
Shooting at Moving and Hidden Targets—Tumour Cell Plasticity and the Notch Signalling Pathway in Head and Neck Squamous Cell Carcinomas

 , ,
, ,  ,
,  and
and 
Simple Summary
Abstract
1. Introduction
1.1. The Mutational Spectrum of Head and Neck Squamous Cell Carcinomas (HNSCC)
1.2. Introducing the Notch Signalling Pathway in HNSCC
1.3. The Tumour Microenvironment (TME) and Intra-Tumour Heterogeneity (ITH) as Moving Targets: Notch, Tumour Heterogeneity and Cellular Dynamics
2. Targeting Oncogenic Pathways in HNSCC
2.1. Current Status of HNSCC Therapy
2.2. Tools for Personalized Medicine in HNSCC and Notch Signalling
2.3. Novel Therapeutic Agents—Overview
2.4. Homing in: NOTCH & NOTCH Signalling as a Target in HNSCC
2.4.1. First Act: Oncogenic NOTCH Mutations Found in Diverse Cancers
2.4.2. Second Act: Loss-of-Function Mutations, but Yet, Gain-Of-Function Activities?

2.4.3. Act 3: What Exactly Drives Activation of Notch Signalling in HNSCC?
2.5. NOTCH and EGFR Signalling
2.5.1. Cetuximab and EGFR Pathway Activity in HNSCC
2.5.2. Crosstalk between Notch and EGFR Signalling
2.6. Immuno-Oncology Drugs: Targeting the Immune Checkpoints
2.7. Cell Cycle Inhibition and Notch Signalling
3. Notch Signalling and Tumour Cell Plasticity
3.1. Tumour Cell Plasticity as a (Moving) Target
3.2. Introducing EMT as a Hallmark of Enhanced Tumour Cell Plasticity
3.3. Visible versus Hidden Forms of EMT in HNSCC
3.4. Hybrid or Partial EMT in HNSSC and Other Epithelial Cancers
3.5. Regulation of EMT in HNSCC by Notch Signalling
3.6. How NOTCH and EMT Signalling May Be Connected
3.7. EMT Regulation within the TME
3.8. Regulation of EMT by Other Pathways in HNSCC
3.9. Notch, EMT and miRNAs
3.10. Multiple Links between EMT; NOTCH Signalling, and Cancer Stem Cells/Stemness
4. Targeting Tumour Angiogenesis in HNSCC
4.1. (Neo-)Angiogenesis in HNSCC
4.2. The Jagged1/Notch Signalling Axis in Angiogenesis
4.3. The DLL4/NOTCH Signalling Axis in Angiogenesis
4.4. The Extracellular Matrix, Notch Signalling, and Angiogenesis
5. Discussion—Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, U.M.; Shimonosono, M.; Flashner, S.; Cruz-Acuña, R.; Gabre, J.T.; Nakagawa, H. Understanding the cellular origin and progression of esophageal cancer using esophageal organoids. Cancer Lett. 2021, 509, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Chen, X. NOTCH and Esophageal Squamous Cell Carcinoma. Adv. Exp. Med. Biol. 2021, 1287, 59–68. [Google Scholar]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Schwaederle, M.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Squamousness: Next-generation sequencing reveals shared molecular features across squamous tumor types. Cell Cycle 2015, 14, 2355–2361. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Kulkarni, A.; Birkeland, A.C.; Michmerhuizen, N.L.; Foltin, S.K.; Mann, J.E.; Hoesli, R.C.; Devenport, S.N.; Jewell, B.M.; Shuman, A.G.; et al. The genomic landscape of UM-SCC oral cavity squamous cell carcinoma cell lines. Oral Oncol. 2018, 87, 144–151. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Bian, W.; Tang, M.; Jiang, H.; Xu, W.; Hao, W.; Sui, Y.; Hou, Y.; Nie, L.; Zhang, H.; Wang, C.; et al. Low-density-lipoprotein-receptor-related protein 1 mediates Notch pathway activation. Dev. Cell 2021, 56, 2902–2919.e8. [Google Scholar] [CrossRef]
- Pogorzelski, M.; Ting, S.; Gauler, T.C.; Breitenbuecher, F.; Vossebein, I.; Hoffarth, S.; Markowetz, J.; Lang, S.; Bergmann, C.; Brandau, S.; et al. Impact of human papilloma virus infection on the response of head and neck cancers to anti-epidermal growth factor receptor antibody therapy. Cell Death Dis. 2014, 5, e1091. [Google Scholar] [CrossRef]
- Koffler, J.; Sharma, S.; Hess, J. Predictive value of epigenetic alterations in head and neck squamous cell carcinoma. Mol. Cell. Oncol. 2014, 1, e954827. [Google Scholar] [CrossRef]
- Nagel, R.; Martens-de Kemp, S.R.; Buijze, M.; Jacobs, G.; Braakhuis, B.J.M.; Brakenhoff, R.H. Treatment response of HPV-positive and HPV-negative head and neck squamous cell carcinoma cell lines. Oral Oncol. 2013, 49, 560–566. [Google Scholar] [CrossRef]
- Rizzo, G.; Black, M.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. Defining the genomic landscape of head and neck cancers through next-generation sequencing. Oral Dis. 2015, 21, e11–e24. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef]
- Zenga, J.; Jackson, R.S.; Graboyes, E.M.; Sinha, P.; Lindberg, M.; Martin, E.J.; Ma, D.; Thorstad, W.L.; Rich, J.T.; Moore, E.J.; et al. Oncologic outcomes of selective neck dissection in HPV-related oropharyngeal squamous cell carcinoma. Laryngoscope 2017, 127, 623–630. [Google Scholar] [CrossRef]
- Zenga, J.; Graboyes, E.M.; Haughey, B.H.; Paniello, R.C.; Mehrad, M.; Lewis, J.S.J.; Thorstad, W.L.; Nussenbaum, B.; Rich, J.T. Definitive Surgical Therapy after Open Neck Biopsy for HPV-Related Oropharyngeal Cancer. Otolaryngol. Neck Surg. Off. J. Am. Acad. Otolaryngol. Neck Surg. 2016, 154, 657–666. [Google Scholar] [CrossRef]
- Mes, S.W.; Te Beest, D.; Poli, T.; Rossi, S.; Scheckenbach, K.; van Wieringen, W.N.; Brink, A.; Bertani, N.; Lanfranco, D.; Silini, E.M.; et al. Prognostic modeling of oral cancer by gene profiles and clinicopathological co-variables. Oncotarget 2017, 8, 59312–59323. [Google Scholar] [CrossRef]
- Brennan, S.; Baird, A.-M.; O’Regan, E.; Sheils, O. The Role of Human Papilloma Virus in Dictating Outcomes in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2021, 8, 677900. [Google Scholar] [CrossRef]
- Ruffin, A.T.; Cillo, A.R.; Tabib, T.; Liu, A.; Onkar, S.; Kunning, S.R.; Lampenfeld, C.; Atiya, H.I.; Abecassis, I.; Kürten, C.H.L.; et al. B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat. Commun. 2021, 12, 3349. [Google Scholar] [CrossRef]
- Wang, J.; Sun, H.; Zeng, Q.; Guo, X.-J.; Wang, H.; Liu, H.-H.; Dong, Z.-Y. HPV-positive status associated with inflamed immune microenvironment and improved response to anti-PD-1 therapy in head and neck squamous cell carcinoma. Sci. Rep. 2019, 9, 13404. [Google Scholar] [CrossRef]
- Mandal, R.; Şenbabaoğ, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.-W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef]
- Xu, K.; Fu, Y.; Han, Y.; Xia, R.; Xu, S.; Duan, S.; Zhang, Z.; Li, J. Fewer tumour-specific PD-1+CD8+ TILs in high-risk “Infiltrating” HPV− HNSCC. Br. J. Cancer 2020, 123, 932–941. [Google Scholar] [CrossRef]
- Outh-Gauer, S.; Morini, A.; Tartour, E.; Lépine, C.; Jung, A.C.; Badoual, C. The Microenvironment of Head and Neck Cancers: Papillomavirus Involvement and Potential Impact of Immunomodulatory Treatments. Head Neck Pathol. 2020, 14, 330–340. [Google Scholar] [CrossRef]
- Tuna, M.; Liu, W.; Amos, C.I.; Mills, G.B. Genome-Wide Profiling of Acquired Uniparental Disomy Reveals Prognostic Factors in Head and Neck Squamous Cell Carcinoma. Neoplasia 2019, 21, 1102–1109. [Google Scholar] [CrossRef]
- Luo, X.-J.; Zheng, M.; Cao, M.-X.; Zhang, W.-L.; Huang, M.-C.; Dai, L.; Tang, Y.-L.; Liang, X.-H. Distinguishable Prognostic miRNA Signatures of Head and Neck Squamous Cell Cancer With or Without HPV Infection. Front. Oncol. 2020, 10, 614487. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef]
- Mao, L. NOTCH Mutations: Multiple Faces in Human Malignancies. Cancer Prev. Res. 2015, 8, 259–261. [Google Scholar] [CrossRef]
- Zhang, T.-H.; Liu, H.-C.; Zhu, L.-J.; Chu, M.; Liang, Y.-J.; Liang, L.-Z.; Liao, G.-Q. Activation of Notch signaling in human tongue carcinoma. J. Oral Pathol. Med. 2011, 40, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Masiero, M.; Banham, A.H.; Harris, A.L. The notch ligand JAGGED1 as a target for anti-tumor therapy. Front. Oncol. 2014, 4, 254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Wu, T.; Li, Q.; Wang, M.-C.; Jing, L.; Ruan, Z.-P.; Yao, Y.; Nan, K.-J.; Guo, H. Notch Signaling Components: Diverging Prognostic Indicators in Lung Adenocarcinoma. Medicine 2016, 95, e3715. [Google Scholar] [CrossRef] [PubMed]
- Hijioka, H.; Setoguchi, T.; Miyawaki, A.; Gao, H.; Ishida, T.; Komiya, S.; Nakamura, N. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int. J. Oncol. 2010, 36, 817–822. [Google Scholar]
- Xiu, M.-X.; Liu, Y.-M.; Kuang, B.-H. The Role of DLLs in Cancer: A Novel Therapeutic Target. Onco. Targets Ther. 2020, 13, 3881–3901. [Google Scholar] [CrossRef]
- Greenwald, I. LIN-12/Notch signaling in C. elegans. Genetics 2005, 191, 1–16. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef]
- Kim, D.E.; Procopio, M.-G.; Ghosh, S.; Jo, S.-H.; Goruppi, S.; Magliozzi, F.; Bordignon, P.; Neel, V.; Angelino, P.; Dotto, G.P. Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation. J. Exp. Med. 2017, 214, 2349–2368. [Google Scholar] [CrossRef]
- Oswald, F.; Täuber, B.; Dobner, T.; Bourteele, S.; Kostezka, U.; Adler, G.; Liptay, S.; Schmid, R.M. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell. Biol. 2001, 21, 7761–7774. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Cai, K.; Xu, P.-P.; Wang, L.; Huang, C.-X.; Fang, Y.; Cheng, S.; Sun, X.-J.; Liu, F.; Huang, J.-Y.; et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-NOTCH-CCL2/CSF1 axis. Signal Transduct. Target. Ther. 2021, 6, 10. [Google Scholar] [CrossRef]
- Wang, L.; Jia, Y.-M.; Zuo, J.; Wang, Y.-D.; Fan, Z.-S.; Feng, L.; Zhang, X.; Han, J.; Lyu, W.-J.; Ni, Z.-Y. Gene mutations of esophageal squamous cell carcinoma based on next-generation sequencing. Chin. Med. J. 2021, 134, 708–715. [Google Scholar] [CrossRef]
- Canté-Barrett, K.; Holtzer, L.; van Ooijen, H.; Hagelaar, R.; Cordo’, V.; Verhaegh, W.; van de Stolpe, A.; Meijerink, J.P.P. A Molecular Test for Quantifying Functional Notch Signaling Pathway Activity in Human Cancer. Cancers 2020, 12, 3142. [Google Scholar] [CrossRef]
- Sun, W.; Gaykalova, D.A.; Ochs, M.F.; Mambo, E.; Arnaoutakis, D.; Liu, Y.; Loyo, M.; Agrawal, N.; Howard, J.; Li, R.; et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 2014, 74, 1091–1104. [Google Scholar] [CrossRef]
- Gerlinger, M.; McGranahan, N.; Dewhurst, S.M.; Burrell, R.A.; Tomlinson, I.; Swanton, C. Cancer: Evolution within a lifetime. Annu. Rev. Genet. 2014, 48, 215–236. [Google Scholar] [CrossRef]
- Lipinski, K.A.; Barber, L.J.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer 2016, 2, 49–63. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Kinker, G.S.; Greenwald, A.C.; Tal, R.; Orlova, Z.; Cuoco, M.S.; McFarland, J.M.; Warren, A.; Rodman, C.; Roth, J.A.; Bender, S.A.; et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 2020, 52, 1208–1218. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Inda, M.A.; van Swinderen, P.; van Brussel, A.; Moelans, C.B.; Verhaegh, W.; van Zon, H.; den Biezen, E.; Bikker, J.W.; van Diest, P.J.; van de Stolpe, A. Heterogeneity in Signaling Pathway Activity within Primary and between Primary and Metastatic Breast Cancer. Cancers 2021, 13, 1345. [Google Scholar] [CrossRef]
- La Rosa, S.; Rubbia-Brandt, L.; Scoazec, J.-Y.; Weber, A. Editorial: Tumor Heterogeneity. Front. Med. 2019, 6, 156. [Google Scholar] [CrossRef]
- Almangush, A.; Bello, I.O.; Heikkinen, I.; Hagström, J.; Haglund, C.; Kowalski, L.P.; Coletta, R.D.; Mäkitie, A.A.; Salo, T.; Leivo, I. Improving Risk Stratification of Early Oral Tongue Cancer with TNM-Immune (TNM-I) Staging System. Cancers 2021, 13, 3235. [Google Scholar] [CrossRef]
- Alabi, R.O.; Elmusrati, M.; Sawazaki-Calone, I.; Kowalski, L.P.; Haglund, C.; Coletta, R.D.; Mäkitie, A.A.; Salo, T.; Leivo, I.; Almangush, A.; et al. Stromal categorization in early oral tongue cancer. Histopathology 2021, 478, 925–932. [Google Scholar]
- Vickman, R.E.; Faget, D.V.; Beachy, P.; Beebe, D.; Bhowmick, N.A.; Cukierman, E.; Deng, W.-M.; Granneman, J.G.; Hildesheim, J.; Kalluri, R.; et al. Deconstructing tumor heterogeneity: The stromal perspective. Oncotarget 2020, 11, 3621–3632. [Google Scholar] [CrossRef]
- Almangush, A.; Alabi, R.O.; Troiano, G.; Coletta, R.D.; Salo, T.; Pirinen, M.; Mäkitie, A.A.; Leivo, I. Clinical significance of tumor-stroma ratio in head and neck cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 480. [Google Scholar] [CrossRef]
- Shang, W.; Zhang, Q.; Huang, Y.; Shanti, R.; Alawi, F.; Le, A.; Jiang, C. Cellular Plasticity-Targeted Therapy in Head and Neck Cancers. J. Dent. Res. 2018, 97, 654–664. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Domínguez, Á.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, C.; Zhang, Z.; Wang, Q.; Wei, H.; Shi, W.; Li, J.; Wang, Z.; Ou, Y.; Wang, W.; et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat. Commun. 2020, 11, 5129. [Google Scholar] [CrossRef]
- Oosting, S.F.; Haddad, R.I. Best Practice in Systemic Therapy for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 815. [Google Scholar] [CrossRef]
- Uppaluri, R.; Campbell, K.M.; Egloff, A.M.; Zolkind, P.; Skidmore, Z.L.; Nussenbaum, B.; Paniello, R.C.; Rich, J.T.; Jackson, R.; Pipkorn, P.; et al. Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase 2 Trial. Clin. Cancer Res. 2020, 26, 5140–5152. [Google Scholar] [CrossRef]
- Yeh, T.-J.; Chan, L.-P.; Tsai, H.-T.; Hsu, C.-M.; Cho, S.-F.; Pan, M.-R.; Liu, Y.-C.; Huang, C.-J.; Wu, C.-W.; Du, J.-S.; et al. The Overall Efficacy and Outcomes of Metronomic Tegafur-Uracil Chemotherapy on Locally Advanced Head and Neck Squamous Cell Carcinoma: A Real-World Cohort Experience. Biology 2021, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Rajendra, A.; Noronha, V.; Joshi, A.; Patil, V.M.; Menon, N.; Prabhash, K. Palliative chemotherapy in head and neck cancer: Balancing between beneficial and adverse effects. Expert Rev. Anticancer Ther. 2020, 20, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Klinghammer, K.; Otto, R.; Raguse, J.-D.; Albers, A.E.; Tinhofer, I.; Fichtner, I.; Leser, U.; Keilholz, U.; Hoffmann, J. Basal subtype is predictive for response to cetuximab treatment in patient-derived xenografts of squamous cell head and neck cancer. Int. J. Cancer 2017, 141, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fukuda, N.; Wang, X.; Urasaki, T.; Ohmoto, A.; Nakano, K.; Yunokawa, M.; Ono, M.; Sato, Y.; Mitani, H.; et al. Efficacy of Nivolumab for Head and Neck Cancer Patients with Primary Sites and Histological Subtypes Excluded from the CheckMate-141 Trial. Cancer Manag. Res. 2020, 12, 4161–4168. [Google Scholar] [CrossRef]
- Zhang, L.; MacIsaac, K.D.; Zhou, T.; Huang, P.-Y.; Xin, C.; Dobson, J.R.; Yu, K.; Chiang, D.Y.; Fan, Y.; Pelletier, M.; et al. Genomic Analysis of Nasopharyngeal Carcinoma Reveals TME-Based Subtypes. Mol. Cancer Res. 2017, 15, 1722–1732. [Google Scholar] [CrossRef]
- Hess, J. Predictive Factors for Outcome and Quality of Life in HPV-Positive and HPV-Negative HNSCC. Recent Results Cancer Res. 2017, 206, 233–242. [Google Scholar]
- Ito, Y.; Kamijo, T.; Yokose, T.; Kawashima, M.; Ogino, T.; Ikeda, H.; Hayashi, R.; Sasaki, S.; Ochiai, A. Microvessel density predicts the radiosensitivity of metastatic head and neck squamous cell carcinoma in cervical lymph nodes. Int. J. Oncol. 2001, 19, 1127–1132. [Google Scholar] [CrossRef]
- Sørensen, B.S.; Busk, M.; Olthof, N.; Speel, E.-J.; Horsman, M.R.; Alsner, J.; Overgaard, J. Radiosensitivity and effect of hypoxia in HPV positive head and neck cancer cells. Radiother. Oncol. 2013, 108, 500–505. [Google Scholar] [CrossRef]
- Jerhammar, F.; Ceder, R.; Garvin, S.; Grénman, R.; Grafström, R.C.; Roberg, K. Fibronectin 1 is a potential biomarker for radioresistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2010, 10, 1244–1251. [Google Scholar] [CrossRef]
- Stausbøl-Grøn, B.; Bentzen, S.M.; Jørgensen, K.E.; Nielsen, O.S.; Bundgaard, T.; Overgaard, J. In vitro radiosensitivity of tumour cells and fibroblasts derived from head and neck carcinomas: Mutual relationship and correlation with clinical data. Br. J. Cancer 1999, 79, 1074–1084. [Google Scholar] [CrossRef]
- Horn, D.; Hess, J.; Freier, K.; Hoffmann, J.; Freudlsperger, C. Targeting EGFR-PI3K-AKT-mTOR signaling enhances radiosensitivity in head and neck squamous cell carcinoma. Expert Opin. Ther. Targets 2015, 19, 795–805. [Google Scholar] [CrossRef]
- Elseragy, A.; Salo, T.; Coletta, R.D.; Kowalski, L.P.; Haglund, C.; Nieminen, P.; Mäkitie, A.A.; Leivo, I.; Almangush, A. A Proposal to Revise the Histopathologic Grading System of Early Oral Tongue Cancer Incorporating Tumor Budding. Am. J. Surg. Pathol. 2019, 43, 703–709. [Google Scholar] [CrossRef]
- Jesinghaus, M.; Steiger, K.; Stögbauer, F.; Haller, B.; Kolk, A.; Straßen, U.; Pickhard, A.; Wirth, M.; Silva, M.; Budczies, J.; et al. Pre-operative cellular dissociation grading in biopsies is highly predictive of post-operative tumour stage and patient outcome in head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 835–846. [Google Scholar] [CrossRef]
- Mahmood, H.; Shaban, M.; Indave, B.I.; Santos-Silva, A.R.; Rajpoot, N.; Khurram, S.A. Use of artificial intelligence in diagnosis of head and neck precancerous and cancerous lesions: A systematic review. Oral Oncol. 2020, 110, 104885. [Google Scholar] [CrossRef]
- Jesinghaus, M.; Boxberg, M.; Konukiewitz, B.; Slotta-Huspenina, J.; Schlitter, A.M.; Steiger, K.; Specht, K.; Wieczorek, K.; Warth, A.; Schmidt, T.; et al. A Novel Grading System Based on Tumor Budding and Cell Nest Size Is a Strong Predictor of Patient Outcome in Esophageal Squamous Cell Carcinoma. Am. J. Surg. Pathol. 2017, 41, 1112–1120. [Google Scholar] [CrossRef]
- Jesinghaus, M.; Brühl, F.; Steiger, K.; Klare, P.; Reiser, M.; Scheiter, A.; Konukiewitz, B.; Kuhn, P.; Münch, S.; Quante, M.; et al. Cellular Dissociation Grading Based on the Parameters Tumor Budding and Cell Nest Size in Pretherapeutic Biopsy Specimens Allows for Prognostic Patient Stratification in Esophageal Squamous Cell Carcinoma Independent from Clinical Staging. Am. J. Surg. Pathol. 2019, 43, 618–627. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris IV, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C.; Gridley, T. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 6, 431–432. [Google Scholar] [CrossRef]
- Misiorek, J.O.; Przybyszewska-Podstawka, A.; Kałafut, J.; Paziewska, B.; Rolle, K.; Rivero-Müller, A.; Nees, M. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells 2021, 10, 94. [Google Scholar] [CrossRef]
- Schleicher, K.; Schramek, D. AJUBA: A regulator of epidermal homeostasis and cancer. Exp. Dermatol. 2021, 30, 546–559. [Google Scholar] [CrossRef]
- Alcolea, M.P.; Greulich, P.; Wabik, A.; Frede, J.; Simons, B.D.; Jones, P.H.; Allegra, E.; Trapasso, S.; Pisani, D.; Puzzo, L.; et al. Notch1 Deficiency Induces Tumor Cell Accumulation Inside the Bronchiolar Lumen and Increases TAZ Expression in an Autochthonous Kras (LSL-G12V) Driven Lung Cancer Mouse Model. Nat. Cell Biol. 2021, 17, 126. [Google Scholar]
- Rothenberg, S.M.; Ellisen, L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Invest. 2012, 122, 1951–1957. [Google Scholar] [CrossRef]
- Gaździcka, J.; Gołąbek, K.; Strzelczyk, J.K.; Ostrowska, Z. Epigenetic Modifications in Head and Neck Cancer. Biochem. Genet. 2020, 58, 213–244. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Lin, S.-J.; Shih, H.-Y.; Chou, C.-H.; Chu, H.-H.; Chiu, C.-C.; Yuh, C.-H.; Yeh, T.-H.; Cheng, Y.-C. Epigenetic regulation of NOTCH1 and NOTCH3 by KMT2A inhibits glioma proliferation. Oncotarget 2017, 8, 63110–63120. [Google Scholar] [CrossRef][Green Version]
- Heuberger, J.; Grinat, J.; Kosel, F.; Liu, L.; Kunz, S.; Vidal, R.O.; Keil, M.; Haybaeck, J.; Robine, S.; Louvard, D.; et al. High Yap and Mll1 promote a persistent regenerative cell state induced by Notch signaling and loss of p53. Proc. Natl. Acad. Sci. USA 2021, 118, e2019699118. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Z.; Ding, X.; Zhang, W.; Li, G.; Liu, L.; Wu, H.; Gu, W.; Wu, Y.; Song, X. A novel Notch1 missense mutation (C1133Y) in the Abruptex domain exhibits enhanced proliferation and invasion in oral squamous cell carcinoma. Cancer Cell Int. 2018, 18, 6. [Google Scholar] [CrossRef]
- Zheng, Y.; Song, A.; Wang, C.; Zhang, W.; Liang, D.; Ding, X.; Li, G.; Zhang, H.; Zhang, W.; Du, Y.; et al. Isoform specific FBXW7 mediates NOTCH1 Abruptex mutation C1133Y deregulation in oral squamous cell carcinoma. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Zhang, M.; Biswas, S.; Qin, X.; Gong, W.; Deng, W.; Yu, H. Does Notch play a tumor suppressor role across diverse squamous cell carcinomas? Cancer Med. 2016, 5, 2048–2060. [Google Scholar] [CrossRef]
- Shah, P.A.; Huang, C.; Li, Q.; Kazi, S.A.; Byers, L.A.; Wang, J.; Johnson, F.M.; Frederick, M.J. NOTCH1 Signaling in Head and Neck Squamous Cell Carcinoma. Cells 2020, 9, 2677. [Google Scholar] [CrossRef]
- Nyman, P.E.; Buehler, D.; Lambert, P.F. Loss of Function of Canonical Notch Signaling Drives Head and Neck Carcinogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 6308–6318. [Google Scholar] [CrossRef]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef]
- Wirth, M.; Jira, D.; Ott, A.; Piontek, G.; Pickhard, A. High NOTCH1 mRNA Expression Is Associated with Better Survival in HNSCC. Int. J. Mol. Sci. 2018, 19, 830. [Google Scholar] [CrossRef] [PubMed]
- Yap, L.F.; Lee, D.; Khairuddin, A.; Pairan, M.F.; Puspita, B.; Siar, C.H.; Paterson, I.C. The opposing roles of NOTCH signalling in head and neck cancer: A mini review. Oral Dis. 2015, 21, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B.; Fukusumi, T.; Califano, J.A.; Zhao, Y.-Y.; et al. The Notch signaling pathway in head and neck squamous cell carcinoma: A meta-analysis. J. Dent. Res. 2017, 9, 645–653. [Google Scholar]
- Porcheri, C.; Meisel, C.T.; Mitsiadis, T. Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 1520. [Google Scholar] [CrossRef]
- Porcheri, C.; Mitsiadis, T.A. Notch in Head and Neck Cancer. Adv. Exp. Med. Biol. 2021, 1287, 81–103. [Google Scholar]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef]
- Srivastava, S.S.; Alam, H.; Patil, S.J.; Shrinivasan, R.; Raikundalia, S.; Chaudhari, P.R.; Vaidya, M.M. Keratin 5/14-mediated cell differentiation and transformation are regulated by TAp63 and Notch-1 in oral squamous cell carcinoma-derived cells. Oncol. Rep. 2018, 39, 2393–2401. [Google Scholar] [CrossRef]
- Ohashi, S.; Natsuizaka, M.; Yashiro-Ohtani, Y.; Kalman, R.A.; Nakagawa, M.; Wu, L.; Klein-Szanto, A.J.; Herlyn, M.; Diehl, J.A.; Katz, J.P.; et al. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology 2010, 139, 2113–2123. [Google Scholar] [CrossRef]
- Ohashi, S.; Natsuizaka, M.; Naganuma, S.; Kagawa, S.; Kimura, S.; Itoh, H.; Kalman, R.A.; Nakagawa, M.; Darling, D.S.; Basu, D.; et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res. 2011, 71, 6836–6847. [Google Scholar] [CrossRef]
- Kagawa, S.; Natsuizaka, M.; Whelan, K.A.; Facompre, N.; Naganuma, S.; Ohashi, S.; Kinugasa, H.; Egloff, A.M.; Basu, D.; Gimotty, P.A.; et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene 2015, 34, 2347–2359. [Google Scholar] [CrossRef]
- Procopio, M.-G.; Laszlo, C.; Al Labban, D.; Kim, D.E.; Bordignon, P.; Jo, S.-H.; Goruppi, S.; Menietti, E.; Ostano, P.; Ala, U.; et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat. Cell Biol. 2015, 17, 1193–1204. [Google Scholar] [CrossRef]
- Homann, N.; Andl, T.; Nees, M.; Schuhmann, A.; Herold-Mende, C.; Bosch, F.X. The significance of aberrant p53 protein in head and neck tumors and its influence on tumor proliferation and differentiation. HNO 1993, 41, 254–260. [Google Scholar]
- Nees, M.; Homann, N.; Discher, H.; Andl, T.; Enders, C.; Herold-Mende, C.; Schuhmann, A.; Bosch, F.X. Expression of Mutated p53 Occurs in Ttimor-distant Epithelia of Head and Neck Cancer Patients: A Possible Molecular Basis for the Development of Multiple Ttamors. Cancer Res. 1993, 53, 4189–4196. [Google Scholar]
- Homann, N.; Nees, M.; Conradt, C.; Dietz, A.; Weidauer, H.; Maier, H.; Bosch, F.X. Overexpression of p53 in tumor-distant epithelia of head and neck cancer patients is associated with an increased incidence of second primary carcinoma. Clin. Cancer Res. 2001, 7, 290–296. [Google Scholar]
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 642352. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Yu, G.-T.; Xiao, T.; Hu, J. The Notch signaling pathway in head and neck squamous cell carcinoma: A meta-analysis. Adv. Clin. Exp. Med. Off. Organ Wroclaw Med. Univ. 2017, 26, 881–887. [Google Scholar] [CrossRef]
- Zou, Y.; Fang, F.; Ding, Y.-J.; Dai, M.-Y.; Yi, X.; Chen, C.; Tao, Z.-Z.; Chen, S.-M. Notch 2 signaling contributes to cell growth, anti-apoptosis and metastasis in laryngeal squamous cell carcinoma. Mol. Med. Rep. 2016, 14, 3517–3524. [Google Scholar] [CrossRef]
- Gan, R.-H.; Lin, L.-S.; Zheng, D.; Zhao, Y.; Ding, L.-C.; Zheng, D.-L.; Lu, Y.-G. High expression of Notch2 drives tongue squamous cell carcinoma carcinogenesis. Exp. Cell Res. 2021, 399, 112452. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef]
- Zhao, Z.-L.; Zhang, L.; Huang, C.-F.; Ma, S.-R.; Bu, L.-L.; Liu, J.-F.; Yu, G.-T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, G.; Zhou, L.; Li, P.; Yun, M.; Shi, Q.; Wang, T.; Wu, X. Notch signalling induces epithelial-mesenchymal transition to promote metastasis in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 42, 2276–2284. [Google Scholar] [CrossRef]
- Broner, E.C.; Trujillo, J.A.; Korzinkin, M.; Subbannayya, T.; Agrawal, N.; Ozerov, I.V.; Zhavoronkov, A.; Rooper, L.; Kotlov, N.; Shen, L.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Oncotarget 2021, 6, 5602–5614. [Google Scholar]
- Matsuura, N.; Tanaka, K.; Yamasaki, M.; Yamashita, K.; Saito, T.; Makino, T.; Yamamoto, K.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; et al. NOTCH3 limits the epithelial-mesenchymal transition and predicts a favorable clinical outcome in esophageal cancer. Cancer Med. 2021, 10, 3986–3996. [Google Scholar] [CrossRef]
- Fukusumi, T.; Guo, T.W.; Ren, S.; Haft, S.; Liu, C.; Sakai, A.; Ando, M.; Saito, Y.; Sadat, S.; Califano, J.A. Reciprocal activation of HEY1 and NOTCH4 under SOX2 control promotes EMT in head and neck squamous cell carcinoma. Int. J. Oncol. 2021, 58, 226–237. [Google Scholar] [CrossRef]
- Lu, Z.; Ren, Y.; Zhang, M.; Fan, T.; Wang, Y.; Zhao, Q.; Liu, H.-M.; Zhao, W.; Hou, G. FLI-06 suppresses proliferation, induces apoptosis and cell cycle arrest by targeting LSD1 and Notch pathway in esophageal squamous cell carcinoma cells. Biomed. Pharmacother. 2018, 107, 1370–1376. [Google Scholar] [CrossRef]
- Kayamori, K.; Katsube, K.-I.; Sakamoto, K.; Ohyama, Y.; Hirai, H.; Yukimori, A.; Ohata, Y.; Akashi, T.; Saitoh, M.; Harada, K.; et al. NOTCH3 Is Induced in Cancer-Associated Fibroblasts and Promotes Angiogenesis in Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0154112. [Google Scholar] [CrossRef]
- Zeng, L.; Nikolaev, A.; Xing, C.; Della Manna, D.L.; Yang, E.S. CHK1/2 Inhibitor Prexasertib Suppresses NOTCH Signaling and Enhances Cytotoxicity of Cisplatin and Radiation in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2020, 19, 1279–1288. [Google Scholar] [CrossRef]
- Taleb, S.; Abbaszadegan, M.R.; Moghbeli, M.; Roudbari, N.H.; Forghanifard, M.M. HES1 as an independent prognostic marker in esophageal squamous cell carcinoma. J. Gastrointest. Cancer 2014, 45, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Matías, G.; Velázquez-Velázquez, C.; Castro-Oropeza, R.; Mantilla-Morales, A.; Ocampo-Sandoval, D.; Burgos-González, A.; Heredia-Gutiérrez, C.; Alvarado-Cabrero, I.; Sánchez-Sandoval, R.; Barco-Bazán, A.; et al. Prevalence of HPV in Mexican Patients with Head and Neck Squamous Carcinoma and Identification of Potential Prognostic Biomarkers. Cancers 2021, 13, 5602. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Zhong, R.; Spiotto, M.T. Notch Signaling and Human Papillomavirus-Associated Oral Tumorigenesis. Adv. Exp. Med. Biol. 2021, 1287, 105–122. [Google Scholar] [PubMed]
- Mountzios, G.; Rampias, T.; Psyrri, A. The mutational spectrum of squamous-cell carcinoma of the head and neck: Targetable genetic events and clinical impact. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Jha, S.K.; Ojha, S.; Sharma, A.; Dholpuria, S.; Raju, V.S.R.; Prasher, P.; Chellappan, D.K.; Gupta, G.; Kumar Singh, S.; et al. The FBXW7-NOTCH interactome: A ubiquitin proteasomal system-induced crosstalk modulating oncogenic transformation in human tissues. Cancer Rep. 2021, 4, e1369. [Google Scholar] [CrossRef] [PubMed]
- Naganawa, Y.; Ishiguro, H.; Kuwabara, Y.; Kimura, M.; Mitsui, A.; Katada, T.; Tanaka, T.; Shiozaki, M.; Fujii, Y.; Takeyama, H. Decreased expression of FBXW7 is correlated with poor prognosis in patients with esophageal squamous cell carcinoma. Exp. Ther. Med. 2010, 1, 841–846. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Hosogane, M.; Okuyama, R.; Aoyama, S.; Onoyama, I.; Nakayama, K.I.; Nakayama, K. Opposing functions of Fbxw7 in keratinocyte growth, differentiation and skin tumorigenesis mediated through negative regulation of c-Myc and Notch. Oncogene 2013, 32, 1921–1932. [Google Scholar] [CrossRef]
- Jedlinski, A.; Ansell, A.; Johansson, A.-C.; Roberg, K. EGFR status and EGFR ligand expression influence the treatment response of head and neck cancer cell lines. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2013, 42, 26–36. [Google Scholar] [CrossRef]
- Leblanc, O.; Vacher, S.; Lecerf, C.; Jeannot, E.; Klijanienko, J.; Berger, F.; Hoffmann, C.; Calugaru, V.; Badois, N.; Chilles, A.; et al. Biomarkers of cetuximab resistance in patients with head and neck squamous cell carcinoma. Cancer Biol. Med. 2020, 17, 208–217. [Google Scholar] [CrossRef]
- Nisa, L.; Barras, D.; Medová, M.; Aebersold, D.M.; Medo, M.; Poliaková, M.; Koch, J.; Bojaxhiu, B.; Eliçin, O.; Dettmer, M.S.; et al. Comprehensive Genomic Profiling of Patient-matched Head and Neck Cancer Cells: A Preclinical Pipeline for Metastatic and Recurrent Disease. Mol. Cancer Res. 2018, 16, 1912–1926. [Google Scholar] [CrossRef]
- Kriegs, M.; Clauditz, T.S.; Hoffer, K.; Bartels, J.; Buhs, S.; Gerull, H.; Zech, H.B.; Bußmann, L.; Struve, N.; Rieckmann, T.; et al. Analyzing expression and phosphorylation of the EGF receptor in HNSCC. Sci. Rep. 2019, 9, 13564. [Google Scholar] [CrossRef]
- de Kort, W.W.B.; Spelier, S.; Devriese, L.A.; van Es, R.J.J.; Willems, S.M. Predictive Value of EGFR-PI3K-AKT-mTOR-Pathway Inhibitor Biomarkers for Head and Neck Squamous Cell Carcinoma: A Systematic Review. Mol. Diagn. Ther. 2021, 25, 123–136. [Google Scholar] [CrossRef]
- Jouan-Hureaux, V.; Boura, C.; Merlin, J.-L.; Faivre, B. Modulation of endothelial cell network formation in vitro by molecular signaling of head and neck squamous cell carcinoma (HNSCC) exposed to cetuximab. Microvasc. Res. 2012, 83, 131–137. [Google Scholar] [CrossRef]
- Peddi, P.; Paryani, B.; Takalkar, A.; Bundrick, P.; Ponugupati, J.; Nair, B.; El-Osta, H. Exceptional response to cetuximab monotherapy in a patient with metastatic oropharyngeal squamous cell carcinoma: A molecular insight. Onco. Targets. Ther. 2016, 9, 705–709. [Google Scholar]
- Khaznadar, S.S.; Khan, M.; Schmid, E.; Gebhart, S.; Becker, E.-T.; Krahn, T.; von Ahsen, O. EGFR overexpression is not common in patients with head and neck cancer. Cell lines are not representative for the clinical situation in this indication. Oncotarget 2018, 9, 28965–28975. [Google Scholar] [CrossRef]
- Fan, W.-L.; Yang, L.-Y.; Hsieh, J.C.-H.; Lin, T.-C.; Lu, M.-Y.J.; Liao, C.-T. Prognostic Genetic Biomarkers Based on Oncogenic Signaling Pathways for Outcome Prediction in Patients with Oral Cavity Squamous Cell Carcinoma. Cancers 2021, 13, 2709. [Google Scholar] [CrossRef]
- Trieu, V.; Pinto, H.; Riess, J.W.; Lira, R.; Luciano, R.; Coty, J.; Boothroyd, D.; Colevas, A.D. Weekly Docetaxel, Cisplatin, and Cetuximab in Palliative Treatment of Patients with Squamous Cell Carcinoma of the Head and Neck. Oncologist 2018, 23, 764. [Google Scholar] [CrossRef]
- Taberna, M.; Oliva, M.; Mesía, R. Cetuximab-Containing Combinations in Locally Advanced and Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 383. [Google Scholar] [CrossRef]
- Lynggaard, C.D.; Therkildsen, M.H.; Kristensen, C.A.; Specht, L. The EXTREME regimen for recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC): Treatment outcome in a single institution cohort. Acta Oncol. 2015, 54, 1071–1075. [Google Scholar] [CrossRef]
- Nakano, K.; Marshall, S.; Taira, S.; Sato, Y.; Tomomatsu, J.; Sasaki, T.; Shimbashi, W.; Fukushima, H.; Yonekawa, H.; Mitani, H.; et al. A comparison of weekly paclitaxel and cetuximab with the EXTREME regimen in the treatment of recurrent/metastatic squamous cell head and neck carcinoma. Oral Oncol. 2017, 73, 21–26. [Google Scholar] [CrossRef]
- Rognoni, C.; Quaglini, S.; Vermorken, J.B.; de Cecco, L.; Licitra, L.; Bossi, P. Cost-effectiveness of Molecular Profile Patient Selection for First-line Treatment of Recurrent/Metastatic Head and Neck Cancer. Clin. Ther. 2019, 41, 2517–2528.e28. [Google Scholar] [CrossRef]
- De Cecco, L.; Giannoccaro, M.; Marchesi, E.; Bossi, P.; Favales, F.; Locati, L.D.; Licitra, L.; Pilotti, S.; Canevari, S. Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer. Genes 2017, 8, 35. [Google Scholar] [CrossRef]
- Schmitz, S.; Bindea, G.; Albu, R.I.; Mlecnik, B.; Machiels, J.-P. Cetuximab promotes epithelial to mesenchymal transition and cancer associated fibroblasts in patients with head and neck cancer. Oncotarget 2015, 6, 34288–34299. [Google Scholar] [CrossRef]
- Siano, M.; Molinari, F.; Martin, V.; Mach, N.; Früh, M.; Freguia, S.; Corradino, I.; Ghielmini, M.; Frattini, M.; Espeli, V. Multicenter Phase II Study of Panitumumab in Platinum Pretreated, Advanced Head and Neck Squamous Cell Cancer. Oncologist 2017, 22, 782. [Google Scholar] [CrossRef]
- Ferris, R.L.; Geiger, J.L.; Trivedi, S.; Schmitt, N.C.; Heron, D.E.; Johnson, J.T.; Kim, S.; Duvvuri, U.; Clump, D.A.; Bauman, J.E.; et al. Phase II trial of post-operative radiotherapy with concurrent cisplatin plus panitumumab in patients with high-risk, resected head and neck cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 2257–2262. [Google Scholar] [CrossRef]
- Siano, M.; Espeli, V.; Mach, N.; Bossi, P.; Licitra, L.; Ghielmini, M.; Frattini, M.; Canevari, S.; De Cecco, L. Gene signatures and expression of miRNAs associated with efficacy of panitumumab in a head and neck cancer phase II trial. Oral Oncol. 2018, 82, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Patel, U.; Pandey, M.; Kannan, S.; Samant, T.A.; Gera, P.; Mittal, N.; Rane, S.; Patil, A.; Noronha, V.; Joshi, A.; et al. Prognostic and predictive significance of nuclear HIF1α expression in locally advanced HNSCC patients treated with chemoradiation with or without nimotuzumab. Br. J. Cancer 2020, 123, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Ang, M.-K.; Montoya, J.E.; Tharavichitkul, E.; Lim, C.; Tan, T.; Wang, L.Y.; Wee, J.; Soong, Y.-L.; Fong, K.-W.; Ng, Q.S.; et al. Phase II study of nimotuzumab (TheraCim-hR3) concurrent with cisplatin/radiotherapy in patients with locally advanced head and neck squamous cell carcinoma. Head Neck 2021, 43, 1641–1651. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Goyal, P.; Agrawal, C.R.; Bothra, S.J.; Jain, P.; Choudhury, K.D.; Gupta, S.K.; Sharma, M.; Bajaj, R.; Upadhyay, A.; et al. Efficacy and tolerability of nimotuzumab in combination with chemotherapy in recurrent and metastatic squamous cell carcinoma of head and neck at a cancer center in Northern India. Indian J. Cancer 2020, 57, 76–83. [Google Scholar] [PubMed]
- Machiels, J.-P.; Specenier, P.; Krauß, J.; Dietz, A.; Kaminsky, M.-C.; Lalami, Y.; Henke, M.; Keilholz, U.; Knecht, R.; Skartved, N.J.; et al. A proof of concept trial of the anti-EGFR antibody mixture Sym004 in patients with squamous cell carcinoma of the head and neck. Cancer Chemother. Pharmacol. 2015, 76, 13–20. [Google Scholar] [CrossRef]
- Fukuoka, S.; Kojima, T.; Koga, Y.; Yamauchi, M.; Komatsu, M.; Komatsuzaki, R.; Sasaki, H.; Yasunaga, M.; Matsumura, Y.; Doi, T.; et al. Preclinical efficacy of Sym004, novel anti-EGFR antibody mixture, in esophageal squamous cell carcinoma cell lines. Oncotarget 2017, 8, 11020–11029. [Google Scholar] [CrossRef]
- Iida, M.; Brand, T.M.; Starr, M.M.; Li, C.; Huppert, E.J.; Luthar, N.; Pedersen, M.W.; Horak, I.D.; Kragh, M.; Wheeler, D.L. Sym004, a novel EGFR antibody mixture, can overcome acquired resistance to cetuximab. Neoplasia 2013, 15, 1196–1206. [Google Scholar] [CrossRef]
- Jones, S.; King, P.J.; Antonescu, C.N.; Sugiyama, M.G.; Bhamra, A.; Surinova, S.; Angelopoulos, N.; Kragh, M.; Pedersen, M.W.; Hartley, J.A.; et al. Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep. 2020, 10, 663. [Google Scholar] [CrossRef]
- Trivedi, S.; Ferris, R.L. Epidermal Growth Factor Receptor-Targeted Therapy for Head and Neck Cancer. Otolaryngol. Clin. North Am. 2021, 54, 743–749. [Google Scholar] [CrossRef]
- Schrader, C.; Boehm, A.; Reiche, A.; Dietz, A.; Mozet, C.; Wichmann, G. Combined effects of lapatinib and cisplatin on colony formation of head and neck squamous cell carcinoma. Anticancer Res. 2012, 32, 3191–3199. [Google Scholar]
- Warren, E.A.K.; Anil, J.; Castro, P.D.; Kemnade, J.; Suzuki, M.; Hegde, M.; Hicks, J.; Yu, W.; Sandulache, V.; Sikora, A.G. Human epidermal growth factor receptor 2 expression in head and neck squamous cell carcinoma: Variation within and across primary tumor sites, and implications for antigen-specific immunotherapy. Head Neck 2021, 43, 1983–1994. [Google Scholar] [CrossRef]
- Guo, Y.; Ahn, M.-J.; Chan, A.; Wang, C.-H.; Kang, J.-H.; Kim, S.-B.; Bello, M.; Arora, R.S.; Zhang, Q.; He, X.; et al. Afatinib versus methotrexate as second-line treatment in Asian patients with recurrent or metastatic squamous cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 3): An open-label, randomised phase III tria. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1831–1839. [Google Scholar]
- Klinghammer, K.; Politz, O.; Eder, T.; Otto, R.; Raguse, J.-D.; Albers, A.; Kaufmann, A.; Tinhofer, I.; Hoffmann, J.; Keller, U.; et al. Combination of copanlisib with cetuximab improves tumor response in cetuximab-resistant patient-derived xenografts of head and neck cancer. Oncotarget 2020, 11, 3688–3697. [Google Scholar] [CrossRef]
- Driehuis, E.; Spelier, S.; Beltrán Hernández, I.; de Bree, R.; M Willems, S.; Clevers, H.; Oliveira, S. Patient-Derived Head and Neck Cancer Organoids Recapitulate EGFR Expression Levels of Respective Tissues and Are Responsive to EGFR-Targeted Photodynamic Therapy. J. Clin. Med. 2019, 8, 1880. [Google Scholar] [CrossRef]
- Schäfer, M.; Semmler, M.L.; Bernhardt, T.; Fischer, T.; Kakkassery, V.; Ramer, R.; Hein, M.; Bekeschus, S.; Langer, P.; Hinz, B.; et al. Small Molecules in the Treatment of Squamous Cell Carcinomas: Focus on Indirubins. Cancers 2021, 13, 1770. [Google Scholar] [CrossRef]
- Li, T.; Wen, H.; Brayton, C.; Das, P.; Smithson, L.A.; Fauq, A.; Fan, X.; Crain, B.J.; Price, D.L.; Golde, T.E.; et al. Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma-secretase. J. Biol. Chem. 2007, 282, 32264–32273. [Google Scholar] [CrossRef]
- Kolev, V.; Mandinova, A.; Guinea-Viniegra, J.; Hu, B.; Lefort, K.; Lambertini, C.; Neel, V.; Dummer, R.; Wagner, E.F.; Dotto, G.P. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat. Cell Biol. 2008, 10, 902–911. [Google Scholar] [CrossRef]
- Kashyap, T.; Pramanik, K.K.; Nath, N.; Mishra, P.; Singh, A.K.; Nagini, S.; Rana, A.; Mishra, R. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3\textgreek{b} signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol. 2018, 86, 234–243. [Google Scholar] [CrossRef]
- Hou, G.; Zhao, Q.; Zhang, M.; Wang, P.; Ye, H.; Wang, Y.; Ren, Y.; Zhang, J.; Lu, Z. LSD1 regulates Notch and PI3K/Akt/mTOR pathways through binding the promoter regions of Notch target genes in esophageal squamous cell carcinoma. Onco. Targets. Ther. 2019, 12, 5215–5225. [Google Scholar] [CrossRef]
- Qin, X.-K.; Du, Y.; Liu, X.-H.; Wang, L. LSD1 Promotes Prostate Cancer Cell Survival by Destabilizing FBXW7 at Post-Translational Level. Front. Oncol. 2020, 10, 616185. [Google Scholar] [CrossRef]
- Y, Z.; Z, W.; X, X.; Y, Z.; W, Z.; Y, D.; J, L.; Z, Z.; W, Z.; H, W.; et al. Membrane-tethered Notch1 exhibits oncogenic property via activation of EGFR-PI3K-AKT pathway in oral squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 5940–5952. [Google Scholar]
- Wang, W.-M.; Zhao, Z.-L.; Ma, S.-R.; Yu, G.-T.; Liu, B.; Zhang, L.; Zhang, W.-F.; Kulkarni, A.B.; Sun, Z.-J.; Zhao, Y.-F. Epidermal growth factor receptor inhibition reduces angiogenesis via hypoxia-inducible factor-1α and Notch1 in head neck squamous cell carcinoma. PLoS ONE 2015, 10, e0119723. [Google Scholar] [CrossRef]
- Secades, P.; de Santa-María, I.S.; Merlo, A.; Suarez, C.; Chiara, M.-D. In vitro study of normoxic epidermal growth factor receptor-induced hypoxia-inducible factor-1-alpha, vascular endothelial growth factor, and BNIP3 expression in head and neck squamous cell carcinoma cell lines: Implications for anti-epidermal growth fac. Head Neck 2015, 37, 1150–1162. [Google Scholar] [CrossRef]
- Stegeman, H.; Span, P.N.; Peeters, W.J.; Verheijen, M.M.; Grénman, R.; Meijer, T.W.; Kaanders, J.H.; Bussink, J. Interaction between hypoxia, AKT and HIF-1 signaling in HNSCC and NSCLC: Implications for future treatment strategies. Futur. Sci. OA 2016, 2, FSO84. [Google Scholar] [CrossRef]
- Coliat, P.; Ramolu, L.; Jégu, J.; Gaiddon, C.; Jung, A.C.; Pencreach, E. Constitutive or Induced HIF-2 Addiction is Involved in Resistance to Anti-EGFR Treatment and Radiation Therapy in HNSCC. Cancers 2019, 11, 1607. [Google Scholar] [CrossRef]
- Wozny, A.-S.; Lauret, A.; Battiston-Montagne, P.; Guy, J.-B.; Beuve, M.; Cunha, M.; Saintigny, Y.; Blond, E.; Magne, N.; Lalle, P.; et al. Differential pattern of HIF-1α expression in HNSCC cancer stem cells after carbon ion or photon irradiation: One molecular explanation of the oxygen effect. Br. J. Cancer 2017, 116, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, S.K.; Kaluz, S.; Wang, D.; Wang, K.; Van Meir, E.G.; Wang, B. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med. Chem. 2013, 5, 553–572. [Google Scholar] [CrossRef] [PubMed]
- de Mendonça Fernandes, G.M.; Galbiatti-Dias, A.L.S.; Ferreira, L.A.M.; Serafim Junior, V.; Rodrigues-Fleming, G.H.; de Oliveira-Cucolo, J.G.; Biselli-Chicote, P.M.; Kawasaki-Oyama, R.S.; Maniglia, J.V.; Pavarino, É.C.; et al. Anti-EGFR treatment effects on laryngeal cancer stem cells. Am. J. Transl. Res. 2021, 13, 143–155. [Google Scholar] [PubMed]
- Wiechec, E.; Hansson, K.T.; Alexandersson, L.; Jönsson, J.-I.; Roberg, K. Hypoxia Mediates Differential Response to Anti-EGFR Therapy in HNSCC Cells. Int. J. Mol. Sci. 2017, 18, 943. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef]
- Mei, Z.; Huang, J.; Qiao, B.; Lam, A.K.-Y. Immune checkpoint pathways in immunotherapy for head and neck squamous cell carcinoma. Int. J. Oral Sci. 2020, 12, 16. [Google Scholar] [CrossRef]
- Puntigam, L.K.; Jeske, S.S.; Götz, M.; Greiner, J.; Laban, S.; Theodoraki, M.-N.; Doescher, J.; Weissinger, S.E.; Brunner, C.; Hoffmann, T.K.; et al. Immune Checkpoint Expression on Immune Cells of HNSCC Patients and Modulation by Chemo- and Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5181. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.J.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Bossi, P.; Alfieri, S.; Strojan, P.; Takes, R.P.; López, F.; Mäkitie, A.; Saba, N.F.; Rodrigo, J.P.; Bradford, C.; Suarez, C.; et al. Safety and Efficacy of Pembrolizumab With Chemoradiotherapy in Locally Advanced Head and Neck Squamous Cell Carcinoma: A Phase IB Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 393, 901–915. [Google Scholar]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.-T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef]
- Saleh, K.; Eid, R.; Haddad, F.G.; Khalife-Saleh, N.; Kourie, H.R. New developments in the management of head and neck —Impact of pembrolizumab. Ther. Clin. Risk Manag. 2018, 14, 295–303. [Google Scholar] [CrossRef]
- Zargar, M.; McFarlane, T.; Chan, K.K.W.; Wong, W.W.L. Cost-Effectiveness of Nivolumab in Recurrent Metastatic Head and Neck Squamous Cell Carcinoma. Oncologist 2018, 23, 225–233. [Google Scholar] [CrossRef]
- Wakasaki, T.; Yasumatsu, R.; Uchi, R.; Taura, M.; Matsuo, M.; Komune, N.; Nakagawa, T. Outcome of chemotherapy following nivolumab treatment for recurrent and/or metastatic head and neck squamous cell carcinoma. Auris. Nasus. Larynx 2020, 47, 116–122. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.J.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.J.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018, 81, 45–51. [Google Scholar] [CrossRef]
- Ferris, R.L.; Licitra, L.; Fayette, J.; Even, C.; Blumenschein, G.J.; Harrington, K.J.; Guigay, J.; Vokes, E.E.; Saba, N.F.; Haddad, R.; et al. Nivolumab in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck: Efficacy and Safety in CheckMate 141 by Prior Cetuximab Use. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5221–5230. [Google Scholar] [CrossRef]
- Li, X.; Fang, Q.; Du, W.; Zhang, X.; Dai, L.; Qiao, Y. Induction chemotherapy combined with immunotherapy in locally advanced head and neck squamous cell carcinoma. BMC Cancer 2021, 21, 622. [Google Scholar] [CrossRef]
- McCusker, M.G.; Orkoulas-Razis, D.; Mehra, R. Potential of Pembrolizumab in Metastatic or Recurrent Head and Neck Cancer: Evidence to Date. Onco Targets Ther. 2020, 13, 3047–3059. [Google Scholar] [CrossRef]
- Soulières, D.; Aguilar, J.L.; Chen, E.; Misiukiewicz, K.; Ernst, S.; Lee, H.J.; Bryant, K.; He, S.; Obasaju, C.K.; Chang, S.-C.; et al. Cetuximab plus platinum-based chemotherapy in head and neck squamous cell carcinoma: A randomized, double-blind safety study comparing cetuximab produced from two manufacturing processes using the EXTREME study regimen. BMC Cancer 2016, 16, 19. [Google Scholar] [CrossRef]
- Szturz, P.; Vermorken, J.B. Immunotherapy in head and neck cancer: Aiming at EXTREME precision. BMC Med. 2017, 15, 110. [Google Scholar] [CrossRef]
- Harrington, K.J.; Ferris, R.L.; Blumenschein, G.J.; Colevas, A.D.; Fayette, J.; Licitra, L.; Kasper, S.; Even, C.; Vokes, E.E.; Worden, F.; et al. Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): Health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1104–1115. [Google Scholar] [CrossRef]
- Kondo, T.; Okamoto, I.; Sato, H.; Koyama, N.; Fushimi, C.; Okada, T.; Masubuchi, T.; Miura, K.; Matsuki, T.; Yamashita, T.; et al. Age-based efficacy and safety of nivolumab for recurrent or metastatic head and neck squamous cell carcinoma: A multicenter retrospective study. Asia. Pac. J. Clin. Oncol. 2020, 16, 340–347. [Google Scholar] [CrossRef]
- Kocikowski, M.; Dziubek, K.; Parys, M. Hyperprogression under Immune Checkpoint-Based Immunotherapy-Current Understanding, The Role of PD-1/PD-L1 Tumour-Intrinsic Signalling, Future Directions and a Potential Large Animal Model. Cancers 2020, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Merlino, D.J.; Johnson, J.M.; Tuluc, M.; Gargano, S.; Stapp, R.; Harshyne JR, L.; Leiby, B.E.; Flanders, A.; Zinner, R.; Axelrod, R.; et al. Discordant Responses Between Primary Head and Neck Tumors and Nodal Metastases Treated With Neoadjuvant Nivolumab: Correlation of Radiographic and Pathologic Treatment Effect. Front. Oncol. 2020, 10, 566315. [Google Scholar] [CrossRef] [PubMed]
- Vasiliadou, I.; Breik, O.; Baker, H.; Leslie, I.; van Sim, R.; Hegarty, G.; Michaelidou, A.; Nathan, K.; Hartley, A.; Good, J.; et al. Safety and Treatment Outcomes of Nivolumab for the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: Retrospective Multicenter Cohort Study. Cancers 2021, 13, 1413. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Shi, Y.; Lv, S.; Dai, T.; Zhang, F.; Liu, S.; Wu, B. Nivolumab vs Pembrolizumab for Treatment of US Patients With Platinum-Refractory Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: A Network Meta-analysis and Cost-effectiveness Analysis. JAMA Netw. Open 2021, 4, e218065. [Google Scholar] [CrossRef]
- Liu, M.; Han, S.; Zheng, B.; Cai, H.; Yang, J.; Zhuang, Q.; Li, N. Cost-Effectiveness Analysis Of Pembrolizumab In The Treatment Of Advanced Recurrent Metastatic Head And Neck Squamous Cell Carcinoma In China And The United States. Cancer Manag. Res. 2019, 11, 9483–9493. [Google Scholar] [CrossRef]
- Yeh, J.; Guddati, A.K. Cost-effectiveness analysis of nivolumab compared to pembrolizumab in the treatment of recurrent or metastatic squamous cell carcinoma of the head and neck. Am. J. Cancer Res. 2020, 10, 1821–1826. [Google Scholar]
- Kornman, K.S.; Polverini, P.J. Clinical application of genetics to guide prevention and treatment of oral diseases. Clin. Genet. 2014, 86, 44–49. [Google Scholar] [CrossRef]
- Chung, C.H.; Bonomi, M.; Steuer, C.E.; Li, J.; Bhateja, P.; Johnson, M.; Masannat, J.; Song, F.; Hernandez-Prera, J.C.; Wenig, B.M.; et al. Concurrent Cetuximab and Nivolumab as a Second-Line or beyond Treatment of Patients with Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results of Phase I/II Study. Cancers 2021, 13, 1180. [Google Scholar] [CrossRef]
- Weidhaas, J.B.; Harris, J.; Schaue, D.; Chen, A.M.; Chin, R.; Axelrod, R.; El-Naggar, A.K.; Singh, A.K.; Galloway, T.J.; Raben, D.; et al. The KRAS-Variant and Cetuximab Response in Head and Neck Squamous Cell Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 483–491. [Google Scholar] [CrossRef]
- Zech, H.B.; Moeckelmann, N.; Boettcher, A.; Muenscher, A.; Binder, M.; Vettorazzi, E.; Bokemeyer, C.; Schafhausen, P.; Betz, C.S.; Busch, C.-J. Phase III study of nivolumab alone or combined with ipilimumab as immunotherapy versus standard of care in resectable head and neck squamous cell carcinoma. Future Oncol. 2020, 16, 3035–3043. [Google Scholar] [CrossRef]
- Machiels, J.-P.; Tao, Y.; Burtness, B.; Tahara, M.; Licitra, L.; Rischin, D.; Waldron, J.; Simon, C.; Gregoire, V.; Harrington, K.; et al. Pembrolizumab given concomitantly with chemoradiation and as maintenance therapy for locally advanced head and neck squamous cell carcinoma: KEYNOTE-412. Future Oncol. 2020, 16, 1235–1243. [Google Scholar] [CrossRef]
- Desilets, A.; Soulières, D. Safety evaluation of pembrolizumab for treating recurrent head and neck squamous cell carcinoma. Expert Opin. Drug Saf. 2020, 19, 927–934. [Google Scholar] [CrossRef]
- Aarstad, H.J.; Heimdal, J.-H.H.; Klementsen, B.; Olofsson, J.; Ulvestad, E.; Abbaszadegan, M.R.; Moghbeli, M.; Riahi, A.; Forghanifard, M.M.; Moghbeli, M.; et al. In vitro humanized 3D microfluidic chip for testing personalized immunotherapeutics for head and neck cancer patients. Sci. Rep. 2020, 12, 111508. [Google Scholar]
- Rodriguez, C.P.; Wu, Q.V.; Voutsinas, J.; Fromm, J.R.; Jiang, X.; Pillarisetty, V.G.; Lee, S.M.; Santana-Davila, R.; Goulart, B.; Baik, C.S.; et al. A Phase II Trial of Pembrolizumab and Vorinostat in Recurrent Metastatic Head and Neck Squamous Cell Carcinomas and Salivary Gland Cancer. Clin. Cancer Res. an Off. J. Am. Assoc. Cancer Res. 2020, 26, 837–845. [Google Scholar] [CrossRef]
- Mao, L.; Zhao, Z.-L.; Yu, G.-T.; Wu, L.; Deng, W.-W.; Li, Y.-C.; Liu, J.-F.; Bu, L.-L.; Liu, B.; Kulkarni, A.B.; et al. γ-Secretase inhibitor reduces immunosuppressive cells and enhances tumour immunity in head and neck squamous cell carcinoma. Int. J. Cancer 2018, 142, 999–1009. [Google Scholar] [CrossRef]
- Zhang, K.; Hong, X.; Song, Z.; Xu, Y.; Li, C.; Wang, G.; Zhang, Y.; Zhao, X.; Zhao, Z.; Zhao, J.; et al. Identification of Deleterious NOTCH Mutation as Novel Predictor to Efficacious Immunotherapy in NSCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3649–3661. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Li, X.; Feng, G.; Hu, S.; Bai, Y. The Impact of NOTCH Pathway Alteration on Tumor Microenvironment and Clinical Survival of Immune Checkpoint Inhibitors in NSCLC. Front. Immunol. 2021, 12, 638763. [Google Scholar] [CrossRef]
- Mazzotta, M.; Filetti, M.; Occhipinti, M.; Marinelli, D.; Scalera, S.; Terrenato, I.; Sperati, F.; Pallocca, M.; Rizzo, F.; Gelibter, A.; et al. Efficacy of immunotherapy in lung cancer with co-occurring mutations in NOTCH and homologous repair genes. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Zhang, Z.; Gu, Y.; Su, X.; Bai, J.; Guan, W.; Ma, J.; Luo, J.; He, J.; Zhang, B.; Geng, M.; et al. Co-Occurring Alteration of NOTCH and DDR Pathways Serves as Novel Predictor to Efficacious Immunotherapy in NSCLC. Front. Oncol. 2021, 11, 659321. [Google Scholar] [CrossRef]
- Cui, Y.; Li, Q.; Li, W.; Wang, Y.; Lv, F.; Shi, X.; Tang, Z.; Shen, Z.; Hou, Y.; Zhang, H.; et al. NOTCH3 is a Prognostic Factor and Is Correlated With Immune Tolerance in Gastric Cancer. Front. Oncol. 2020, 10, 574937. [Google Scholar] [CrossRef]
- Long, J.; Wang, D.; Yang, X.; Wang, A.; Lin, Y.; Zheng, M.; Zhang, H.; Sang, X.; Wang, H.; Hu, K.; et al. Identification of NOTCH4 mutation as a response biomarker for immune checkpoint inhibitor therapy. BMC Med. 2021, 19, 154. [Google Scholar] [CrossRef]
- Jiang, P.; Li, Y.; Xu, Z.; He, S. A signature of 17 immune-related gene pairs predicts prognosis and immune status in HNSCC patients. Transl. Oncol. 2021, 14, 100924. [Google Scholar] [CrossRef]
- Mathieu, M.; Cotta-Grand, N.; Daudelin, J.-F.; Thébault, P.; Labrecque, N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol. Cell Biol. 2013, 91, 82–88. [Google Scholar] [CrossRef]
- Pan, T.; Liu, Z.; Yin, J.; Zhou, T.; Liu, J.; Qu, H. Notch Signaling Pathway Was Involved in Regulating Programmed Cell Death 1 Expression during Sepsis-Induced Immunosuppression. Mediat. Inflamm. 2015, 2015, 539841. [Google Scholar] [CrossRef]
- Hanna, G.J.; Lizotte, P.; Cavanaugh, M.; Kuo, F.C.; Shivdasani, P.; Frieden, A.; Chau, N.G.; Schoenfeld, J.D.; Lorch, J.H.; Uppaluri, R.; et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Zandberg, D.P.; Algazi, A.P.; Jimeno, A.; Good, J.S.; Fayette, J.; Bouganim, N.; Ready, N.E.; Clement, P.M.; Even, C.; Jang, R.W.; et al. Durvalumab for recurrent or metastatic head and neck squamous cell carcinoma: Results from a single-arm, phase II study in patients with ≥25% tumour cell PD-L1 expression who have progressed on platinum-based chemotherapy. Eur. J. Cancer 2019, 107, 142–152. [Google Scholar] [CrossRef]
- Ferris, R.L.; Haddad, R.; Even, C.; Tahara, M.; Dvorkin, M.; Ciuleanu, T.E.; Clement, P.M.; Mesia, R.; Kutukova, S.; Zholudeva, L.; et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 942–950. [Google Scholar] [CrossRef]
- Carlisle, J.W.; Steuer, C.E.; Owonikoko, T.K.; Saba, N.F. An update on the immune landscape in lung and head and neck cancers. CA. Cancer J. Clin. 2020, 70, 505–517. [Google Scholar] [CrossRef]
- Siu, L.L.; Even, C.; Mesía, R.; Remenar, E.; Daste, A.; Delord, J.-P.; Krauss, J.; Saba, N.F.; Nabell, L.; Ready, N.E.; et al. Safety and Efficacy of Durvalumab With or Without Tremelimumab in Patients With PD-L1-Low/Negative Recurrent or Metastatic HNSCC: The Phase 2 CONDOR Randomized Clinical Trial. JAMA Oncol. 2019, 5, 195–203. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Hanna, G.J.; Jo, V.Y.; Rawal, B.; Chen, Y.-H.; Catalano, P.S.; Lako, A.; Ciantra, Z.; Weirather, J.L.; Criscitiello, S.; et al. Neoadjuvant Nivolumab or Nivolumab Plus Ipilimumab in Untreated Oral Cavity Squamous Cell Carcinoma: A Phase 2 Open-Label Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1563–1570. [Google Scholar] [CrossRef]
- Kao, H.-F.; Lou, P.-J. Immune checkpoint inhibitors for head and neck squamous cell carcinoma: Current landscape and future directions. Head Neck 2019, 41 (Suppl. S1), 4–18. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Gostian, A.O.; Eckstein, M.; Rutzner, S.; von der Grün, J.; Illmer, T.; Hautmann, M.G.; Klautke, G.; Laban, S.; Brunner, T.; et al. Safety and efficacy of single cycle induction treatment with cisplatin/docetaxel/ durvalumab/tremelimumab in locally advanced HNSCC: First results of CheckRad-CD8. JAMA Oncol. 2020, 8, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Masarwy, R.; Kampel, L.; Horowitz, G.; Gutfeld, O.; Muhanna, N. Neoadjuvant PD-1/PD-L1 Inhibitors for Resectable Head and Neck Cancer: A Systematic Review and Meta-analysis. JAMA Otolaryngol. Head Neck Surg. 2021, 147, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef]
- Ferrarotto, R.; William, W.N.J.; Tseng, J.E.; Marur, S.; Shin, D.M.; Murphy, B.; Cohen, E.E.W.; Thomas, C.Y.; Willey, R.; Cosaert, J.; et al. Randomized phase II trial of cixutumumab alone or with cetuximab for refractory recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2018, 82, 83–90. [Google Scholar] [CrossRef]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef]
- Lee, D.H. Update of early phase clinical trials in cancer immunotherapy. BMB Rep. 2021, 54, 70–88. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Niehr, F.; Weichert, W.; Stenzinger, A.; Budach, V.; Tinhofer, I. CCI-779 (Temsirolimus) exhibits increased anti-tumor activity in low EGFR expressing HNSCC cell lines and is effective in cells with acquired resistance to cisplatin or cetuximab. J. Transl. Med. 2015, 13, 106. [Google Scholar] [CrossRef][Green Version]
- Dunn, L.A.; Fury, M.G.; Xiao, H.; Baxi, S.S.; Sherman, E.J.; Korte, S.; Pfister, C.; Haque, S.; Katabi, N.; Ho, A.L.; et al. A phase II study of temsirolimus added to low-dose weekly carboplatin and paclitaxel for patients with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2533–2538. [Google Scholar] [CrossRef]
- Liu, X.; Kambrick, S.; Fu, S.; Naing, A.; Subbiah, V.; Blumenschein, G.R.; Glisson, B.S.; Kies, M.S.; Tsimberidou, A.M.; Wheler, J.J.; et al. Advanced malignancies treated with a combination of the VEGF inhibitor bevacizumab, anti-EGFR antibody cetuximab, and the mTOR inhibitor temsirolimus. Oncotarget 2016, 7, 23227–23238. [Google Scholar] [CrossRef]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K.; et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: A multicentre, multigroup, phase 2 trial. Lancet. Oncol. 2019, 20, 1295–1305. [Google Scholar] [CrossRef]
- Wang, T.-H.; Chen, C.-C.; Leu, Y.-L.; Lee, Y.-S.; Lian, J.-H.; Hsieh, H.-L.; Chen, C.-Y. Palbociclib induces DNA damage and inhibits DNA repair to induce cellular senescence and apoptosis in oral squamous cell carcinoma. J. Formos. Med. Assoc. 2021, 120, 1695–1705. [Google Scholar] [CrossRef]
- Gadsden, N.J.; Fulcher, C.D.; Li, D.; Shrivastava, N.; Thomas, C.; Segall, J.E.; Prystowsky, M.B.; Schlecht, N.F.; Gavathiotis, E.; Ow, T.J. Palbociclib Renders Human Papilloma Virus-Negative Head and Neck Squamous Cell Carcinoma Vulnerable to the Senolytic Agent Navitoclax. Mol. Cancer Res. 2021, 19, 862–873. [Google Scholar] [CrossRef]
- Robinson, A.M.; Rathore, R.; Redlich, N.J.; Adkins, D.R.; VanArsdale, T.; Van Tine, B.A.; Michel, L.S. Cisplatin exposure causes c-Myc-dependent resistance to CDK4/6 inhibition in HPV-negative head and neck squamous cell carcinoma. Cell Death Dis. 2019, 10, 867. [Google Scholar] [CrossRef]
- Ham, J.C.; van Meerten, E.; Fiets, W.E.; Beerepoot, L.V.; Jeurissen, F.J.F.; Slingerland, M.; Jonker, M.A.; Husson, O.; van der Graaf, W.T.A.; van Herpen, C.M.L. Methotrexate plus or minus cetuximab as first-line treatment in a recurrent or metastatic (R/M) squamous cell carcinoma population of the head and neck (SCCHN), unfit for cisplatin combination treatment, a phase Ib-randomized phase II study Commence. Head Neck 2020, 42, 828–838. [Google Scholar] [CrossRef]
- Sukari, A.; Nagasaka, M.; Diab, M.; Al Sibai, K.; Atassi, B.; Elayoubi, J.A.; Kim, S.; Küçük, Ö. Cetuximab and methotrexate in recurrent or metastatic head and neck squamous cell carcinoma-A single institution analysis of 54 patients. Clin. Otolaryngol. Off. J. ENT-UK Off. J. Neth. Soc. Oto-Rhino-Laryngol. Cerv.-Fac. Surg. 2019, 44, 639–643. [Google Scholar] [CrossRef]
- Xu, C.; Nikolova, O.; Basom, R.S.; Mitchell, R.M.; Shaw, R.; Moser, R.D.; Park, H.; Gurley, K.E.; Kao, M.C.; Green, C.L.; et al. Functional Precision Medicine Identifies Novel Druggable Targets and Therapeutic Options in Head and Neck Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2828–2843. [Google Scholar] [CrossRef]
- Gerullis, H.; Wawroschek, F.; Köhne, C.-H.; Ecke, T.H. Vinflunine in the treatment of advanced urothelial cancer: Clinical evidence and experience. Ther. Adv. Urol. 2017, 9, 28–35. [Google Scholar] [CrossRef]
- Lee, J.W.; Parameswaran, J.; Sandoval-Schaefer, T.; Eoh, K.J.; Yang, D.-H.; Zhu, F.; Mehra, R.; Sharma, R.; Gaffney, S.G.; Perry, E.B.; et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3430–3442. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Idris, A.; Johnson, N.W.; Fallaha, S.; Clarke, D.T.W.; Martin, D.; Morgan, I.M.; Gabrielli, B.; McMillan, N.A.J. Aurora kinases are a novel therapeutic target for HPV-positive head and neck cancers. Oral Oncol. 2018, 86, 105–112. [Google Scholar] [CrossRef]
- Tang, P.A.; Siu, L.L.; Chen, E.X.; Hotte, S.J.; Chia, S.; Schwarz, J.K.; Pond, G.R.; Johnson, C.; Colevas, A.D.; Synold, T.W.; et al. Phase II study of ispinesib in recurrent or metastatic squamous cell carcinoma of the head and neck. Invest. New Drugs 2008, 26, 257–264. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef]
- Jung, A.R.; Jung, C.-H.; Noh, J.K.; Lee, Y.C.; Eun, Y.-G. Epithelial-mesenchymal transition gene signature is associated with prognosis and tumor microenvironment in head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 3652. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, P. TGFβ and matrix-regulated epithelial to mesenchymal transition. Biochim. Biophys. Acta 2014, 1840, 2621–2634. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Si, J.; Yue, J.; Ma, S. The mechanisms and reversal strategies of tumor radioresistance in esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2021, 147, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, L.; Liu, F.-Y.F.; Li, P.; He, J.; Kirkwood, C.L.; Sohn, J.; Chan, J.M.; Magner, W.J.; Kirkwood, K.L.; et al. Linking Cancer Stem Cell Plasticity to Therapeutic Resistance-Mechanism and Novel Therapeutic Strategies in Esophageal Cancer. Cells 2021, 9, 105401. [Google Scholar]
- Haensel, D.; Dai, X. Epithelial-to-mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev. Dyn. 2018, 247, 473. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Invest. 2009, 119, 1417–1419. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Zidar, N.; Boštjančič, E.; Malgaj, M.; Gale, N.; Dovšak, T.; Didanovič, V. The role of epithelial-mesenchymal transition in squamous cell carcinoma of the oral cavity. Virchows Arch. 2018, 472, 237–245. [Google Scholar] [CrossRef]
- Kiełbus, M.; Czapiński, J.; Kałafut, J.; Woś, J.; Stepulak, A.; Rivero-Müller, A. Genetically Engineered Lung Cancer Cells for Analyzing Epithelial–Mesenchymal Transition. Cells 2019, 8, 1644. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Zheng, X.; Dai, F.; Feng, L.; Zou, H.; Feng, L.; Xu, M. Communication between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights into Cancer Progression. Front. Oncol. 2021, 11, 617597. [Google Scholar] [CrossRef]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; d’Hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; Sol, A.d.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Hu, F.-W.; Yu, C.-C.; Tsai, L.-L.; Yu, C.-H.; Wu, B.-C.; Chen, Y.-W.; Huang, P.-I.; Lo, W.-L. Epithelial–mesenchymal transition transcription factor ZEB1/ZEB2 co-expression predicts poor prognosis and maintains tumor-initiating properties in head and neck cancer. Oral Oncol. 2013, 49, 34–41. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Aggarwal, V.; Montoya, C.A.; Donnenberg, V.S.; Sant, S.; Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int. J. Cancer 2018, 142, 102113. [Google Scholar]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Alabi, R.O.; Elmusrati, M.; Sawazaki-Calone, I.; Kowalski, L.P.; Haglund, C.; Coletta, R.D.; Mäkitie, A.A.; Salo, T.; Leivo, I.; Almangush, A.; et al. Staging and grading of oral squamous cell carcinoma: An update. Histopathology 2020, 43, 703–709. [Google Scholar]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Que, J.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; Lebleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Bunz, F. EMT and back again: Visualizing the dynamic phenotypes of metastasis. Cancer Res. 2020, 80, 153–155. [Google Scholar] [CrossRef]
- Daniel, Y.; Lelou, E.; Aninat, C.; Corlu, A.; Cabillic, F. Interplay between Metabolism Reprogramming and Epithelial-to-Mesenchymal Transition in Cancer Stem Cells. Cancers 2021, 13, 1973. [Google Scholar] [CrossRef]
- Liao, T.-T.; Yang, M.-H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.K.; Goldman, A.; Pietilä, M.; Mani, S.A.; Sengupta, S.; Ben-Jacob, E.; Levine, H.; Onuchic, J.N. Notch-Jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J. R. Soc. Interface 2016, 13, 20151106. [Google Scholar] [CrossRef]
- Parikh, A.S.; Puram, S.V.; Faquin, W.C.; Richmon, J.D.; Emerick, K.S.; Deschler, D.G.; Varvares, M.A.; Tirosh, I.; Bernstein, B.E.; Lin, D.T. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncol. 2019, 99, 104458. [Google Scholar] [CrossRef]
- Puram, S.V.; Parikh, A.S.; Tirosh, I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol. Cell. Oncol. 2018, 5, e1448244. [Google Scholar] [CrossRef]
- Qi, Z.; Liu, Y.; Mints, M.; Mullins, R.; Sample, R.; Law, T.; Barrett, T.; Mazul, A.L.; Jackson, R.S.; Kang, S.Y.; et al. Single-Cell Deconvolution of Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 1230. [Google Scholar] [CrossRef]
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Invest. 2016, 96, 508–516. [Google Scholar] [CrossRef]
- Inamura, N.; Kimura, T.; Wang, L.; Yanagi, H.; Tsuda, M.; Tanino, M.; Nishihara, H.; Fukuda, S.; Tanaka, S. Notch1 regulates invasion and metastasis of head and neck squamous cell carcinoma by inducing EMT through c-Myc. Auris. Nasus. Larynx 2017, 44, 447–457. [Google Scholar] [CrossRef]
- Fukusumi, T.; Guo, T.W.; Sakai, A.; Ando, M.; Ren, S.; Haft, S.; Liu, C.; Amornphimoltham, P.; Gutkind, J.S.; Califano, J.A. The NOTCH4-HEY1 pathway induces epithelial mesenchymal transition in head and neck squamous cell carcinoma. Clin. Cancer Res. 2018, 24, 619. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, S.; Qi, D.; Zheng, S.; Guo, J.; Zhang, S.; Weng, Z. Inhibition of Notch Signaling by a γ-Secretase Inhibitor Attenuates Hepatic Fibrosis in Rats. PLoS ONE 2012, 7, e46512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Z.; Zhang, M.; Gross, N.; Gong, L.; Zhang, S.; Lei, D.; Zeng, Q.; Luo, X.; Li, G.; et al. High Notch1 expression affects chemosensitivity of head and neck squamous cell carcinoma to paclitaxel and cisplatin treatment. Biomed. Pharmacother. 2019, 118, 109306. [Google Scholar] [CrossRef]
- Gan, R.-H.; Wei, H.; Xie, J.; Zheng, D.-P.; Luo, E.-L.; Huang, X.-Y.; Xie, J.; Zhao, Y.; Ding, L.-C.; Su, B.-H.; et al. Notch1 regulates tongue cancer cells proliferation, apoptosis and invasion. Cell Cycle 2018, 17, 216–224. [Google Scholar] [CrossRef]
- Mk, H.; Prince, S.; Mohan, A.M.; Krishnan, K.V.; Devi, A. Association of Notch4 with metastasis in human oral squamous cell carcinoma. Life Sci. 2016, 156, 38–46. [Google Scholar] [CrossRef]
- Villarejo, A.; Cortés-Cabrera, Á.; Molina-Ortíz, P.; Portillo, F.; Cano, A. Differential Role of Snail1 and Snail2 Zinc Fingers in E-cadherin Repression and Epithelial to Mesenchymal Transition. J. Biol. Chem. 2014, 289, 930. [Google Scholar] [CrossRef]
- Ota, I.; Masui, T.; Kurihara, M.; Yook, J.-I.; Mikami, S.; Kimura, T.; Shimada, K.; Konishi, N.; Yane, K.; Yamanaka, T.; et al. Snail-induced EMT promotes cancer stem cell-like properties in head and neck cancer cells. Oncol. Rep. 2016, 35, 261–266. [Google Scholar] [CrossRef]
- Ishida, T.; Hijioka, H.; Kume, K.; Miyawaki, A.; Nakamura, N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol. Lett. 2013, 6, 1201–1206. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudas, J.; Ingruber, J.; Glueckert, R.; Sprung, S.; Fleischer, F.; Cidlinsky, N.; Dejaco, D.; Kofler, B.; Giotakis, A.I.; et al. Slug Is A Surrogate Marker of Epithelial to Mesenchymal Transition (EMT) in Head and Neck Cancer. J. Clin. Med. 2020, 9, 2061. [Google Scholar] [CrossRef]
- Fahim, Y.; Yousefi, M.; Izadpanah, M.H.; Forghanifard, M.M. TWIST1 correlates with Notch signaling pathway to develop esophageal squamous cell carcinoma. Mol. Cell. Biochem. 2020, 474, 181–188. [Google Scholar] [CrossRef]
- White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. [Google Scholar] [CrossRef]
- Zeng, Q.; Li, S.; Chepeha, D.B.; Giordano, T.J.; Li, J.; Zhang, H.; Polverini, P.J.; Nor, J.; Kitajewski, J.; Wang, C.Y. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 2005, 8, 13–23. [Google Scholar] [CrossRef]
- Zhang, T.; Liang, L.; Liu, X.; Wu, J.-N.; Chen, J.; Su, K.; Zheng, Q.; Huang, H.; Liao, G.-Q. TGFβ1-Smad3-Jagged1-Notch1-Slug signaling pathway takes part in tumorigenesis and progress of tongue squamous cell carcinoma. J. oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2016, 45, 486–493. [Google Scholar] [CrossRef]
- Yan, M.; Plowman, G.D. Delta-like 4/Notch signaling and its therapeutic implications. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 7243–7246. [Google Scholar] [CrossRef]
- Khelil, M.; Griffin, H.; Bleeker, M.C.G.; Steenbergen, R.D.M.; Zheng, K.; Saunders-Wood, T.; Samuels, S.; Rotman, J.; Vos, W.; van den Akker, B.E.; et al. Delta-Like Ligand-Notch1 Signaling Is Selectively Modulated by HPV16 E6 to Promote Squamous Cell Proliferation and Correlates with Cervical Cancer Prognosis. Cancer Res. 2021, 81, 1909–1921. [Google Scholar] [CrossRef]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef]
- Lin, J.-T.; Chen, M.-K.; Yeh, K.-T.; Chang, C.-S.; Chang, T.-H.; Lin, C.-Y.; Wu, Y.-C.; Su, B.-W.; Lee, K.-D.; Chang, P.-J. Association of high levels of Jagged-1 and Notch-1 expression with poor prognosis in head and neck cancer. Ann. Surg. Oncol. 2010, 17, 2976–2983. [Google Scholar] [CrossRef]
- Zhang, H.; Song, Y.; Du, Z.; Li, X.; Zhang, J.; Chen, S.; Chen, F.; Li, T.; Zhan, Q. Exome sequencing identifies new somatic alterations and mutation patterns of tongue squamous cell carcinoma in a Chinese population. J. Pathol. 2020, 251, 353–364. [Google Scholar] [CrossRef]
- Jing, P.; Zhou, S.; Xu, P.; Cui, P.; Liu, X.; Liu, X.; Liu, X.; Wang, H.; Xu, W. PDK1 promotes metastasis by inducing epithelial–mesenchymal transition in hypopharyngeal carcinoma via the Notch1 signaling pathway. Exp. Cell Res. 2020, 386, 111746. [Google Scholar] [CrossRef]
- Li, J.-Y.; Huang, W.-X.; Zhou, X.; Chen, J.; Li, Z. Numb inhibits epithelial-mesenchymal transition via RBP-Jκ-dependent Notch1/PTEN/FAK signaling pathway in tongue cancer. BMC Cancer 2019 191 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Li, B.; Chen, M.; Lu, M.; Xin-Xiang, J.; Meng-Xiong, P.; Jun-Wu, M. Glutaredoxin 3 promotes migration and invasion via the Notch signalling pathway in oral squamous cell carcinoma. Free Radic. Res. 2018, 52, 390–401. [Google Scholar] [CrossRef]
- Weaver, A.N.; Burch, M.B.; Cooper, T.S.; Della Manna, D.L.; Wei, S.; Ojesina, A.I.; Rosenthal, E.L.; Yang, E.S. Notch Signaling Activation Is Associated with Patient Mortality and Increased FGF1-Mediated Invasion in Squamous Cell Carcinoma of the Oral Cavity. Mol. Cancer Res. 2016, 14, 883–891. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef]
- Joo, Y.-H.; Jung, C.-K.; Kim, M.-S.; Sun, D.-I. Relationship between vascular endothelial growth factor and Notch1 expression and lymphatic metastasis in tongue cancer. Otolaryngol. Neck Surg. Off. J. Am. Acad. Otolaryngol. Neck Surg. 2009, 140, 512–518. [Google Scholar] [CrossRef]
- Kanamoto, A.; Ninomiya, I.; Harada, S.; Tsukada, T.; Okamoto, K.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Valproic acid inhibits irradiation-induced epithelial-mesenchymal transition and stem cell-like characteristics in esophageal squamous cell carcinoma. Int. J. Oncol. 2016, 49, 1859–1869. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotoh, K.; Asaoka, T.; Wada, H.; et al. A Histone Deacetylase Inhibitor Suppresses Epithelial-Mesenchymal Transition and Attenuates Chemoresistance in Biliary Tract Cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef]
- Dennis, M.; Wang, G.; Luo, J.; Lin, Y.; Dohadwala, M.; Abemayor, E.; Elashoff, D.A.; Sharma, S.; Dubinett, S.M.; St John, M.A. Snail controls the mesenchymal phenotype and drives erlotinib resistance in oral epithelial and head and neck squamous cell carcinoma cells. Otolaryngol. Neck Surg. Off. J. Am. Acad. Otolaryngol. Neck Surg. 2012, 147, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Montoya, C.A.; Donnenberg, V.S.; Sant, S.; Antony, J.; Thiery, J.P.; Huang, R.Y.-J.; Ardila, D.C.; Aggarwal, V.; Singh, M.; et al. SLUG-related partial epithelial-to-mesenchymal transition is a transcriptomic prognosticator of head and neck cancer survival. Biochemistry. 2019, 8, 41004. [Google Scholar]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.-I. Therapy resistance mediated by exosomes. Mol. Cancer 2019, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Kirave, P.; Gondaliya, P.; Kulkarni, B.; Rawal, R.; Garg, R.; Jain, A.; Kalia, K. Exosome mediated miR-155 delivery confers cisplatin chemoresistance in oral cancer cells via epithelial-mesenchymal transition. Oncotarget 2020, 11, 1157–1171. [Google Scholar] [CrossRef]
- Lin, X.-J.; He, C.-L.; Sun, T.; Duan, X.-J.; Sun, Y.; Xiong, S.-J. hsa-miR-485-5p reverses epithelial to mesenchymal transition and promotes cisplatin-induced cell death by targeting PAK1 in oral tongue squamous cell carcinoma. Int. J. Mol. Med. 2017, 40, 83–89. [Google Scholar] [CrossRef]
- Jiang, J.; Zheng, M.; Zhang, M.; Yang, X.; Li, L.; Wang, S.-S.; Wu, J.-S.; Yu, X.-H.; Wu, J.-B.; Pang, X.; et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma Through miR-642b-3p. Neoplasia 2019, 21, 216–229. [Google Scholar] [CrossRef]
- Liu, W.; Shi, X.; Wang, B. microRNA-133a exerts tumor suppressive role in oral squamous cell carcinoma through the Notch signaling pathway via downregulation of CTBP2. Cancer Gene Ther. 2021, 1–11. [Google Scholar] [CrossRef]
- Lombardo, Y.; Faronato, M.; Filipovic, A.; Vircillo, V.; Magnani, L.; Coombes, R.C. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. 2014, 16, R62. [Google Scholar] [CrossRef]
- Zheng, X.; Yu, C.; Xu, M. Linking Tumor Microenvironment to Plasticity of Cancer Stem Cells: Mechanisms and Application in Cancer Therapy. Front. Oncol. 2021, 11, 678333. [Google Scholar] [CrossRef]
- Moghbeli, M.; Abbaszadegan, M.R.; Golmakani, E.; Forghanifard, M.M. Correlation of Wnt and NOTCH pathways in esophageal squamous cell carcinoma. J. Cell Commun. Signal. 2016, 10, 129–135. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Chang, W.H.; Lai, A.G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 2019, 121, 666–678. [Google Scholar] [CrossRef]
- Upadhyay, P.; Nair, S.; Kaur, E.; Aich, J.; Dani, P.; Sethunath, V.; Gardi, N.; Chandrani, P.; Godbole, M.; Sonawane, K.; et al. Notch pathway activation is essential for maintenance of stem-like cells in early tongue cancer. Oncotarget 2016, 7, 50437–50449. [Google Scholar] [CrossRef]
- Lee, S.H.; Hong, H.S.; Liu, Z.X.; Kim, R.H.; Kang, M.K.; Park, N.-H.; Shin, K.-H. TNFα enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 424, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A mechanism-based computational model to capture the interconnections among epithelial-mesenchymal transition, cancer stem cells and Notch-Jagged signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5. [Google Scholar] [CrossRef]
- Gross, S.J.; Webb, A.M.; Peterlin, A.D.; Durrant, J.R.; Judson, R.J.; Raza, Q.; Kitajewski, J.K.; Kushner, E.J.; Meurette, O.; Mehlen, P.; et al. Soluble Notch ligand and receptor peptides act antagonistically during angiogenesis. Cancer Cell 2015, 6, 789–805. [Google Scholar]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef]
- Kumar, A.; Desai, S.; Pandey, B.N. Endothelial Dll4-Notch signaling in tumor microenvironment: Is there any hidden therapeutic opportunity? Transl. Lung Cancer Res. 2013, 2, 439–441. [Google Scholar]
- Tetzlaff, F.; Fischer, A. Control of Blood Vessel Formation by Notch Signaling. Adv. Exp. Med. Biol. 2018, 1066, 319–338. [Google Scholar]
- Carla, C.; Daris, F.; Cecilia, B.; Francesca, B.; Francesca, C.; Paolo, F. Angiogenesis in head and neck cancer: A review of the literature. J. Oncol. 2012, 2012, 358472. [Google Scholar] [CrossRef][Green Version]
- Brzozowa, M.; Wojnicz, R.; Kowalczyk-Ziomek, G.; Helewski, K. The Notch ligand Delta-like 4 (DLL4) as a target in angiogenesis-based cancer therapy? Contemp. Oncol. 2013, 17, 234–237. [Google Scholar] [CrossRef]
- Dufraine, J.; Funahashi, Y.; Kitajewski, J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene 2008, 27, 5132–5137. [Google Scholar] [CrossRef]
- Pedrosa, A.-R.; Trindade, A.; Carvalho, C.; Graça, J.; Carvalho, S.; Peleteiro, M.C.; Adams, R.H.; Duarte, A. Endothelial Jagged1 promotes solid tumor growth through both pro-angiogenic and angiocrine functions. Oncotarget 2015, 6, 24404–24423. [Google Scholar] [CrossRef]
- Gao, W.; Sweeney, C.; Walsh, C.; Rooney, P.; McCormick, J.; Veale, D.J.; Fearon, U. Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann. Rheum. Dis. 2013, 72, 1080–1088. [Google Scholar] [CrossRef]
- Hicks, C.; Johnston, S.H.; diSibio, G.; Collazo, A.; Vogt, T.F.; Weinmaster, G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell Biol. 2000, 2, 515–520. [Google Scholar] [CrossRef]
- Saiki, W.; Ma, C.; Okajima, T.; Takeuchi, H. Current Views on the Roles of O-Glycosylation in Controlling Notch-Ligand Interactions. Biomolecules 2021, 11, 309. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, H.; Liang, Y.; Liang, L.; Zheng, G.-S.; Huang, H.; Wu, J.; Liao, G. Suppression of tongue squamous cell carcinoma growth by inhibition of Jagged1 in vitro and in vivo. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2013, 42, 322–331. [Google Scholar] [CrossRef]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G.; Ridgway, J.; et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef]
- Trindade, A.; Djokovic, D.; Gigante, J.; Mendonça, L.; Duarte, A. Endothelial Dll4 overexpression reduces vascular response and inhibits tumor growth and metastasization in vivo. BMC Cancer 2017, 17, 189. [Google Scholar] [CrossRef]
- Trindade, A.; Duarte, A. Notch Signaling Function in the Angiocrine Regulation of Tumor Development. Cells 2020, 9, 2467. [Google Scholar] [CrossRef]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.-C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef]
- Yan, M.; Callahan, C.A.; Beyer, J.C.; Allamneni, K.P.; Zhang, G.; Ridgway, J.B.; Niessen, K.; Plowman, G.D. Chronic DLL4 blockade induces vascular neoplasms. Nature 2010, 463, E6–E7. [Google Scholar] [CrossRef]
- Djokovic, D.; Trindade, A.; Gigante, J.; Pinho, M.; Harris, A.L.; Duarte, A. Incomplete Dll4/Notch signaling inhibition promotes functional angiogenesis supporting the growth of skin papillomas. BMC Cancer 2015, 15, 608. [Google Scholar] [CrossRef]
- Klose, R.; Berger, C.; Moll, I.; Adam, M.G.; Schwarz, F.; Mohr, K.; Augustin, H.G.; Fischer, A. Soluble Notch ligand and receptor peptides act antagonistically during angiogenesis. Cardiovasc. Res. 2015, 107, 153–163. [Google Scholar] [CrossRef]
- Gross, S.J.; Webb, A.M.; Peterlin, A.D.; Durrant, J.R.; Judson, R.J.; Raza, Q.; Kitajewski, J.K.; Kushner, E.J. Notch regulates vascular collagen IV basement membrane through modulation of lysyl hydroxylase 3 trafficking. Angiogenesis 2021, 24, 789–805. [Google Scholar] [CrossRef]
- Antfolk, D.; Sjöqvist, M.; Cheng, F.; Isoniemi, K.; Duran, C.L.; Rivero-Muller, A.; Antila, C.; Niemi, R.; Landor, S.; Bouten, C.V.C.; et al. Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc. Natl. Acad. Sci. USA 2017, 201703057. [Google Scholar] [CrossRef]
- Estrach, S.; Cailleteau, L.; Franco, C.A.; Gerhardt, H.; Stefani, C.; Lemichez, E.; Gagnoux-Palacios, L.; Meneguzzi, G.; Mettouchi, A. Laminin-binding integrins induce Dll4 expression and Notch signaling in endothelial cells. Circ. Res. 2011, 109, 172–182. [Google Scholar] [CrossRef]
- Shahoumi, L.A.; Yeudall, W.A. Targeted therapies for non-HPV-related head and neck cancer: Challenges and opportunities in the context of predictive, preventive, and personalized medicine. EPMA J. 2019, 10, 291–305. [Google Scholar] [CrossRef]
- Macha, M.A.; Wani, N.A.; Ganai, R.A.; Bhat, A.A.; Hamid, A.; Hashem, S.; Haris, M.; Chauhan, S.S.; Zargar, M.A.; Batra, S.K. Recent Advances in Head and Neck Tumor Microenvironment-Based Therapy. Adv. Exp. Med. Biol. 2020, 1296, 11–31. [Google Scholar]
- Troy, J.D.; Weissfeld, J.L.; Youk, A.O.; Thomas, S.; Wang, L.; Grandis, J.R. Expression of EGFR, VEGF, and NOTCH1 suggest differences in tumor angiogenesis in HPV-positive and HPV-negative head and neck squamous cell carcinoma. Head Neck Pathol. 2013, 7, 344–355. [Google Scholar] [CrossRef]
- Lubin, D.J.; Mick, R.; Shroff, S.G.; Stashek, K.; Furth, E.E. The notch pathway is activated in neoplastic progression in esophageal squamous cell carcinoma. Hum. Pathol. 2018, 72, 66–70. [Google Scholar] [CrossRef]
- Rettig, E.M.; Bishop, J.A.; Agrawal, N.; Chung, C.H.; Sharma, R.; Zamuner, F.; Li, R.J.; Koch, W.M.; Califano, J.A.; Guo, T.; et al. HEY1 is expressed independent of NOTCH1 and is associated with poor prognosis in head and neck squamous cell carcinoma. Oral Oncol. 2018, 82, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, Z.; Fan, X.; Xu, X.; Chen, M.; Chang, B.; Zhang, Y. Notch3 overexpression associates with poor prognosis in human non-small-cell lung cancer. Med. Oncol. 2013, 30, 595. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Juillerat-Jeanneret, L.; Golshayan, D. Notch Antagonists: Potential Modulators of Cancer and Inflammatory Diseases. J. Med. Chem. 2016, 59, 7719–7737. [Google Scholar] [CrossRef] [PubMed]
- Rettig, E.M.; Chung, C.H.; Bishop, J.A.; Howard, J.D.; Sharma, R.; Li, R.J.; Douville, C.; Karchin, R.; Izumchenko, E.; Sidransky, D.; et al. Cleaved NOTCH1 Expression Pattern in Head and Neck Squamous Cell Carcinoma Is Associated with NOTCH1 Mutation, HPV Status, and High-Risk Features. Cancer Prev. Res. 2015, 8, 287–295. [Google Scholar] [CrossRef]







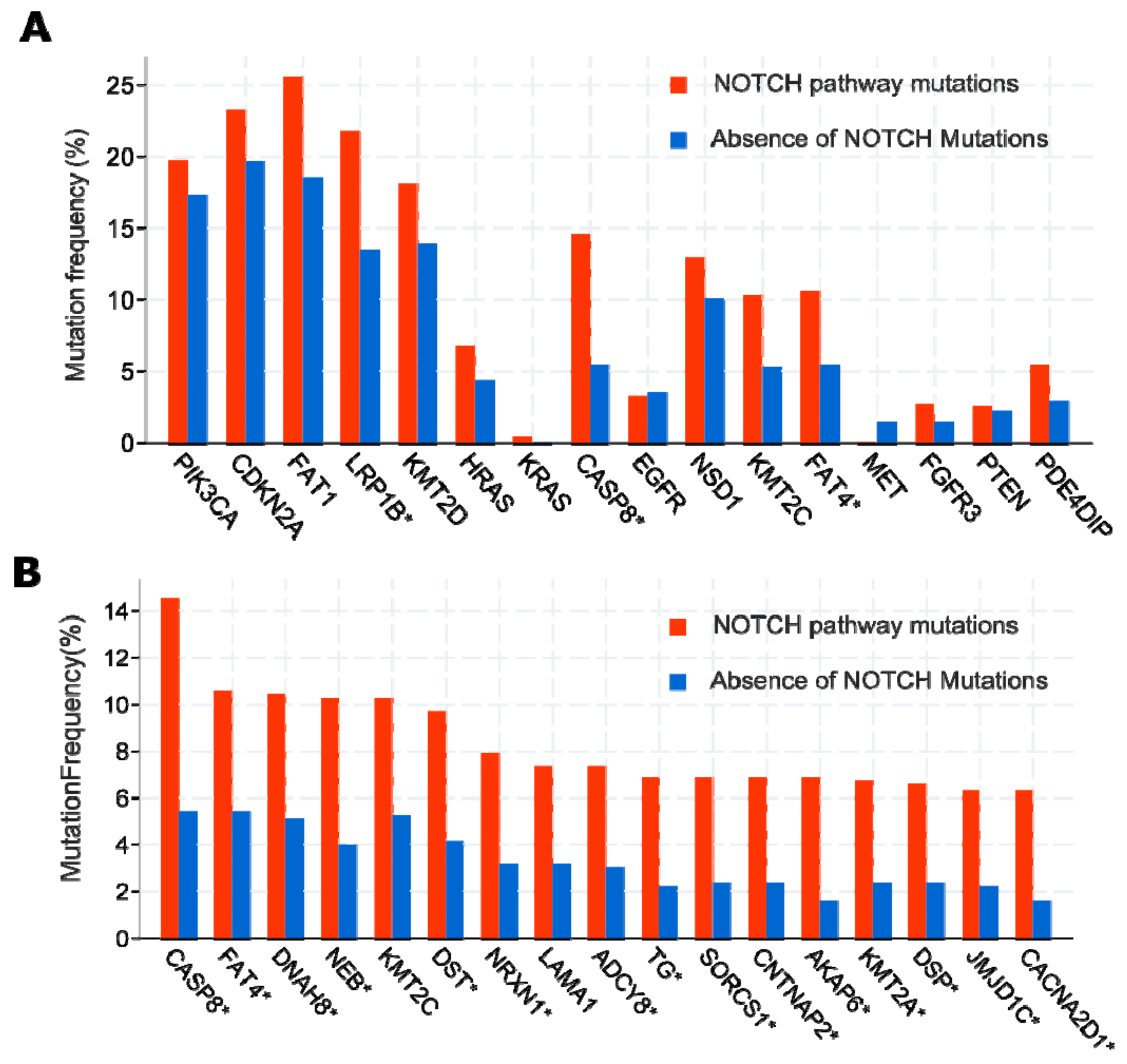{kind=link}
{kind=link}
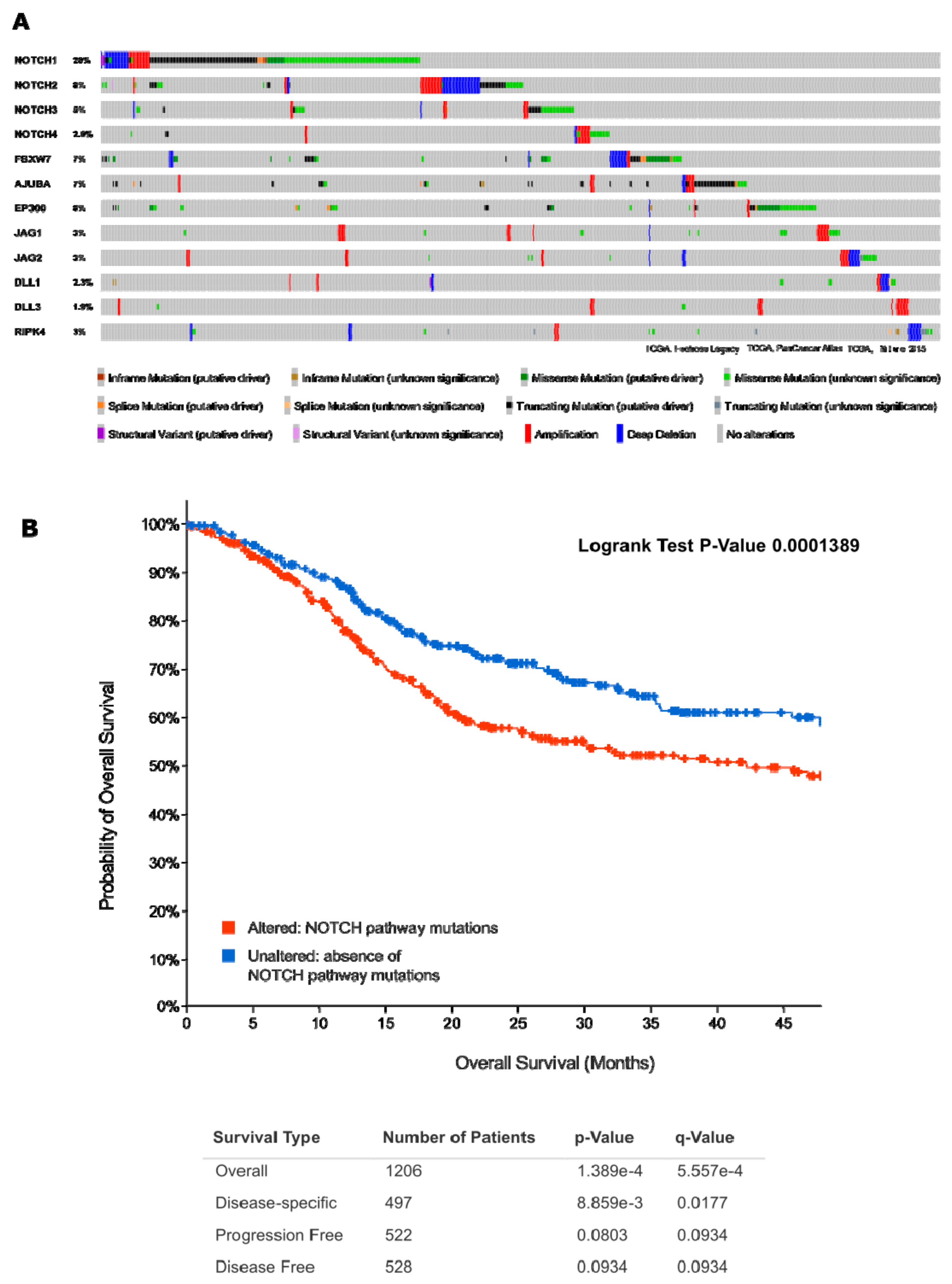{kind=link}
{kind=link}
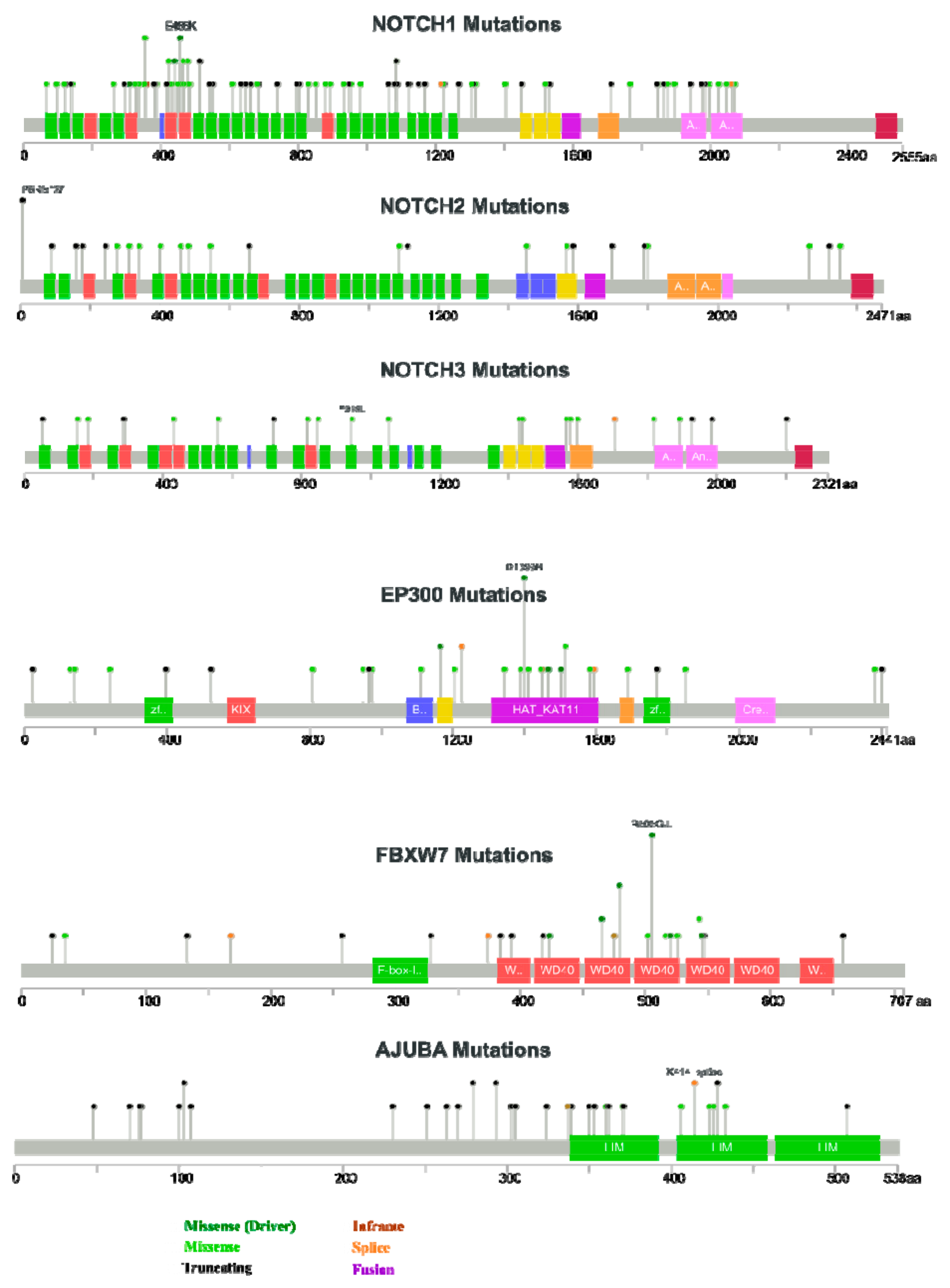{kind=link}
{kind=link}
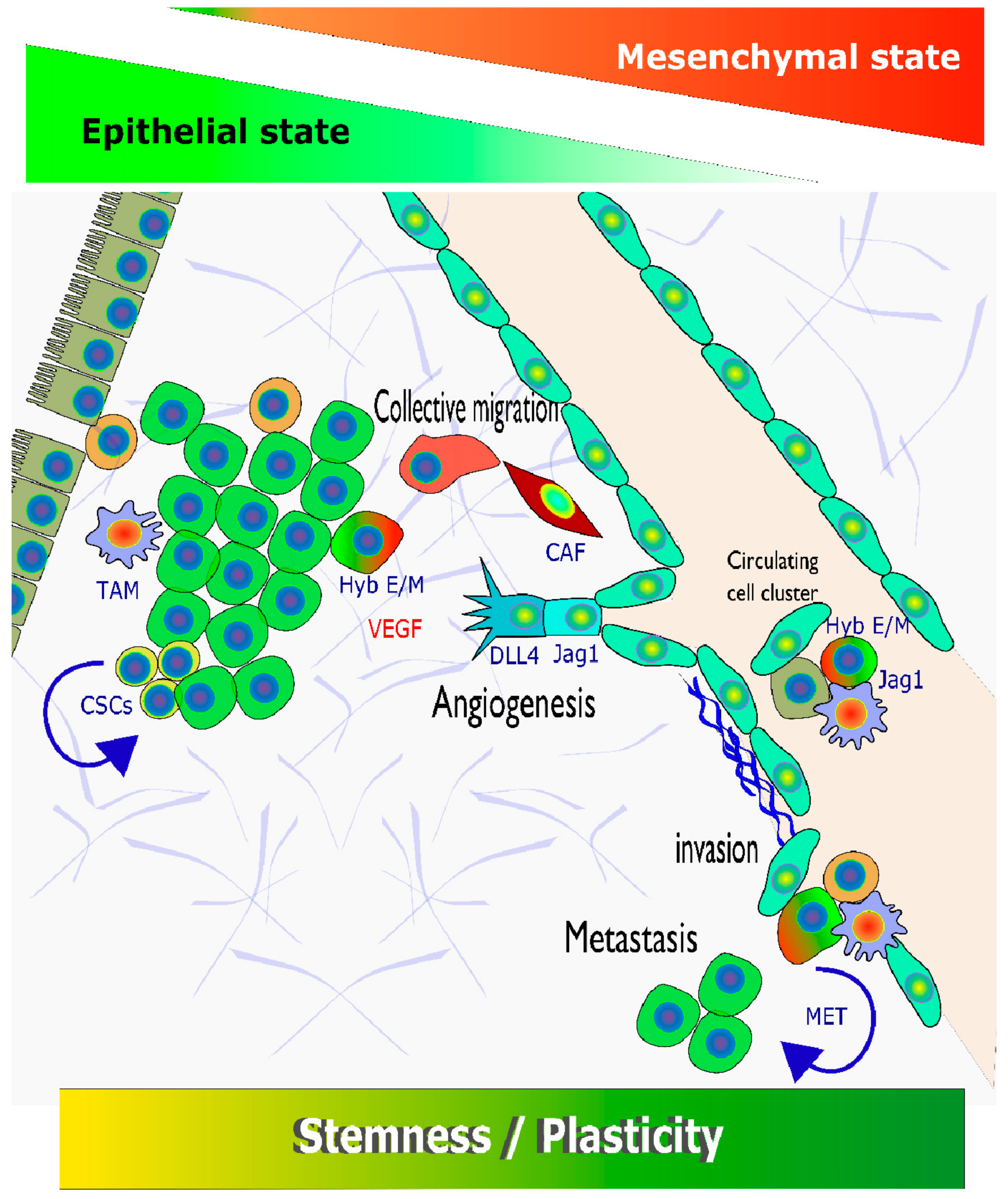{kind=link}
| Intervention/Treatment | Molecular Target | ClinicalTrials.gov Identifier |
|---|---|---|
| Alisertib (MLN8237) | Aurora A kinase | NCT01045421 |
| Axitinib (AG-013736) | VEGFR | NCT01469546, NCT02762513 |
| EGFR; tubulin; DNA | NCT00736944 | |
| Cetuximab + Abraxane (protein-bound Paclitaxel) + Cisplatin + 5-FU + Radiation | EGFR; DNA | NCT01218048 |
| Cetuximab + Cisplatin or carboplatin + Surgery + Post-surgical radiation | EGFR | NCT01012258 |
| Cetuximab + concomitant boost radiotherapy | EGFR; nucleic acids | NCT01581970 |
| Cetuximab + Cyclophosphamide | EGFR; tubulin; DNA | NCT01437449 |
| Cetuximab + Docetaxel + Cisplatin + Carboplatin | EGFR; TLR9 | NCT01040832 |
| Cetuximab + EMD 1201081 | EGFR; IGF-1R | NCT00957853 |
| Cetuximab + IMC-A12 + Surgical tumour resection | EGFR; tubulin | NCT01412229 |
| Cetuximab + Nab-paclitaxel + Carboplatin | EGFR; PD-1; DNA | NCT03349710 |
| Cetuximab + Nivolumab + Cisplatin + Radiotherapy | EGFR; PARP1 | NCT01758731 |
| Cetuximab + Olaparib + Radiation Therapy | EGFR; tubulin; DNA | NCT02124707 |
| Cetuximab + Paclitaxel + Carboplatin | EGFR; tubulin; DNA | NCT01566435 |
| Cetuximab + Paclitaxel albumin-stabilized nanoparticle + Cisplatin + 5-FU + Intensity modulated radiation | EGFR; | NCT00768664 |
| Dacomitinib (PF-00299804) | ALK1 | NCT01458392 |
| Dalantercept | PDK; DNA | NCT01386632 |
| DCA (dichloroacetate) + Cisplatin + Radiation | PD-L1 | NCT02207530 |
| Durvalumab (MEDI4736) | PD-L1; CTLA4 | NCT02319044 |
| Durvalumab (MEDI4736) + Tremelimumab | PD-L1; CTLA4 | NCT02369874 |
| Durvalumab (MEDI4736) + Tremelimumab + Standard of Care | EGFR; DNA | NCT00304278 |
| Erlotinib (Tarceva) + Intra-arterial Cisplatin + Radiation Therapy | immune system | NCT02163057 |
| INO-3112 (DNA Vaccine) | immune system; DNA; COX1/2 | NCT00210470 |
| IRX-2 + Cyclophosphamide + Indomethacin + Zinc + Omeprazole | KSP | NCT00095628 |
| Ispinesib | EGFR; DNA | NCT01044433 |
| Lapatinib ditosylate + Capecitabine (5-FU precursor) | Eg5; immune system | NCT01059643 |
| Litronesib (Y2523355) + Pegfilgrastim | Respiratory complex I; DNA | NCT02325401 |
| Metformin + Cisplatin + Radiation Therapy | TLR8; DNA | NCT01836029 |
| Motolimod (VTX-2337) + Carboplatin + Cisplatin + 5-FU | tubulin; EGFR; DNA | NCT00343083 |
| Paclitaxel + Erbitux + Carboplatin + Radiation | CDKs; DNA | NCT03194373 |
| Palbociclib + Carboplatin (5-FU prodrug) | EGFR | NCT00446446 |
| Panitumumab | EGFR; DNA | NCT00756444 |
| Panitumumab + Cisplatin + 5-FU | EGFR; DNA; tubulin | NCT00454779 |
| Panitumumab + Cisplatin + Docetaxel | PD-1 | NCT02255097 |
| Pembrolizumab | PD-1; BTK | NCT02454179 |
| Pembrolizumab + Acalabrutinib | PD-1 | NCT03057613 |
| Pembrolizumab + Radiation: IMRT 60-66Gy | PD-1; immune system | NCT02626000 |
| Pembrolizumab + Talimogene laherparepvec | HDAC | NCT00084682 |
| Romidepsin | immune system | NCT02013050 |
| SGX942 | EGFR | NCT01417936 |
| Sym004 (Futuximab and Modotuximab) | PDE5 | NCT01697800 |
| Tadalafil | DNA; tubulin | NCT00270790 |
| Taxol (Paclitaxel) + Carboplatin + Amifostine + Radiotherapy | mTOR; tubulin; DNA | NCT01016769 |
| Temsirolimus + Weekly Paclitaxel + Carboplatin | 4-1BB; MS4A1 | NCT01307267 |
| Utomilumab (PF-05082566) + Rituximab | tubulin; DHFR | NCT02347332 |
| Vinflunine + Methotrexate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kałafut, J.; Czerwonka, A.; Anameriç, A.; Przybyszewska-Podstawka, A.; Misiorek, J.O.; Rivero-Müller, A.; Nees, M. Shooting at Moving and Hidden Targets—Tumour Cell Plasticity and the Notch Signalling Pathway in Head and Neck Squamous Cell Carcinomas. Cancers 2021, 13, 6219. https://doi.org/10.3390/cancers13246219
Kałafut J, Czerwonka A, Anameriç A, Przybyszewska-Podstawka A, Misiorek JO, Rivero-Müller A, Nees M. Shooting at Moving and Hidden Targets—Tumour Cell Plasticity and the Notch Signalling Pathway in Head and Neck Squamous Cell Carcinomas. Cancers. 2021; 13(24):6219. https://doi.org/10.3390/cancers13246219
Chicago/Turabian StyleKałafut, Joanna, Arkadiusz Czerwonka, Alinda Anameriç, Alicja Przybyszewska-Podstawka, Julia O. Misiorek, Adolfo Rivero-Müller, and Matthias Nees. 2021. "Shooting at Moving and Hidden Targets—Tumour Cell Plasticity and the Notch Signalling Pathway in Head and Neck Squamous Cell Carcinomas" Cancers 13, no. 24: 6219. https://doi.org/10.3390/cancers13246219
APA StyleKałafut, J., Czerwonka, A., Anameriç, A., Przybyszewska-Podstawka, A., Misiorek, J. O., Rivero-Müller, A., & Nees, M. (2021). Shooting at Moving and Hidden Targets—Tumour Cell Plasticity and the Notch Signalling Pathway in Head and Neck Squamous Cell Carcinomas. Cancers, 13(24), 6219. https://doi.org/10.3390/cancers13246219