2019, 43, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Baumgaertner, P.; Devevre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Grabmeier-Pfistershammer, K.; Steinberger, P.; Rieger, A.; Leitner, J.; Kohrgruber, N. Identification of PD-1 as a unique marker for failing immune reconstitution in HIV-1-infected patients on treatment. J. Acquir. Immune Defic. Syndr. 2011, 56, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4 + T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef]
- Beshir, A.B.; Argueta, C.E.; Menikarachchi, L.C.; Gascon, J.A.; Fenteany, G. Locostatin Disrupts Association of Raf Kinase Inhibitor Protein with Binding Proteins by Modifying a Conserved Histidine Residue in the Ligand-Binding Pocket. Forum Immunopathol. Dis. Ther. 2011, 2, 47–58. [Google Scholar] [CrossRef]
- McCormack, J.E.; Callahan, J.E.; Kappler, J.; Marrack, P.C. Profound deletion of mature T cells in vivo by chronic exposure to exogenous superantigen. J. Immunol. 1993, 150, 3785–3792. [Google Scholar]
- Kappler, J.; Herman, W.A.; Clements, J.; Marrack, P. Mutations defining functional regions of the superantigen staphylococcal enterotoxin B. J. Exp. Med. 1992, 175, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.L.; Jenkins, M.K.; Schwartz, R.H. Clonal expansion versus functional clonal inactivation: A costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.E. Sepsis, systemic inflammatory response, and multiple organ dysfunction: The mystery continues. Am. Surg. 2012, 78, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.T. Interrogating Raf-1 Kinase Inhibitor Protein (RKIP) as a Novel Therapeutic Target for Modulating Inflammatory Responses. Ph.D. Thesis, University of Connecticut, Storrs, CT, USA, 2016. [Google Scholar]
- Todd, J.; Fishaut, M.; Kapral, F.; Welch, T. Toxic-shock syndrome associated with phage-group-I Staphylococci. Lancet 1978, 2, 1116–1118. [Google Scholar] [CrossRef]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. American College of Chest Physicians/Society of Bone RC. Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome). JAMA 1992, 268, 3452–3455. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef]
- Noble, A.; Truman, J.P.; Vyas, B.; Vukmanovic-Stejic, M.; Hirst, W.J.; Kemeny, D.M. The balance of protein kinase C and calcium signaling directs T cell subset development. J. Immunol. 2000, 164, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Isakov, N.; Altman, A. Protein Kinase C (PKC) in T cell activation. Annu. Rev. Immunol. 2002, 20, 761–794. [Google Scholar] [CrossRef]
- Tada, Y.; Nagasawa, K.; Yamauchi, Y.; Tsukamoto, H.; Niho, Y. A defect in the protein kinase C system in T cells from patients with systemic lupus erythematosus. Clin. Immunol. Immunopathol. 1998, 60, 220–231. [Google Scholar] [CrossRef]
- Iori, E.; Marescotti, M.C.; Vedovato, M.; Ceolotto, G.; Avogaro, A.; Tiengo, A.; Del Prato, S.; Trevisan, R. In situ protein Kinase C activity is increased in cultured fibroblasts from Type 1 diabetic patients with nephropathy. Diabetologia 2003, 46, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bonavida, B. A New Linkage between the Tumor Suppressor RKIP and Autophagy: Targeted Therapeutics. Crit. Rev. Oncog. 2018, 23, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Bi, Y.R.; Li, Y.; Fu, R.; Lv, W.C.; Jiang, N.; Xu, Y.; Ren, B.X.; Chen, Y.D.; Xie, H.; et al. A potent CBP/p300-Snail interaction inhibitor suppresses tumor growth and metastasis in wild-type p53-expressing cancer. Sci. Adv. 2020, 6, 8500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Li, Q.; Meng, D. Bach1: Function, Regulation, and Involvement in Disease. Oxid. Med. Cell Longev. 2018, 2018, 1347969. [Google Scholar] [CrossRef] [PubMed]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach Proteins Belong to a Novel Family of BTB-Basic Leucine Zipper Transcription Factors That Interact with MafK and Regulate Transcription through the NF-E2 Site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in Pathophysiology: A Matter of Scavenging, Metabolism and Trafficking across Cell Membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef]
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 862–872. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra III, A.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef]
- Gehling, V.S.; Vaswani, R.G.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; Campbell, R.; Lee, C.; et al. Discovery, design, and synthesis of indole-based EZH2 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3644–3649. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.P.; Bingham, P.; Brooun, A.; Collins, M.; Deng, Y.L.; Dinh, D.; Fan, C.; Gajiwala, K.S.; Grantner, R.; Gukasyan, H.J.; et al. Optimization of orally bioavailable enhancer of zeste homolog 2 (EZH2) inhibitors using ligand and property- based design strategies: Identification of development candidate (R)-5,8- Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J. Med. Chem. 2018, 61, 650–665. [Google Scholar]
- Zhu, M.R.; Du, D.H.; Hu, J.C.; Li, L.C.; Liu, J.Q.; Ding, H.; Kong, X.Q.; Jiang, H.L.; Chen, K.X.; Luo, C. Development of a high-throughput fluorescence polarization assay for the discovery of EZH2-EED interaction inhibitors. Acta Pharm. Sin. 2018, 39, 302–310. [Google Scholar] [CrossRef]
- Wang, X.; Cao, W.; Zhang, J.; Yan, M.; Xu, Q.; Wu, X.; Wan, L.; Zhang, Z.; Zhang, C.; Qin, X.; et al. A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. EMBO J. 2017, 36, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef]
- Ménoret, A.; McAleer, J.P.; Ngoi, S.M.; Ray, S.; Eddy, N.A.; Fenteany, G.; Lee, S.J.; Rossi, R.J.; Mukherji, B.; Allen, D.L.; et al. The oxazolidinone derivative locostatin induces cytokine appeasement. J. Immunol. 2009, 183, 7489–7496. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wenzel, S. Interactions of RKIP with inflammatory signaling pathways. Crit. Rev. Oncog. 2014, 19, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, N.; Zhou, K.; Su, F.; Jiang, Y.; Shou, J.; Liu, H.; Ma, C.; Qian, Y.; Wang, K.; et al. RKIP mediates autoimmune inflammation by positively regulating IL-17R signaling. EMBO Rep. 2018, 19, e44951. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression of RKIP | Impact/Functions | References | |
|---|---|---|---|
| Adenocarcinomas | Decreased RKIP expression | RKIP increases progression, metastasis, and invasion leading to a poor prognosis | Wei et al., 2015; Wei et al., 2014 |
| Colon Cancer | Reduction of RKIP | Amplifies radio-resistance and chemoresistance | Zaravinos et al., 2018; Lee et al., 2016 |
| Prostate Cancer | RKIP is downregulated | Enhances metastasis in prostate cancer-associated cell lines | Beach et al., 2008 |
| Pancreatic Cancer | RKIP is induced | Prevents the invasive metastasis of pancreatic cancer cells | Kim and Kim, 2012 |
| Gliomas | Low expression of RKIP | Does not affect cell proliferation, and enhances cell migration | Martinho et al., 2012 |
| Renal Cell Carcinoma | High RKIP expression | Induces cell survival and progression-free survival | Papale et al., 2017 |
| Gastric Cancer | Low levels of RKIP expression | Negatively correlated with depth of tumor invasion | Wang et al., 2010 |
| Lung Cancer | Decreased levels in invasive cancers | Greater advantage in survival | Huerta-Yepez et al., 2011 |
| Leukemia | Loss of RKIP expression is common | RKIP inhibits proliferation of myeloid cells | Zebisch et al., 2019 |
| Multiple Myeloma | RKIP is overexpressed | Enhances tumor progression | Shvartsur et al., 2017 |
| Expression of RKIP | Impact/Functions | References | |
|---|---|---|---|
| SIRS (systemic inflammatory response syndrome) | RKIP expression is decreased | IFNy production is decreased leading to induction of SIRS | Wright and Vella, 2013 |
| AITP (acute idiopathic thrombocytopenic purpura) | RKIP activation is increased (when PKC expression increases) | Increases the functions of T cells progression and proliferation | Wu et al., 2005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touboul, R.; Baritaki, S.; Zaravinos, A.; Bonavida, B. RKIP Pleiotropic Activities in Cancer and Inflammatory Diseases: Role in Immunity. Cancers 2021, 13, 6247. https://doi.org/10.3390/cancers13246247
Touboul R, Baritaki S, Zaravinos A, Bonavida B. RKIP Pleiotropic Activities in Cancer and Inflammatory Diseases: Role in Immunity. Cancers. 2021; 13(24):6247. https://doi.org/10.3390/cancers13246247
Chicago/Turabian StyleTouboul, Roni, Stavroula Baritaki, Apostolos Zaravinos, and Benjamin Bonavida. 2021. "RKIP Pleiotropic Activities in Cancer and Inflammatory Diseases: Role in Immunity" Cancers 13, no. 24: 6247. https://doi.org/10.3390/cancers13246247
APA StyleTouboul, R., Baritaki, S., Zaravinos, A., & Bonavida, B. (2021). RKIP Pleiotropic Activities in Cancer and Inflammatory Diseases: Role in Immunity. Cancers, 13(24), 6247. https://doi.org/10.3390/cancers13246247