
Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities


Simple Summary
Abstract
1. Non-Small Cell Lung Cancer (NSCLC)
2. Signal Transducer and Activator of Transcription 3 (STAT3)
3. STAT3 Signaling
4. Tumor-Promoting Effects of STAT3 Activation in NSCLC
4.1. Angiogenesis
4.2. Cell Survival
4.3. Promoting Cancer Cell Stemness
4.4. Drug Resistance in Oncogene-Addicted Cells
4.5. Immune Modulation via Tumor-Cell Intrinsic STAT3 Activation
4.6. Immune Modulation via Tumor-Cell Extrinsic STAT3 Activation
5. Role of STAT3-Activating Cytokines and Growth Factors in NSCLC
6. STAT3 as a Therapeutic Target
6.1. Targeting Upstream Signaling Mechanisms
6.2. Target SH2 Domain of STAT3
6.3. Promote Degradation of STAT mRNA
6.4. Interfere with STAT3–DNA Binding
6.5. Challenges Associated with STAT3 Inhibition
7. Therapeutic Opportunities
7.1. STAT3 as a Biomarker to Identify Patients with Aberrant EGFR Signaling
7.2. Targeting STAT3 in Tyrosine Kinase Inhibitor Resistant NSCLC
7.3. Targeting STAT3 to Overcome Chemo- and Radiotherapy Resistance
7.4. Combining STAT3 Inhibition with Immune Checkpoint Blockade
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Sabanathan, S.; Richardson, J.; Mearns, A.J.; Goulden, C. Results of surgical treatment of stage I and II lung cancer. J. Cardiovasc. Surg. 1996, 37, 169–172. [Google Scholar]
- Nesbitt, J.C.; Putnam, J.B., Jr.; Walsh, G.L.; Roth, J.A.; Mountain, C.F. Survival in early-stage non-small cell lung cancer. Ann. Thorac. Surg. 1995, 60, 466–472. [Google Scholar] [CrossRef]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef]
- Walters, S.; Maringe, C.; Coleman, M.P.; Peake, M.D.; Butler, J.; Young, N.; Bergström, S.; Hanna, L.; Jakobsen, E.; Kölbeck, K.; et al. Lung cancer survival and stage at diagnosis in Australia, Canada, Denmark, Norway, Sweden and the UK: A population-based study, 2004–2007. Thorax 2013, 68, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-G.; Shih, J.-Y. Management of acquired resistance to EGFR TKI–targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Addeo, A.; Passaro, A.; Malapelle, U.; Luigi Banna, G.; Subbiah, V.; Friedlaender, A. Immunotherapy in non-small cell lung cancer harbouring driver mutations. Cancer Treat. Rev. 2021, 96, 102179. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, N.; Bazhenova, L. New strategies in immunotherapy for lung cancer: Beyond PD-1/PD-L1. Ther. Adv. Respir. Dis. 2018, 12, 1753466618794133. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [PubMed]
- Dudka, A.A.; Sweet, S.M.M.; Heath, J.K. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. 2010, 70, 3391–3401. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Sen, B.; Saigal, B.; Diao, L.; Wang, J.; Nanjundan, M.; Cascone, T.; Mills, G.B.; Heymach, J.V.; Johnson, F.M. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6852–6861. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef]
- Wormald, S.; Hilton, D.J. Inhibitors of cytokine signal transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.-S.; Cho, Y.-Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006, 16, 196–202. [Google Scholar] [CrossRef]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK–STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Zheng, Z.; Song, L.; Cantor, A.; Bepler, G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8288–8294. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ceja, S.G.; Reyes-Maldonado, E.; Vázquez-Manríquez, M.E.; López-Luna, J.J.; Belmont, A.; Gutiérrez-Castellanos, S. Differential expression of STAT5 and Bcl-xL, and high expression of Neu and STAT3 in non-small-cell lung carcinoma. Lung Cancer 2006, 54, 163–168. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, Y.; Li, Y.; Lv, T.; Liu, J.; Wang, X. Prognostic significance of STAT3 expression and its correlation with chemoresistance of non-small cell lung cancer cells. Acta Histochem. 2012, 114, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.J.; Jin, F.G.; Liu, T.G.; Fu, E.Q.; Xie, Y.H.; Sun, R.L. Overexpression of STAT3 potentiates growth, survival, and radioresistance of non-small-cell lung cancer (NSCLC) cells. J. Surg. Res. 2011, 171, 675–683. [Google Scholar] [CrossRef]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer 2012, 11, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Z. MicroRNA-19a functions as an oncogenic microRNA in non-small cell lung cancer by targeting the suppressor of cytokine signaling 1 and mediating STAT3 activation. Int. J. Mol. Med. 2015, 35, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Im, J.-Y.; Kim, B.-K.; Lee, K.-W.; Chun, S.-Y.; Kang, M.-J.; Won, M. DDIAS promotes STAT3 activation by preventing STAT3 recruitment to PTPRM in lung cancer cells. Oncogenesis 2020, 9, 1. [Google Scholar] [CrossRef]
- Kluge, A.; Dabir, S.; Vlassenbroeck, I.; Eisenberg, R.; Dowlati, A. Protein inhibitor of activated STAT3 expression in lung cancer. Mol. Oncol. 2011, 5, 256–264. [Google Scholar] [CrossRef]
- Schlessinger, K.; Levy, D.E. Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res. 2005, 65, 5828–5834. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Simons, M. Integrative signaling in angiogenesis. Mol. Cell. Biochem. 2004, 264, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Camps, C.; Sirera, R. Angiogenesis in non-small cell lung cancer: The prognostic impact of neoangiogenesis and the cytokines VEGF and bFGF in tumours and blood. Lung Cancer 2006, 51, 143–158. [Google Scholar] [CrossRef]
- Bačić, I.; Karlo, R.; Zadro, A.Š.; Zadro, Z.; Skitarelić, N.; Antabak, A. Tumor angiogenesis as an important prognostic factor in advanced non-small cell lung cancer (Stage IIIA). Oncol. Lett. 2018, 15, 2335–2339. [Google Scholar] [CrossRef]
- Zhao, M.; Gao, F.H.; Wang, J.Y.; Liu, F.; Yuan, H.H.; Zhang, W.Y.; Jiang, B. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer 2011, 73, 366–374. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lee, H.; Pal, S.; Jove, V.; Deng, J.; Zhang, W.; Hoon, D.S.; Wakabayashi, M.; Forman, S.; Yu, H. B cells promote tumor progression via STAT3 regulated-angiogenesis. PLoS ONE 2013, 8, e64159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Qu, Z.; Sun, F.; Han, L.; Li, L.; Yan, S.; Stabile, L.P.; Chen, L.F.; Siegfried, J.M.; Xiao, G. Myeloid STAT3 Promotes Lung Tumorigenesis by Transforming Tumor Immunosurveillance into Tumor-Promoting Inflammation. Cancer Immunol. Res. 2017, 5, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Lai, W.W.; Chen, H.H.W.; Liu, H.S.; Su, W.C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309. [Google Scholar] [CrossRef]
- Liang, L.; Hui, K.; Hu, C.; Wen, Y.; Yang, S.; Zhu, P.; Wang, L.; Xia, Y.; Qiao, Y.; Sun, W.; et al. Autophagy inhibition potentiates the anti-angiogenic property of multikinase inhibitor anlotinib through JAK2/STAT3/VEGFA signaling in non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 71. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Mi, Z.; Jiang, X.; Sun, L.; Zheng, B.; Wang, J.; Meng, M.; Zhang, L.; Wang, Z.; et al. STAT3/miR-135b/NF-κB axis confers aggressiveness and unfavorable prognosis in non-small-cell lung cancer. Cell Death Dis. 2021, 12, 493. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zheng, Y.; Tan, J.; Tian, R.; Shen, P.; Cai, W.; Liao, H. MiR-199a-5p–HIF-1α-STAT3 Positive Feedback Loop Contributes to the Progression of Non-Small Cell Lung Cancer. Front. Cell Dev. Biol. 2021, 8, 1931. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef]
- Shirogane, T.; Fukada, T.; Muller, J.M.; Shima, D.T.; Hibi, M.; Hirano, T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity 1999, 11, 709–719. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ray, R.M.; Johnson, L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 2005, 392, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.V.; Greulich, H.; Sellers, W.R.; Meyerson, M.; Frank, D.A. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006, 66, 3162–3168. [Google Scholar] [CrossRef] [PubMed]
- Akca, H.; Tani, M.; Hishida, T.; Matsumoto, S.; Yokota, J. Activation of the AKT and STAT3 pathways and prolonged survival by a mutant EGFR in human lung cancer cells. Lung Cancer 2006, 54, 25–33. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zeng, H.; Ying, K. The combination of stem cell markers CD133 and ABCG2 predicts relapse in stage I non-small cell lung carcinomas. Med. Oncol. 2011, 28, 1458–1462. [Google Scholar] [CrossRef] [PubMed]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung cancer stem cells-origin, characteristics and therapy. Stem Cell Investig. 2018, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Qi, X.-W.; Yan, G.-N.; Zhang, Q.-B.; Xu, C.; Bian, X.-W. Is CD133 Expression a Prognostic Biomarker of Non-Small-Cell Lung Cancer? A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e100168. [Google Scholar] [CrossRef]
- Akunuru, S.; James Zhai, Q.; Zheng, Y. Non-small cell lung cancer stem/progenitor cells are enriched in multiple distinct phenotypic subpopulations and exhibit plasticity. Cell Death Dis. 2012, 3, e352. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, S.I.; Kim, R.K.; Cho, E.W.; Kim, I.G. Tescalcin/c-Src/IGF1Rβ-mediated STAT3 activation enhances cancer stemness and radioresistant properties through ALDH1. Sci. Rep. 2018, 8, 10711. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef]
- Hsu, H.-S.; Huang, P.-I.; Chang, Y.-L.; Tzao, C.; Chen, Y.-W.; Shih, H.-C.; Hung, S.-C.; Chen, Y.-C.; Tseng, L.-M.; Chiou, S.-H. Cucurbitacin I inhibits tumorigenic ability and enhances radiochemosensitivity in nonsmall cell lung cancer-derived CD133-positive cells. Cancer 2011, 117, 2970–2985. [Google Scholar] [CrossRef]
- Ho, J.N.; Kang, G.Y.; Lee, S.S.; Kim, J.; Bae, I.H.; Hwang, S.G.; Um, H.D. Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci. 2010, 101, 1417–1423. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. BBI608 inhibits cancer stemness and reverses cisplatin resistance in NSCLC. Cancer Lett. 2018, 428, 117–126. [Google Scholar] [CrossRef]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-Addicted Non-Small-Cell Lung Cancer: Treatment Opportunities and Future Perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- Riely, G.J.; Pao, W.; Pham, D.; Li, A.R.; Rizvi, N.; Venkatraman, E.S.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M.; Miller, V.A. Clinical Course of Patients with Non–Small Cell Lung Cancer and Epidermal Growth Factor Receptor Exon 19 and Exon 21 Mutations Treated with Gefitinib or Erlotinib. Clin. Cancer Res. 2006, 12, 839. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Jänne, P.A. Mechanisms of Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 2895. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Busund, L.T.; Kilvær, T.L.; Andersen, S.; Richardsen, E.; Paulsen, E.E.; Hald, S.; Khanehkenari, M.R.; Cooper, W.A.; Kao, S.C.; et al. The Role of Tumor-Infiltrating Lymphocytes in Development, Progression, and Prognosis of Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok Jedd, D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, H.; Qin, Y.; Roberts, J.; Cummings, O.W.; Yan, C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007, 67, 8494–8503. [Google Scholar] [CrossRef] [PubMed]
- Ihara, S.; Kida, H.; Arase, H.; Tripathi, L.P.; Chen, Y.A.; Kimura, T.; Yoshida, M.; Kashiwa, Y.; Hirata, H.; Fukamizu, R.; et al. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis. Cancer Res. 2012, 72, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.A. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, B.; Jiang, R.; Li, J.; Wang, B. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell. Immunol. 2017, 311, 86–93. [Google Scholar] [CrossRef]
- Yuan, F.; Fu, X.; Shi, H.; Chen, G.; Dong, P.; Zhang, W. Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PLoS ONE 2014, 9, e107063. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Liu, X.; Xue, S.; Chen, D.; Zou, J.; Jiang, H. TGR5 deficiency activates antitumor immunity in non-small cell lung cancer via restraining M2 macrophage polarization. Acta Pharm. Sin. B 2021, in press. [Google Scholar] [CrossRef]
- Wölfle, S.J.; Strebovsky, J.; Bartz, H.; Sähr, A.; Arnold, C.; Kaiser, C.; Dalpke, A.H.; Heeg, K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 2011, 41, 413–424. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A critical role for Stat3 signaling in immune tolerance. Immunity 2003, 19, 425–436. [Google Scholar] [CrossRef]
- Brayer, J.; Cheng, F.; Wang, H.; Horna, P.; Vicente-Suarez, I.; Pinilla-Ibarz, J.; Sotomayor, E.M. Enhanced CD8 T cell cross-presentation by macrophages with targeted disruption of STAT3. Immunol. Lett. 2010, 131, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Kortylewski, M.; Kujawski, M.; Zhang, C.; Reckamp, K.; Armstrong, B.; Wang, L.; Kowolik, C.; Deng, J.; Figlin, R.; et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010, 70, 7455–7464. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Herrmann, A.; Yang, C.; Wang, L.; Liu, Y.; Salcedo, R.; Yu, H. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009, 69, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, X.; Huang, G.; Cheng, L.; Lv, J.; Zheng, D.; Lin, S.; Wang, S.; Wu, Q.; Long, Y.; et al. Myeloid-derived suppressor cells promote lung cancer metastasis by CCL11 to activate ERK and AKT signaling and induce epithelial-mesenchymal transition in tumor cells. Oncogene 2021, 40, 1476–1489. [Google Scholar] [CrossRef]
- Rébé, C.; Végran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation: A key factor in tumor immunoescape. Jakstat 2013, 2, e23010. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005, 65, 9525–9535. [Google Scholar] [CrossRef]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Du, H.; Li, Y.; Qu, P.; Yan, C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid-derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am. J. Pathol. 2011, 179, 2131–2141. [Google Scholar] [CrossRef]
- Sumida, K.; Ohno, Y.; Ohtake, J.; Kaneumi, S.; Kishikawa, T.; Takahashi, N.; Taketomi, A.; Kitamura, H. IL-11 induces differentiation of myeloid-derived suppressor cells through activation of STAT3 signalling pathway. Sci. Rep. 2015, 5, 13650. [Google Scholar] [CrossRef] [PubMed]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras–Mutant Lung Cancer. Cancer Res. 2016, 76, 3189. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Huang, X.; Li, F.-R. Impaired dendritic cell functions in lung cancer: A review of recent advances and future perspectives. Cancer Commun. 2019, 39, 43. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Hoffmann, H.; Dienemann, H.; Schnabel, P.A.; Enk, A.H.; Ring, S.; Mahnke, K. Non-small cell lung cancer induces an immunosuppressive phenotype of dendritic cells in tumor microenvironment by upregulating B7-H3. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Nefedova, Y.; Cheng, P.; Gilkes, D.; Blaskovich, M.; Beg, A.A.; Sebti, S.M.; Gabrilovich, D.I. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J. Immunol. 2005, 175, 4338–4346. [Google Scholar] [CrossRef] [PubMed]
- Melillo, J.A.; Song, L.; Bhagat, G.; Blazquez, A.B.; Plumlee, C.R.; Lee, C.; Berin, C.; Reizis, B.; Schindler, C. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol. 2010, 184, 2638–2645. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef]
- Mitchell, K.G.; Diao, L.; Karpinets, T.; Negrao, M.V.; Tran, H.T.; Parra, E.R.; Corsini, E.M.; Reuben, A.; Federico, L.; Bernatchez, C.; et al. Neutrophil expansion defines an immunoinhibitory peripheral and intratumoral inflammatory milieu in resected non-small cell lung cancer: A descriptive analysis of a prospectively immunoprofiled cohort. J. ImmunoTher. Cancer 2020, 8, e000405. [Google Scholar] [CrossRef]
- Knaapen, A.M.; Schins, R.P.F.; Polat, D.; Becker, A.; Borm, P.J.A. Mechanisms of neutrophil-induced DNA damage in respiratory tract epithelial cells. Mol. Cell. Biochem. 2002, 234, 143–151. [Google Scholar] [CrossRef]
- Hattar, K.; Franz, K.; Ludwig, M.; Sibelius, U.; Wilhelm, J.; Lohmeyer, J.; Savai, R.; Subtil, F.S.B.; Dahlem, G.; Eul, B.; et al. Interactions between neutrophils and non-small cell lung cancer cells: Enhancement of tumor proliferation and inflammatory mediator synthesis. Cancer Immunol. Immunother. 2014, 63, 1297–1306. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Zhang, L.; Snow, J.W.; Jones, D.M.; Smith, A.M.; El Kasmi, K.C.; Liu, F.; Goldsmith, M.A.; Link, D.C.; Murray, P.J.; et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood 2006, 108, 3682–3690. [Google Scholar] [CrossRef]
- Zhang, H.; Nguyen-Jackson, H.; Panopoulos, A.D.; Li, H.S.; Murray, P.J.; Watowich, S.S. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 2010, 116, 2462–2471. [Google Scholar] [CrossRef]
- Cacalano, N.A. Regulation of Natural Killer Cell Function by STAT3. Front. Immunol. 2016, 7, 128. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Straka, E.; Kudweis, P.; Biaggio, M.; Poli, V.; Strobl, B.; Müller, M.; Sexl, V. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood 2014, 124, 2370–2379. [Google Scholar] [CrossRef]
- Kortylewski, M.; Yu, H. Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 2008, 20, 228–233. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Liu, J.; Zhang, Z.; Mao, X.; Zhou, H. MicroRNA-130a enhances the killing ability of natural killer cells against non-small cell lung cancer cells by targeting signal transducers and activators of transcription 3. Biochem. Biophys. Res. Commun. 2020, 523, 481–486. [Google Scholar] [CrossRef]
- Chou, W.-C.; Levy, D.E.; Lee, C.-K. STAT3 positively regulates an early step in B-cell development. Blood 2006, 108, 3005–3011. [Google Scholar] [CrossRef]
- Ding, C.; Chen, X.; Dascani, P.; Hu, X.; Bolli, R.; Zhang, H.-G.; McLeish, K.R.; Yan, J. STAT3 Signaling in B Cells Is Critical for Germinal Center Maintenance and Contributes to the Pathogenesis of Murine Models of Lupus. J. Immunol. 2016, 196, 4477–4486. [Google Scholar] [CrossRef]
- Herrmann, A.; Lahtz, C.; Nagao, T.; Song, J.Y.; Chan, W.C.; Lee, H.; Yue, C.; Look, T.; Mülfarth, R.; Li, W.; et al. CTLA4 Promotes Tyk2-STAT3-Dependent B-cell Oncogenicity. Cancer Res. 2017, 77, 5118–5128. [Google Scholar] [CrossRef]
- Zhang, C.; Xin, H.; Zhang, W.; Yazaki, P.J.; Zhang, Z.; Le, K.; Li, W.; Lee, H.; Kwak, L.; Forman, S.; et al. CD5 Binds to Interleukin-6 and Induces a Feed-Forward Loop with the Transcription Factor STAT3 in B Cells to Promote Cancer. Immunity 2016, 44, 913–923. [Google Scholar] [CrossRef]
- Zhou, J.; Min, Z.; Zhang, D.; Wang, W.; Marincola, F.; Wang, X. Enhanced frequency and potential mechanism of B regulatory cells in patients with lung cancer. J. Transl. Med. 2014, 12, 304. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, Q.; Tang, K.; Sun, Y.; Zhang, K.; Zhang, Y.; Luo, S.; Zhang, H.; Ye, D.; Huang, B. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett. 2015, 364, 118–124. [Google Scholar] [CrossRef]
- Yu, P.; Lee, Y.; Liu, W.; Krausz, T.; Chong, A.; Schreiber, H.; Fu, Y.X. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 2005, 201, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Flavell, R.A. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 2007, 445, 766–770. [Google Scholar] [CrossRef]
- Kinjyo, I.; Inoue, H.; Hamano, S.; Fukuyama, S.; Yoshimura, T.; Koga, K.; Takaki, H.; Himeno, K.; Takaesu, G.; Kobayashi, T.; et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J. Exp. Med. 2006, 203, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Santner-Nanan, B.; Hu, M.; Skarratt, K.; Lee, C.H.; Stormon, M.; Wong, M.; Fuller, S.J.; Nanan, R. IL-10 Potentiates Differentiation of Human Induced Regulatory T Cells via STAT3 and Foxo1. J. Immunol. 2015, 195, 3665. [Google Scholar] [CrossRef]
- Kong, L.Y.; Wei, J.; Sharma, A.K.; Barr, J.; Abou-Ghazal, M.K.; Fokt, I.; Weinberg, J.; Rao, G.; Grimm, E.; Priebe, W.; et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol. Immunother. 2009, 58, 1023–1032. [Google Scholar] [CrossRef]
- Reuben, A.; Zhang, J.; Chiou, S.-H.; Gittelman, R.M.; Li, J.; Lee, W.-C.; Fujimoto, J.; Behrens, C.; Liu, X.; Wang, F.; et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Commun. 2020, 11, 603. [Google Scholar] [CrossRef]
- Hiraoka, K.; Miyamoto, M.; Cho, Y.; Suzuoki, M.; Oshikiri, T.; Nakakubo, Y.; Itoh, T.; Ohbuchi, T.; Kondo, S.; Katoh, H. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br. J. Cancer 2006, 94, 275–280. [Google Scholar] [CrossRef]
- Kawai, O.; Ishii, G.; Kubota, K.; Murata, Y.; Naito, Y.; Mizuno, T.; Aokage, K.; Saijo, N.; Nishiwaki, Y.; Gemma, A.; et al. Predominant infiltration of macrophages and CD8(+) T Cells in cancer nests is a significant predictor of survival in stage IV nonsmall cell lung cancer. Cancer 2008, 113, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Prado-Garcia, H.; Aguilar-Cazares, D.; Flores-Vergara, H.; Mandoki, J.J.; Lopez-Gonzalez, J.S. Effector, memory and naïve CD8+ T cells in peripheral blood and pleural effusion from lung adenocarcinoma patients. Lung Cancer 2005, 47, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Trojan, A.; Urosevic, M.; Dummer, R.; Giger, R.; Weder, W.; Stahel, R.A. Immune activation status of CD8+ T cells infiltrating non-small cell lung cancer. Lung Cancer 2004, 44, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.M.; Yu, C.R.; Golestaneh, N.; Amadi-Obi, A.; Lee, Y.S.; Eseonu, A.; Mahdi, R.M.; Egwuagu, C.E. STAT3 protein promotes T-cell survival and inhibits interleukin-2 production through up-regulation of Class O Forkhead transcription factors. J. Biol. Chem. 2011, 286, 30888–30897. [Google Scholar] [CrossRef]
- Schmetterer, K.G.; Neunkirchner, A.; Wojta-Stremayr, D.; Leitner, J.; Steinberger, P.; Pickl, W.F. STAT3 governs hyporesponsiveness and granzyme B-dependent suppressive capacity in human CD4+ T cells. FASEB J. 2015, 29, 759–771. [Google Scholar] [CrossRef]
- Austin, J.W.; Lu, P.; Majumder, P.; Ahmed, R.; Boss, J.M. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J. Immunol. 2014, 192, 4876–4886. [Google Scholar] [CrossRef] [PubMed]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010, 70, 9599–9610. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Wang, L.; Cao, L.; Wang, H.; Liu, B.; Zhang, Q.; Meng, Z.; Wu, X.; Zhou, Q.; Xu, K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 2017, 8, 76116–76128. [Google Scholar] [CrossRef]
- Fan, J.; Xu, G.; Chang, Z.; Zhu, L.; Yao, J. miR-210 transferred by lung cancer cell-derived exosomes may act as proangiogenic factor in cancer-associated fibroblasts by modulating JAK2/STAT3 pathway. Clin. Sci. 2020, 134, 807–825. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef]
- Teramoto, K.; Igarashi, T.; Kataoka, Y.; Ishida, M.; Hanaoka, J.; Sumimoto, H.; Daigo, Y. Clinical significance of PD-L1-positive cancer-associated fibroblasts in pN0M0 non-small cell lung cancer. Lung Cancer 2019, 137, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.X.; Cao, Z.B.; He, T.T.; Huang, T.; Xiang, C.L.; Liu, Y. TGFβ1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. OncoTargets Ther. 2019, 12, 5323–5334. [Google Scholar] [CrossRef]
- Barrera, L.; Montes-Servín, E.; Barrera, A.; Ramírez-Tirado, L.A.; Salinas-Parra, F.; Bañales-Méndez, J.L.; Sandoval-Ríos, M.; Arrieta, Ó. Cytokine profile determined by data-mining analysis set into clusters of non-small-cell lung cancer patients according to prognosis. Ann. Oncol. 2015, 26, 428–435. [Google Scholar] [CrossRef]
- Pine, S.R.; Mechanic, L.E.; Enewold, L.; Chaturvedi, A.K.; Katki, H.A.; Zheng, Y.-L.; Bowman, E.D.; Engels, E.A.; Caporaso, N.E.; Harris, C.C. Increased levels of circulating interleukin 6, interleukin 8, C-reactive protein, and risk of lung cancer. J. Natl. Cancer Inst. 2011, 103, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, H.; Tomida, M.; Akiyama, H.; Nakajima, Y.; Okada, D.; Yoshino, N.; Takiguchi, Y.; Tanzawa, H. Serum hepatocyte growth factor and interleukin-6 are effective prognostic markers for non-small cell lung cancer. Anticancer Res. 2012, 32, 3251–3258. [Google Scholar]
- Song, L.; Smith, M.A.; Doshi, P.; Sasser, K.; Fulp, W.; Altiok, S.; Haura, E.B. Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J. Thorac. Oncol. 2014, 9, 974–982. [Google Scholar] [CrossRef]
- Carpagnano, G.E.; Resta, O.; Foschino-Barbaro, M.P.; Gramiccioni, E.; Carpagnano, F. Interleukin-6 is increased in breath condensate of patients with non-small cell lung cancer. Int. J. Biol. Markers 2002, 17, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kimura, H.; Yokota, S.; Yamamoto, Y.; Hashimoto, T.; Nakagawa, M.; Ito, M.; Ogura, T. Effect of IL-6 elevation in malignant pleural effusion on hyperfibrinogenemia in lung cancer patients. Jpn. J. Clin. Oncol. 2000, 30, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Hsiao, C.F.; Yeh, Y.M.; Chang, G.C.; Tsai, Y.H.; Chen, Y.M.; Huang, M.S.; Chen, H.L.; Li, Y.J.; Yang, P.C.; et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int. J. Cancer 2013, 132, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.M.; Mariano, V.S.; Pastrez, P.R.A.; Pinto, M.C.; Castro, A.G.; Syrjanen, K.J.; Longatto-Filho, A. High systemic IL-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS ONE 2017, 12, e0181125. [Google Scholar] [CrossRef]
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 Activates STAT3 Activity in Non-Small–Cell Lung Cancer Cells and IL-6 Neutralizing Antibodies Can Suppress JAK1-STAT3 Signaling. Mol. Cancer Ther. 2011, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, H.; Iizasa, T.; Koh, E.; Suzuki, M.; Otsuji, M.; Chang, H.; Motohashi, S.; Yokoi, S.; Hiroshima, K.; Tagawa, M.; et al. Correlation between interleukin 6 production and tumor proliferation in non-small cell lung cancer. Cancer Immunol. Immunother. 2004, 53, 786–792. [Google Scholar] [CrossRef]
- Rice, S.J.; Liu, X.; Zhang, J.; Jia, B.; Zheng, H.; Belani, C.P. Advanced NSCLC patients with high IL-6 levels have altered peripheral T cell population and signaling. Lung Cancer 2019, 131, 58–61. [Google Scholar] [CrossRef]
- Kang, D.H.; Park, C.K.; Chung, C.; Oh, I.J.; Kim, Y.C.; Park, D.; Kim, J.; Kwon, G.C.; Kwon, I.; Sun, P.; et al. Baseline Serum Interleukin-6 Levels Predict the Response of Patients with Advanced Non-small Cell Lung Cancer to PD-1/PD-L1 Inhibitors. Immune Netw. 2020, 20, e27. [Google Scholar] [CrossRef] [PubMed]
- Keegan, A.; Ricciuti, B.; Garden, P.; Cohen, L.; Nishihara, R.; Adeni, A.; Paweletz, C.; Supplee, J.; Jänne, P.A.; Severgnini, M.; et al. Plasma IL-6 changes correlate to PD-1 inhibitor responses in NSCLC. J. Immunother. Cancer 2020, 8, e000678. [Google Scholar] [CrossRef]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef]
- Li, L.; Han, R.; Xiao, H.; Lin, C.; Wang, Y.; Liu, H.; Li, K.; Chen, H.; Sun, F.; Yang, Z.; et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin. Cancer Res. 2014, 20, 2714–2726. [Google Scholar] [CrossRef]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Lu, G.; Yao, Y.; Gu, W. An Autocrine IL-6/IGF-1R Loop Mediates EMT and Promotes Tumor Growth in Non-small Cell Lung Cancer. Int. J. Biol. Sci. 2019, 15, 1882–1891. [Google Scholar] [CrossRef]
- Schütz, A.; Röser, K.; Klitzsch, J.; Lieder, F.; Aberger, F.; Gruber, W.; Mueller, K.M.; Pupyshev, A.; Moriggl, R.; Friedrich, K. Lung Adenocarcinomas and Lung Cancer Cell Lines Show Association of MMP-1 Expression With STAT3 Activation. Transl. Oncol. 2015, 8, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, X.; Tsai, Y.; Yang, L.; Chuang, K.H.; Keng, P.C.; Lee, S.O.; Chen, Y. IL-6 Mediates Macrophage Infiltration after Irradiation via Up-regulation of CCL2/CCL5 in Non-small Cell Lung Cancer. Radiat. Res. 2017, 187, 50–59. [Google Scholar] [CrossRef]
- Naqash, A.R.; Yang, L.V.; Sanderlin, E.J.; Atwell, D.C.; Walker, P.R. Interleukin-6 as one of the potential mediators of immune-related adverse events in non-small cell lung cancer patients treated with immune checkpoint blockade: Evidence from a case report. Acta Oncol. 2018, 57, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Shi, Q.; Williams, L.A.; Mao, L.; Cleeland, C.S.; Komaki, R.R.; Mobley, G.M.; Liao, Z. Inflammatory cytokines are associated with the development of symptom burden in patients with NSCLC undergoing concurrent chemoradiation therapy. Brain Behav. Immun. 2010, 24, 968–974. [Google Scholar] [CrossRef]
- Ke, W.; Zhang, L.; Dai, Y. The role of IL-6 in immunotherapy of non-small cell lung cancer (NSCLC) with immune-related adverse events (irAEs). Thorac. Cancer 2020, 11, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.-I.; Wang, Y.-C.; Hung, C.-Y.; Yu, C.-H.; Su, W.-C.; Chang, W.-C.; Hung, J.-J. Positive feedback regulation between IL10 and EGFR promotes lung cancer formation. Oncotarget 2016, 7, 20840–20854. [Google Scholar] [CrossRef]
- Zeni, E.; Mazzetti, L.; Miotto, D.; Lo Cascio, N.; Maestrelli, P.; Querzoli, P.; Pedriali, M.; De Rosa, E.; Fabbri, L.M.; Mapp, C.E.; et al. Macrophage expression of interleukin-10 is a prognostic factor in nonsmall cell lung cancer. Eur. Respir. J. 2007, 30, 627. [Google Scholar] [CrossRef]
- Vahl, J.M.; Friedrich, J.; Mittler, S.; Trump, S.; Heim, L.; Kachler, K.; Balabko, L.; Fuhrich, N.; Geppert, C.-I.; Trufa, D.I.; et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br. J. Cancer 2017, 117, 1644–1655. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, Y.; Liu, R.; Qi, J.; Hou, Y.; Chang, J.; Ren, L. Upregulation of IL-11, an IL-6 Family Cytokine, Promotes Tumor Progression and Correlates with Poor Prognosis in Non-Small Cell Lung Cancer. Cell Physiol. Biochem. 2018, 45, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, J.; Lv, X.; Yang, Q.; Yao, S.; Zhang, D.; Chen, J. Clinical value of serum and exhaled breath condensate inflammatory factor IL-11 levels in non-small cell lung cancer: Clinical value of IL-11 in non-small cell lung cancer. Int. J. Biol. Markers 2021, 36, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Pan, B.; Che, D.; Cao, J.; Shen, J.; Jin, S.; Zhou, Y.; Liu, F.; Gu, K.; Man, Y.; Shang, L.; et al. Interleukin-17 levels correlate with poor prognosis and vascular endothelial growth factor concentration in the serum of patients with non-small cell lung cancer. Biomarkers 2015, 20, 232–239. [Google Scholar] [CrossRef]
- Chen, X.; Wan, J.; Liu, J.; Xie, W.; Diao, X.; Xu, J.; Zhu, B.; Chen, Z. Increased IL-17-producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer 2010, 69, 348–354. [Google Scholar] [CrossRef]
- Pan, B.; Shen, J.; Cao, J.; Zhou, Y.; Shang, L.; Jin, S.; Cao, S.; Che, D.; Liu, F.; Yu, Y. Interleukin-17 promotes angiogenesis by stimulating VEGF production of cancer cells via the STAT3/GIV signaling pathway in non-small-cell lung cancer. Sci. Rep. 2015, 5, 16053. [Google Scholar] [CrossRef]
- Numasaki, M.; Watanabe, M.; Suzuki, T.; Takahashi, H.; Nakamura, A.; McAllister, F.; Hishinuma, T.; Goto, J.; Lotze, M.T.; Kolls, J.K.; et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J. Immunol. 2005, 175, 6177–6189. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Han, J.; Fan, J.; Duan, L.; Guo, M.; Lv, Z.; Hu, G.; Chen, L.; Wu, F.; Tao, X.; et al. IL-17 induces EMT via Stat3 in lung adenocarcinoma. Am. J. Cancer Res. 2016, 6, 440–451. [Google Scholar]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Cao, J.; Jin, S.; Lv, L.; Qi, L.; Liu, F.; Geng, J.; Yu, Y. Interleukin-22 promotes lung cancer cell proliferation and migration via the IL-22R1/STAT3 and IL-22R1/AKT signaling pathways. Mol. Cell. Biochem. 2016, 415, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guillon, A.; Gueugnon, F.; Mavridis, K.; Dalloneau, E.; Jouan, Y.; Diot, P.; Heuzé-Vourc’h, N.; Courty, Y.; Si-Tahar, M. Interleukin-22 receptor is overexpressed in nonsmall cell lung cancer and portends a poor prognosis. Eur. Respir. J. 2016, 47, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, J.; Chen, J.; Jin, S.; Yao, J.; Yu, T.; Wang, W.; Guo, R. IL-22 Confers EGFR-TKI Resistance in NSCLC via the AKT and ERK Signaling Pathways. Front. Oncol. 2019, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, N.; Caetano, M.S.; Cumpian, A.M.; Unver, N.; De la Garza Ramos, C.; Noble, O.; Daliri, S.; Hernandez, B.J.; Gutierrez, B.A.; Evans, S.E.; et al. IL22 Promotes Kras-Mutant Lung Cancer by Induction of a Protumor Immune Response and Protection of Stemness Properties. Cancer Immunol. Res. 2018, 6, 788–797. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Wei, H.; Zheng, C.; Sun, R.; Zhang, J.; Tian, Z. Antiapoptotic activity of autocrine interleukin-22 and therapeutic effects of interleukin-22-small interfering RNA on human lung cancer xenografts. Clin. Cancer Res. 2008, 14, 6432–6439. [Google Scholar] [CrossRef]
- Larochette, V.; Miot, C.; Poli, C.; Beaumont, E.; Roingeard, P.; Fickenscher, H.; Jeannin, P.; Delneste, Y. IL-26, a Cytokine With Roles in Extracellular DNA-Induced Inflammation and Microbial Defense. Front. Immunol. 2019, 10, 204. [Google Scholar] [CrossRef]
- Niu, Y.; Ye, L.; Peng, W.; Wang, Z.; Wei, X.; Wang, X.; Li, Y.; Zhang, S.; Xiang, X.; Zhou, Q. IL-26 promotes the pathogenesis of malignant pleural effusion by enhancing CD4+IL-22+ T-cell differentiation and inhibiting CD8+ T-cell cytotoxicity. J. Leukoc. Biol. 2021, 110, 39–52. [Google Scholar] [CrossRef]
- Wang, H.; Si, S.n.; Jiang, M.; Chen, L.; Huang, K.; Yu, W. Leukemia inhibitory factor is involved in the pathogenesis of NSCLC through activation of the STAT3 signaling pathway. Oncol. Lett. 2021, 22, 663. [Google Scholar] [CrossRef]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer 2017, 16, 2234–2245. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Weissfeld, L.A.; Singh-Kaw, P.; Weyant, R.J.; Testa, J.R.; Landreneau, R.J. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. 1997, 57, 433–439. [Google Scholar] [PubMed]
- Hosoda, H.; Izumi, H.; Tukada, Y.; Takagiwa, J.; Chiaki, T.; Yano, M.; Arai, H. Plasma hepatocyte growth factor elevation may be associated with early metastatic disease in primary lung cancer patients. Ann. Thorac. Cardiovasc. Surg. 2012, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Sakamori, Y.; Ozasa, H.; Yagi, Y.; Ajimizu, H.; Yasuda, Y.; Funazo, T.; Nomizo, T.; Yoshida, H.; Nagai, H.; et al. Clinical impact of high serum hepatocyte growth factor in advanced non-small cell lung cancer. Oncotarget 2017, 8, 71805–71816. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; Vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Shao, Y.F.; Zhao, X.; Geng, Y.T.; Wang, K.; Yin, Y.M. Expression and clinical significance of leptin, the functional receptor of leptin (OB-Rb) and HER-2 in non-small-cell lung cancer: A retrospective analysis. J. Cancer Res. Clin. Oncol. 2011, 137, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liu, Q.; Zhang, N.; Zheng, L.; Sang, M.; Feng, J.; Zhang, J.; Wu, X.; Shan, B. Leptin promotes metastasis by inducing an epithelial-mesenchymal transition in A549 lung cancer cells. Oncol. Res. 2013, 21, 165–171. [Google Scholar] [CrossRef]
- Zheng, X.J.; Yang, Z.X.; Dong, Y.J.; Zhang, G.Y.; Sun, M.F.; An, X.K.; Pan, L.H.; Zhang, S.L. Downregulation of leptin inhibits growth and induces apoptosis of lung cancer cells via the Notch and JAK/STAT3 signaling pathways. Biol. Open 2016, 5, 794–800. [Google Scholar] [CrossRef]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef]
- Rigas, J.R.; Schuster, M.; Orlov, S.V.; Milovanovic, B.; Prabhash, K.; Smith, J.T. Efect of ALD518, a humanized anti-IL-6 antibody, on lean body mass loss and symptoms in patients with advanced non-small cell lung cancer (NSCLC): Results of a phase II randomized, double-blind safety and efficacy trial. J. Clin. Oncol. 2010, 28, 7622. [Google Scholar] [CrossRef]
- Schuster, M.; Rigas, J.R.; Orlov, S.V.; Milovanovic, B.; Prabhash, K.; Smith, J.T. ALD518, a humanized anti-IL-6 antibody, treats anemia in patients with advanced non-small cell lung cancer (NSCLC): Results of a phase II, randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 2010, 28, 7631. [Google Scholar] [CrossRef]
- Park, J.S.; Hong, M.H.; Chun, Y.J.; Kim, H.R.; Cho, B.C. A phase Ib study of the combination of afatinib and ruxolitinib in EGFR mutant NSCLC with progression on EGFR-TKIs. Lung Cancer 2019, 134, 46–51. [Google Scholar] [CrossRef]
- Giaccone, G.; Sanborn, R.E.; Waqar, S.N.; Martinez-Marti, A.; Ponce, S.; Zhen, H.; Kennealey, G.; Erickson-Viitanen, S.; Schaefer, E. A Placebo-Controlled Phase II Study of Ruxolitinib in Combination With Pemetrexed and Cisplatin for First-Line Treatment of Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer and Systemic Inflammation. Clin. Lung Cancer 2018, 19, e567–e574. [Google Scholar] [CrossRef]
- Yu, H.A.; Perez, L.; Chang, Q.; Gao, S.P.; Kris, M.G.; Riely, G.J.; Bromberg, J. A Phase 1/2 Trial of Ruxolitinib and Erlotinib in Patients with EGFR-Mutant Lung Adenocarcinomas with Acquired Resistance to Erlotinib. J. Thorac. Oncol. 2017, 12, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Lorusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist 2013, 18, 819–820. [Google Scholar] [CrossRef]
- Barbie, D.A.; Spira, A.; Kelly, K.; Humeniuk, R.; Kawashima, J.; Kong, S.; Koczywas, M. Phase 1B Study of Momelotinib Combined With Trametinib in Metastatic, Kirsten Rat Sarcoma Viral Oncogene Homolog-Mutated Non-Small-Cell Lung Cancer After Platinum-Based Chemotherapy Treatment Failure. Clin. Lung Cancer 2018, 19, e853–e859. [Google Scholar] [CrossRef] [PubMed]
- Padda, S.K.; Reckamp, K.L.; Koczywas, M.; Neal, J.W.; Kawashima, J.; Kong, S.; Huang, D.B.; Kowalski, M.; Wakelee, H.A. A phase 1b study of erlotinib and momelotinib for the treatment of EGFR-mutated, tyrosine kinase inhibitor-naive metastatic non-small cell lung cancer. Cancer Chemother. Pharmacol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Johnson, F.M.; Bekele, B.N.; Feng, L.; Wistuba, I.; Tang, X.M.; Tran, H.T.; Erasmus, J.J.; Hwang, L.-L.; Takebe, N.; Blumenschein, G.R.; et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4609–4615. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Costa, D.B.; Heist, R.S.; Garcia, E.; Lindeman, N.I.; Sholl, L.M.; Oxnard, G.R.; Johnson, B.E.; Hammerman, P.S. Treatment-related toxicities in a phase II trial of dasatinib in patients with squamous cell carcinoma of the lung. J. Thorac. Oncol. 2013, 8, 1434–1437. [Google Scholar] [CrossRef]
- Creelan, B.C.; Gray, J.E.; Tanvetyanon, T.; Chiappori, A.A.; Yoshida, T.; Schell, M.J.; Antonia, S.J.; Haura, E.B. Phase 1 trial of dasatinib combined with afatinib for epidermal growth factor receptor- (EGFR-) mutated lung cancer with acquired tyrosine kinase inhibitor (TKI) resistance. Br. J. Cancer 2019, 120, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Liu, S.V.; Crawford, J.; Torres, T.; Chen, V.; Thompson, J.; Tan, M.; Esposito, G.; Subramaniam, D.S.; Giaccone, G. A Phase I Trial of Dasatinib and Osimertinib in TKI Naïve Patients With Advanced EGFR-Mutant Non-Small-Cell Lung Cancer. Front. Oncol. 2021, 11, 3550. [Google Scholar] [CrossRef]
- Gold, K.A.; Lee, J.J.; Harun, N.; Tang, X.; Price, J.; Kawedia, J.D.; Tran, H.T.; Erasmus, J.J.; Blumenschein, G.R.; William, W.N.; et al. A phase I/II study combining erlotinib and dasatinib for non-small cell lung cancer. Oncologist 2014, 19, 1040–1041. [Google Scholar] [CrossRef]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Hong, D.S.; Burris, H.A., III; Naing, A.; Jones, S.F.; Falchook, G.; Bricmont, P.; Elekes, A.; Rock, E.P.; Kurzrock, R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Pirker, R.; Filipits, M. Cetuximab in non-small-cell lung cancer. Transl. Lung Cancer Res. 2012, 1, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Rajan, A.; Giaccone, G. Tyrosine kinase inhibitors in lung cancer. Hematol. Oncol. Clin. N. Am. 2012, 26, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Verlicchi, A.; Gkountakos, A.; Pilotto, S.; Santarpia, M.; Chaib, I.; Ramirez Serrano, J.L.; Viteri, S.; Morales-Espinosa, D.; Dazzi, C.; et al. Molecular Bases for Combinatorial Treatment Strategies in Patients with KRAS Mutant Lung Adenocarcinoma and Squamous Cell Lung Carcinoma. Pulm. Ther. 2016, 2, 1–18. [Google Scholar] [CrossRef]
- Ma, C.; Wei, S.; Song, Y. T790M and acquired resistance of EGFR TKI: A literature review of clinical reports. J. Thorac. Dis. 2011, 3, 10–18. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef]
- Taverna, J.A.; Hung, C.N.; DeArmond, D.T.; Chen, M.; Lin, C.L.; Osmulski, P.A.; Gaczynska, M.E.; Wang, C.M.; Lucio, N.D.; Chou, C.W.; et al. Single-Cell Proteomic Profiling Identifies Combined AXL and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 2020, 80, 1551–1563. [Google Scholar] [CrossRef]
- Mohrherr, J.; Haber, M.; Breitenecker, K.; Aigner, P.; Moritsch, S.; Voronin, V.; Eferl, R.; Moriggl, R.; Stoiber, D.; Győrffy, B.; et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int. J. Cancer 2019, 145, 3376–3388. [Google Scholar] [CrossRef]
- Hu, Y.; Hong, Y.; Xu, Y.; Liu, P.; Guo, D.H.; Chen, Y. Inhibition of the JAK/STAT pathway with ruxolitinib overcomes cisplatin resistance in non-small-cell lung cancer NSCLC. Apoptosis 2014, 19, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Takigawa, N.; Ninomiya, T.; Ochi, N.; Yasugi, M.; Honda, Y.; Kubo, T.; Ichihara, E.; Hotta, K.; Tanimoto, M.; et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer 2014, 83, 30–36. [Google Scholar] [CrossRef]
- Song, L.; Morris, M.; Bagui, T.; Lee, F.Y.; Jove, R.; Haura, E.B. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006, 66, 5542–5548. [Google Scholar] [CrossRef]
- Kelley, M.J.; Jha, G.; Shoemaker, D.; Herndon, J.E., II; Gu, L.; Barry, W.T.; Crawford, J.; Ready, N. Phase II Study of Dasatinib in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Cancer Investig. 2017, 35, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.M.; Bharadwaj, U.; Eckols, T.K.; Kolosov, M.; Kasembeli, M.M.; Fridley, C.; Siller, R.; Tweardy, D.J. Small-molecule targeting of signal transducer and activator of transcription (STAT) 3 to treat non-small cell lung cancer. Lung Cancer 2015, 90, 182–190. [Google Scholar] [CrossRef]
- Brambilla, L.; Lahiri, T.; Cammer, M.; Levy, D.E. STAT3 Inhibitor OPB-51602 Is Cytotoxic to Tumor Cells through Inhibition of Complex I and ROS Induction. iScience 2020, 23, 101822. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Dong, H.; Mo, J.; Zhang, Y.; Huang, J.; Ouyang, S.; Shi, S.; Zhu, K.; Qu, X.; Hu, W.; et al. A novel STAT3 inhibitor W2014-S regresses human non-small cell lung cancer xenografts and sensitizes EGFR-TKI acquired resistance. Theranostics 2021, 11, 824–840. [Google Scholar] [CrossRef]
- Jing, N.; Tweardy, D.J. Targeting Stat3 in cancer therapy. Anticancer Drugs 2005, 16, 601–607. [Google Scholar] [CrossRef]
- Fletcher, S.; Drewry, J.A.; Shahani, V.M.; Page, B.D.; Gunning, P.T. Molecular disruption of oncogenic signal transducer and activator of transcription 3 (STAT3) protein. Biochem. Cell Biol. 2009, 87, 825–833. [Google Scholar] [CrossRef]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.E.; Murphy, T.F.; Shu, P.; Huang, H.F.; Meyenhofer, M.; Barton, A. Novel single-stranded oligonucleotides that inhibit signal transducer and activator of transcription 3 induce apoptosis in vitro and in vivo in prostate cancer cell lines. Mol. Cancer 2004, 3, 1183–1191. [Google Scholar]
- Hecker, M.; Wagner, A.H. Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 2017, 144, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Wei, H.; Tian, Z. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol. Rep. 2007, 17, 1377–1382. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.; Zhang, J.; Wang, L.; Wei, H.; Tian, Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer 2007, 7, 149. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-Human Trial of a STAT3 Decoy Oligonucleotide in Head and Neck Tumors: Implications for Cancer Therapy. Cancer Discov. 2012, 2, 694. [Google Scholar] [CrossRef] [PubMed]
- Njatcha, C.; Farooqui, M.; Kornberg, A.; Johnson, D.E.; Grandis, J.R.; Siegfried, J.M. STAT3 Cyclic Decoy Demonstrates Robust Antitumor Effects in Non-Small Cell Lung Cancer. Mol. Cancer 2018, 17, 1917–1926. [Google Scholar] [CrossRef]
- Njatcha, C.; Farooqui, M.; Almotlak, A.A.; Siegfried, J.M. Prevention of Tobacco Carcinogen-Induced Lung Tumor Development by a Novel STAT3 Decoy Inhibitor. Cancer Prev. Res. 2020, 13, 735–746. [Google Scholar] [CrossRef]
- Jing, N.; Li, Y.; Xu, X.; Sha, W.; Li, P.; Feng, L.; Tweardy, D.J. Targeting Stat3 with G-quartet oligodeoxynucleotides in human cancer cells. DNA Cell Biol. 2003, 22, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, P.; Garcia, G.E.; Zhu, Q.; Yuan, P.; Feng, L.; Mao, L.; Jing, N. Inhibition of Stat3 activation and tumor growth suppression of non-small cell lung cancer by G-quartet oligonucleotides. Int. J. Oncol. 2007, 31, 129–136. [Google Scholar] [CrossRef]
- Najjar, I.; Fagard, R. STAT1 and pathogens, not a friendly relationship. Biochimie 2010, 92, 425–444. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Chaib, I.; Karachaliou, N.; Pilotto, S.; Codony Servat, J.; Cai, X.; Li, X.; Drozdowskyj, A.; Servat, C.C.; Yang, J.; Hu, C.; et al. Co-activation of STAT3 and YES-Associated Protein 1 (YAP1) Pathway in EGFR-Mutant NSCLC. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Chen, T.H.; Feng, W.; Jänne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef]
- Dowlati, A.; Kluge, A.; Nethery, D.; Halmos, B.; Kern, J.A. SCH66336, inhibitor of protein farnesylation, blocks signal transducer and activators of transcription 3 signaling in lung cancer and interacts with a small molecule inhibitor of epidermal growth factor receptor/human epidermal growth factor receptor 2. Anticancer Drugs 2008, 19, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Kluge, A.; Dabir, S.; Kern, J.; Nethery, D.; Halmos, B.; Ma, P.; Dowlati, A. Cooperative interaction between protein inhibitor of activated signal transducer and activator of transcription-3 with epidermal growth factor receptor blockade in lung cancer. Int. J. Cancer 2009, 125, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, A.; Nethery, D.; Kern, J.A. Combined inhibition of epidermal growth factor receptor and JAK/STAT pathways results in greater growth inhibition in vitro than single agent therapy. Mol. Cancer 2004, 3, 459–463. [Google Scholar]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Chen, J.S.; Liao, W.Y.; Ho, C.C.; Hsu, C.L.; Yang, C.Y.; Chen, K.Y.; Lee, J.H.; Lin, Z.Z.; Shih, J.Y.; et al. Clinical outcomes and secondary epidermal growth factor receptor (EGFR) T790M mutation among first-line gefitinib, erlotinib and afatinib-treated non-small cell lung cancer patients with activating EGFR mutations. Int. J. Cancer 2019, 144, 2887–2896. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, W. Overcoming acquired resistance of gefitinib in lung cancer cells without T790M by AZD9291 or Twist1 knockdown in vitro and in vivo. Arch. Toxicol. 2019, 93, 1555–1571. [Google Scholar] [CrossRef]
- Chiu, H.C.; Chou, D.L.; Huang, C.T.; Lin, W.H.; Lien, T.W.; Yen, K.J.; Hsu, J.T. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem. Pharmacol. 2011, 81, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, L.; Sun, Y.; Yu, W.; Wang, X. Targeting STAT3 signaling overcomes gefitinib resistance in non-small cell lung cancer. Cell Death Dis. 2021, 12, 561. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, H.; Guan, L.; Lai, C.; Yu, W.; Lai, M. LL1, a novel and highly selective STAT3 inhibitor, displays anti-colorectal cancer activities in vitro and in vivo. Br. J. Pharmacol. 2020, 177, 298–313. [Google Scholar] [CrossRef]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef]
- Bian, C.; Liu, Z.; Li, D.; Zhen, L. PI3K/AKT inhibition induces compensatory activation of the MET/STAT3 pathway in non-small cell lung cancer. Oncol. Lett. 2018, 15, 9655–9662. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.O.; Lee, Y.H.; Park, J.A.; Kim, J.H.; Hong, S.E.; Kim, H.A.; Kim, E.K.; Noh, W.C.; Kim, B.H.; Ye, S.K.; et al. Blockage of Stat3 enhances the sensitivity of NSCLC cells to PI3K/mTOR inhibition. Biochem. Biophys. Res. Commun. 2014, 444, 502–508. [Google Scholar] [CrossRef]
- Barré, B.; Vigneron, A.; Perkins, N.; Roninson, I.B.; Gamelin, E.; Coqueret, O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol. Med. 2007, 13, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.; Takemura, K.; Kihara, M.; Nishimura, M.; Ueda, N.; Naito, S.; Lee, E.; Shimizu, E.; Yamauchi, A. Overexpression of constitutive signal transducer and activator of transcription 3 mRNA in cisplatin-resistant human non-small cell lung cancer cells. Oncol. Rep. 2005, 13, 217–222. [Google Scholar]
- Vatsyayan, R.; Chaudhary, P.; Lelsani, P.C.; Singhal, P.; Awasthi, Y.C.; Awasthi, S.; Singhal, S.S. Role of RLIP76 in doxorubicin resistance in lung cancer. Int. J. Oncol. 2009, 34, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Rathos, M.J.; Khanwalkar, H.; Joshi, K.; Manohar, S.M.; Joshi, K.S. Potentiation of in vitro and in vivoantitumor efficacy of doxorubicin by cyclin-dependent kinase inhibitor P276-00 in human non-small cell lung cancer cells. BMC Cancer 2013, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lu, X.; Wang, G. STAT3 inhibitor enhances chemotherapy drug efficacy by modulating mucin 1 expression in non-small cell lung carcinoma. Trop. J. Pharm. Res. 2017, 16, 1513–1521. [Google Scholar] [CrossRef][Green Version]
- Codony-Servat, C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; de Los Llanos Gil, M.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in EGFR mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017, 8, 47305–47316. [Google Scholar] [CrossRef]
- Timmerman, R.; Paulus, R.; Galvin, J.; Michalski, J.; Straube, W.; Bradley, J.; Fakiris, A.; Bezjak, A.; Videtic, G.; Johnstone, D.; et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA 2010, 303, 1070–1076. [Google Scholar] [CrossRef]
- Curran, W.J., Jr.; Paulus, R.; Langer, C.J.; Komaki, R.; Lee, J.S.; Hauser, S.; Movsas, B.; Wasserman, T.; Rosenthal, S.A.; Gore, E.; et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: Randomized phase III trial RTOG 9410. J. Natl. Cancer Inst. 2011, 103, 1452–1460. [Google Scholar] [CrossRef]
- Bradley, J.D.; Paulus, R.; Komaki, R.; Masters, G.; Blumenschein, G.; Schild, S.; Bogart, J.; Hu, C.; Forster, K.; Magliocco, A.; et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): A randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015, 16, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Spiro, S.G.; Silvestri, G.A. One hundred years of lung cancer. Am. J. Respir. Crit. Care Med. 2005, 172, 523–529. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Li, R.; Park, D.; Xie, M.; Sica, G.L.; Cao, Y.; Xiao, Z.-Q.; Deng, X. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol. Cancer Ther. 2014, 13, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Pawelczyk, K.; Piotrowska, A.; Ciesielska, U.; Jablonska, K.; Gletzel-Plucinska, N.; Grzegrzolka, J.; Podhorska-Okolow, M.; Dziegiel, P.; Nowinska, K. Role of PD-L1 Expression in Non-Small Cell Lung Cancer and Their Prognostic Significance according to Clinicopathological Factors and Diagnostic Markers. Int. J. Mol. Sci. 2019, 20, 824. [Google Scholar] [CrossRef]
- Mino-Kenudson, M. Programmed cell death ligand-1 (PD-L1) expression by immunohistochemistry: Could it be predictive and/or prognostic in non-small cell lung cancer? Cancer Biol. Med. 2016, 13, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.Y.; Huang, J.A.; Chen, Y.; Chen, C.; Zhang, X.G. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med. Oncol. 2011, 28, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Li, W.; Chen, K.; Xie, Y.; Liu, Q.; Yao, M.; Duan, W.; Zhou, X.; Liang, R.; Tao, M. B7-H1 and B7-H3 are independent predictors of poor prognosis in patients with non-small cell lung cancer. Oncotarget 2015, 6, 3452–3461. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Jia, M.; Feng, W.; Kang, S.; Zhang, Y.; Shen, J.; He, J.; Jiang, L.; Wang, W.; Guo, Z.; Peng, G.; et al. Evaluation of the efficacy and safety of anti-PD-1 and anti-PD-L1 antibody in the treatment of non-small cell lung cancer (NSCLC): A meta-analysis. J. Thorac. Dis. 2015, 7, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef]
- Koh, J.; Jang, J.-Y.; Keam, B.; Kim, S.; Kim, M.-Y.; Go, H.; Kim, T.M.; Kim, D.-W.; Kim, C.-W.; Jeon, Y.K.; et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1α and STAT3. Oncoimmunology 2015, 5, e1108514. [Google Scholar] [CrossRef] [PubMed]
- Roussel, H.; De Guillebon, E.; Biard, L.; Mandavit, M.; Gibault, L.; Fabre, E.; Antoine, M.; Hofman, P.; Beau-Faller, M.; Blons, H.; et al. Composite biomarkers defined by multiparametric immunofluorescence analysis identify ALK-positive adenocarcinoma as a potential target for immunotherapy. Oncoimmunology 2017, 6, e1286437. [Google Scholar] [CrossRef]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS–Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef]
- Tang, D.; Zhao, D.; Wu, Y.; Yao, R.; Zhou, L.; Lu, L.; Gao, W.; Sun, Y. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J. Cell Mol. Med. 2018, 22, 3847–3856. [Google Scholar] [CrossRef]
- Abdelhamed, S.; Ogura, K.; Yokoyama, S.; Saiki, I.; Hayakawa, Y. AKT-STAT3 Pathway as a Downstream Target of EGFR Signaling to Regulate PD-L1 Expression on NSCLC cells. J. Cancer 2016, 7, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Wu, S.; Standifer, N.; Jure-Kunkel, M.; de los Reyes, M.; Shrestha, Y.; Halpin, R.; Rothstein, R.; Mulgrew, K.; Blackmore, S.; et al. Resistance to Durvalumab and Durvalumab plus Tremelimumab Is Associated with Functional STK11 Mutations in Patients with Non–Small Cell Lung Cancer and Is Reversed by STAT3 Knockdown. Cancer Discov. 2021, 11, 2828–2845. [Google Scholar] [CrossRef]
- Cavazzoni, A.; Digiacomo, G.; Alfieri, R.; La Monica, S.; Fumarola, C.; Galetti, M.; Bonelli, M.; Cretella, D.; Barili, V.; Zecca, A.; et al. Pemetrexed Enhances Membrane PD-L1 Expression and Potentiates T Cell-Mediated Cytotoxicity by Anti-PD-L1 Antibody Therapy in Non-Small-Cell Lung Cancer. Cancers 2020, 12, 666. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
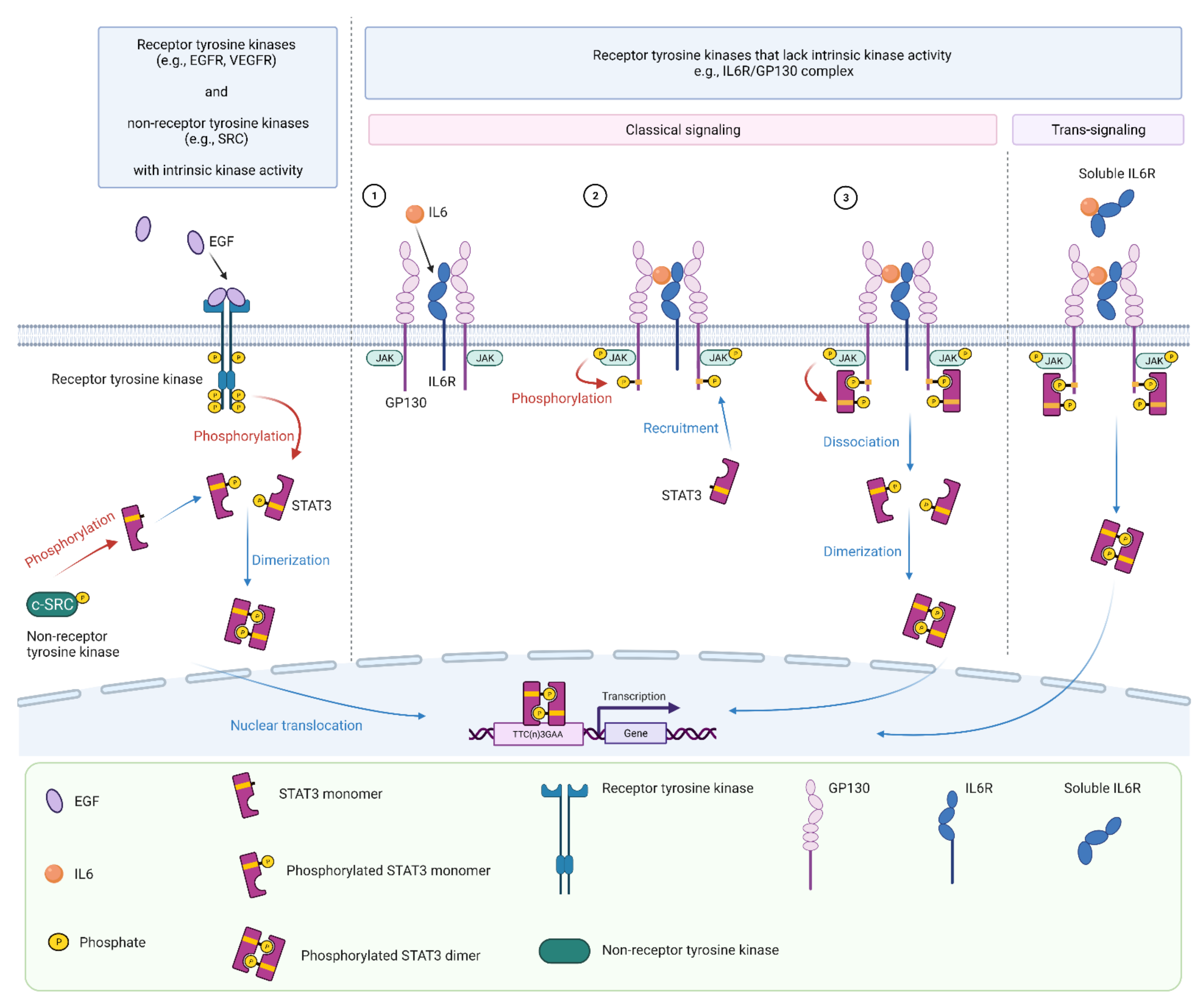{kind=link}
{kind=link}
{kind=link}
| Mechanism | Drug | Phase | Toxicities | Responses | Outcome | References |
|---|---|---|---|---|---|---|
| Inhibit IL6/JAK/STAT3 signaling | Siltuximab a | I/II | Hepatic function abnormalities, neutropenia | No objective responses | Completed | NCT00841191 [208] |
| ALD518 a | II | No dose limiting toxicity | Reduction of anemia and cachexia | Completed | NCT00866970 [209,210] | |
| Inhibit receptor/non-receptor tyrosine kinases | Ruxolitinib b + Afatinib c | I | Diarrhea, anemia, paronychia, acneiform rash, oral mucositis | ORR 23.3% Median PFS 4.9 months | Completed | NCT02145637 [211] |
| Ruxolitinib b + Pemetrexed/Cisplatin d | II | No dose limiting toxicity with the combination | Response rate: 31% (ruxolitinib) vs. 35% (placebo) | Terminated (no clinical benefit) | NCT02119650 [212] | |
| Ruxolitinib b + Erlotinib c | I/II | Anemia, diarrhea, liver function derangement | ORR 5% Median PFS 2.2 months | Completed | NCT02155465 [213] | |
| AZD1480 b | I | Pleiotropic neurologic toxicity | No responses seen | Completed | NCT01219543 [214] | |
| AZD4205 b + Osimertinib c | I/II | Not reported | Completed | NCT03450330 | ||
| Itacitinib b + Pembrolizumab e | II | Ongoing, N/A | NCT03425006 | |||
| Itacitinib2 + Osimertinib c | I/II | Ongoing, N/A | NCT02917993 | |||
| Momelotinib b + Trametinib c | I | GI toxicity, fatigue, liver function derangement | No objective responses Median PFS 3.6 months | Terminated (no clinical benefit) | NCT02258607 [215] | |
| Momelotinib b + Erlotinib c | I | Neutropenia, diarrhea, skin toxicity, fatigue | ORR 54.5% Median PFS 9.2 months | Terminated (no benefit over erlotinib monotherapy) | NCT02206763 [216] | |
| Pacritinib b + Erlotinib c | I/II | Not reported | Terminated (due to drug shortage) | NCT02342353 | ||
| Dasatinib f | II | Fatigue, dyspnea | ORR 3% | Completed | NCT00459342 [217] | |
| Dasatinib f | II | ≥Grade 3 toxicity: dyspnea, fatigue, AST elevation, anorexia, nausea | Terminated (safety concerns) | NCT01491633 [218] | ||
| Dasatinib f + Afatinib c | I | New or increased pleural effusions | No objective responses | Completed | NCT01999985 [219] | |
| Dasatinib f + Osimertinib c | I | Pleural effusions, myelosuppression, rash, transaminitis | ORR: 90% Median PFS 19.4 months | Completed (prematurely closed due to slow accrual) | NCT02954523 [220] | |
| Dasatinib f + Erlotinib c | I/II | Not reported | ORR: 15% Median PFS 3.3 months | Completed | NCT00826449 [221] | |
| Block STAT3 dimerization | OPB-51620 g | I | Fatigue, GI toxicity, early-onset peripheral neuropathy | Completed | NCT01184807 [222] | |
| OPB-31121 g | I | GI toxicity | No objective responses | Completed | NCT00955812 [223] | |
| C188-9 (TTI-101) g | I | Recruiting | NCT03195699 | |||
| Promote degradation of STAT3 mRNA | AZD9150 h + anti-PDL1 e | II | Ongoing, N/A | NCT02983578 | ||
| AZD9150 h + anti-PDL1 e | II | Ongoing, N/A | NCT03334617 | |||
| AZD9150 h + anti-PDL1 e | I/II | Ongoing, N/A | NCT03421353 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parakh, S.; Ernst, M.; Poh, A.R. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. https://doi.org/10.3390/cancers13246228
Parakh S, Ernst M, Poh AR. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers. 2021; 13(24):6228. https://doi.org/10.3390/cancers13246228
Chicago/Turabian StyleParakh, Sagun, Matthias Ernst, and Ashleigh R. Poh. 2021. "Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities" Cancers 13, no. 24: 6228. https://doi.org/10.3390/cancers13246228
APA StyleParakh, S., Ernst, M., & Poh, A. R. (2021). Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers, 13(24), 6228. https://doi.org/10.3390/cancers13246228