
Interplay between Genome, Metabolome and Microbiome in Colorectal Cancer

 , , , ,
, , , ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Data Generation
2.3. Analyses
3. Results
3.1. Genetic Associations of Adenoma and Colorectal Cancer
3.2. Genetic Associations of Selected Microbiome and Metabolome Traits
3.3. Mendelian Randomization
3.4. Multiomic Integration
3.5. Polygenic Risk Scores and Predictive Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stewart, B.; Wild, C. World Cancer Report 2014; IARC Publications: Lyon, France, 2014. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Cross, A.J.; Ferrucci, L.M.; Risch, A.; Graubard, B.I.; Ward, M.H.; Park, Y.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R. A Large Prospective Study of Meat Consumption and Colorectal Cancer Risk: An Investigation of Potential Mechanisms Underlying this Association. Cancer Res. 2010, 70, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Levin, B.; Lieberman, D.A.; McFarland, B.; Andrews, K.S.; Brooks, D.; Bond, J.; Dash, C.; Giardiello, F.M.; Glick, S.; Johnson, D.; et al. Screening and Surveillance for the Early Detection of Colorectal Cancer and Adenomatous Polyps, 2008: A Joint Guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J. Clin. 2008, 58, 130–160. [Google Scholar] [CrossRef] [PubMed]
- Abulí, A.; Fernández-Rozadilla, C.; Alonso-Espinaco, V.; Muñoz, J.; Gonzalo, V.; Bessa, X.; González, D.; Clofent, J.; Cubiella, J.; Morillas, J.D.; et al. Case-control study for colorectal cancer genetic susceptibility in EPICOLON: Previously identified variants and mucins. BMC Cancer 2011, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Abulí, A.; Bessa, X.; González, J.R.; Ruiz–Ponte, C.; Cáceres, A.; Muñoz, J.; Gonzalo, V.; Balaguer, F.; Fernández–Rozadilla, C.; González, D.; et al. Susceptibility Genetic Variants Associated With Colorectal Cancer Risk Correlate With Cancer Phenotype. Gastroenterol. 2010, 139, 788–796. [Google Scholar] [CrossRef]
- Burns, M.B.; Montassier, E.; Abrahante, J.; Priya, S.; Niccum, D.E.; Khoruts, A.; Starr, T.K.; Knights, D.; Blekhman, R. Colorectal cancer mutational profiles correlate with defined microbial communities in the tumor microenvironment. PLoS Genet. 2018, 14, e1007376. [Google Scholar] [CrossRef] [PubMed]
- Law, P.J.; The PRACTICAL Consortium; Timofeeva, M.; Fernandez-Rozadilla, C.; Broderick, P.; Studd, J.; Fernandez-Tajes, J.; Farrington, S.; Svinti, V.; Palles, C.; et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, M.N.; Newcomb, P.A.; Makar, K.W.; Burnett-Hartman, A.N.; Potter, J.D.; Upton, M.P.; Zhu, L.-C.; Rosenfeld, M.E.; Schwartz, S.M.; Rutter, C.M. Blood lipids and colorectal polyps: Testing an etiologic hypothesis using phenotypic measurements and Mendelian randomization. Cancer Causes Control 2015, 26, 467–473. [Google Scholar] [CrossRef]
- Rodriguez-Broadbent, H.; Law, P.J.; Sud, A.; Palin, K.; Tuupanen, S.; Gylfe, A.; Hänninen, U.A.; Cajuso, T.; Tanskanen, T.; Kondelin, J.; et al. Mendelian randomisation implicates hyperlipidaemia as a risk factor for colorectal cancer. Int. J. Cancer 2017, 140, 2701–2708. [Google Scholar] [CrossRef]
- Cornish, A.J.; Law, P.J.; Timofeeva, M.; Palin, K.; Farrington, S.M.; Palles, C.; A Jenkins, M.; Casey, G.; Brenner, H.; Chang-Claude, J.; et al. Modifiable pathways for colorectal cancer: A mendelian randomisation analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 55–62. [Google Scholar] [CrossRef]
- Ibáñez-Sanz, G.; Díez-Villanueva, A.; Alonso, M.H.; Moranta, F.R.; Pérez-Gómez, B.; Bustamante, M.; Martin, V.; Llorca, J.; Amiano, P.; Ardanaz, E.; et al. Risk Model for Colorectal Cancer in Spanish Population Using Environmental and Genetic Factors: Results from the MCC-Spain study. Sci. Rep. 2017, 7, 43263. [Google Scholar] [CrossRef]
- Thomas, M.; Sakoda, L.C.; Hoffmeister, M.; Rosenthal, E.A.; Lee, J.K.; van Duijnhoven, F.J.; Platz, E.A.; Wu, A.H.; Dampier, C.H.; de la Chapelle, A.; et al. Genome-wide Modeling of Polygenic Risk Score in Colorectal Cancer Risk. Am. J. Hum. Genet. 2020, 107, 432–444. [Google Scholar] [CrossRef]
- Cubiella, J.; Clos-Garcia, M.; Alonso, C.; Martinez-Arranz, I.; Perez-Cormenzana, M.; Barrenetxea, Z.; Berganza-Granda, J.; Rodríguez-Llopis, I.; D’Amato, M.; Bujanda, L.; et al. Targeted UPLC-MS Metabolic Analysis of Human Faeces Reveals Novel Low-Invasive Candidate Markers for Colorectal Cancer. Cancers 2018, 10, 300. [Google Scholar] [CrossRef] [PubMed]
- Clos-Garcia, M.; Garcia, K.; Alonso, C.; Iruarrizaga-Lejarreta, M.; D’Amato, M.; Crespo, A.; Iglesias, A.; Cubiella, J.; Bujanda, L.; Falcón-Pérez, J.M. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers 2020, 12, 1142. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Garay, J.A.R.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host–microbiota maladaptation in colorectal cancer. Nat. Cell Biol. 2020, 585, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Dalal, N.; Jalandra, R.; Sharma, M.; Prakash, H.; Makharia, G.K.; Solanki, P.R.; Singh, R.; Kumar, A. Omics technologies for improved diagnosis and treatment of colorectal cancer: Technical advancement and major perspectives. Biomed. Pharmacother. 2020, 131, 110648. [Google Scholar] [CrossRef]
- Moskowitz, J.E.; Doran, A.G.; Lei, Z.; Busi, S.B.; Hart, M.L.; Franklin, C.L.; Sumner, L.W.; Keane, T.M.; Amos-Landgraf, J.M. Integration of genomics, metagenomics, and metabolomics to identify interplay between susceptibility alleles and microbiota in adenoma initiation. BMC Cancer 2020, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cubiella, J.; on Behalf of the COLONPREDICT Study Investigators; Vega, P.; Salve, M.; Díaz-Ondina, M.; Alves, M.T.; Quintero, E.; Álvarez-Sánchez, V.; Fernández-Bañares, F.; Boadas, J.; et al. Development and external validation of a faecal immunochemical test-based prediction model for colorectal cancer detection in symptomatic patients. BMC Med. 2016, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, 1–29. [Google Scholar] [CrossRef]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Eviroment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008. [Google Scholar]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Arnol, D.; Bredikhin, D.; Deloro, Y.; Velten, B.; Marioni, J.C.; Stegle, O. MOFA+: A statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience 2019, 8, 082. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Forrest, M.E.; Saiakhova, A.; Beard, L.; Buchner, D.A.; Scacheri, P.C.; LaFramboise, T.; Markowitz, S.; Khalil, A.M. Colon Cancer-Upregulated Long Non-Coding RNA lincDUSP Regulates Cell Cycle Genes and Potentiates Resistance to Apoptosis. Sci. Rep. 2018, 8, 7324. [Google Scholar] [CrossRef] [PubMed]
- Benlahfid, M.; Traboulsi, W.; Sergent, F.; Benharouga, M.; Elhattabi, K.; Erguibi, D.; Karkouri, M.; Elattar, H.; Fadil, A.; Fahmi, Y.; et al. Endocrine gland-derived vascular endothelial growth factor (EG-VEGF) and its receptor PROKR2 are associated to human colorectal cancer progression and peritoneal carcinomatosis. Cancer Biomark. 2018, 21, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Scrimieri, F.; Ghosh, S.; Zhong, J.; Kim, M.-S.; Ren, Y.R.; Morgan, R.A.; Iacobuzio-Donahue, C.A.; Pandey, A.; Kern, S.E. FAM190A Deficiency Creates a Cell Division Defect. Am. J. Pathol. 2013, 183, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.U.; Park, J.T. Functional evaluation of alternative splicing in the FAM190A gene. Genes Genom. 2018, 41, 193–199. [Google Scholar] [CrossRef] [PubMed]
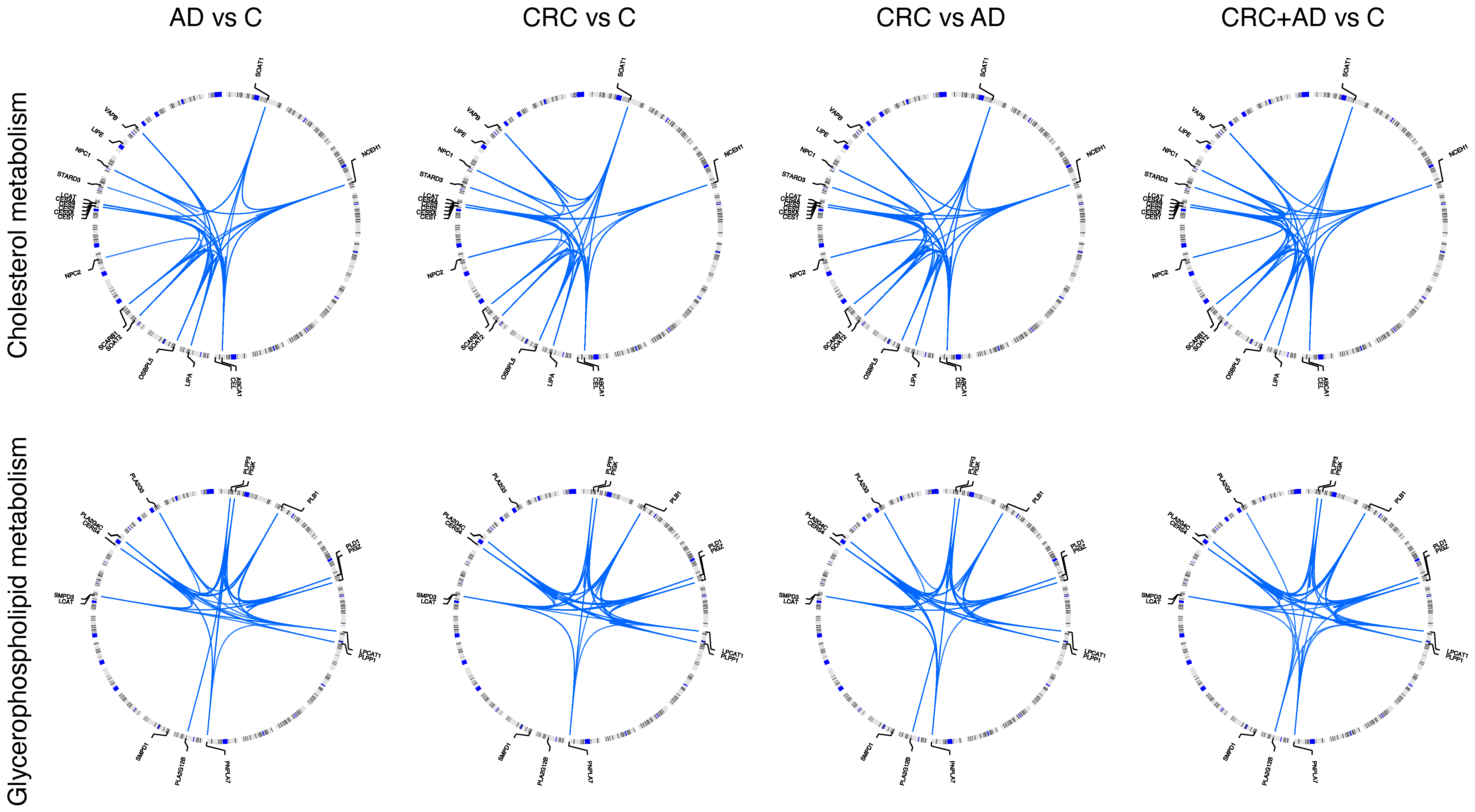

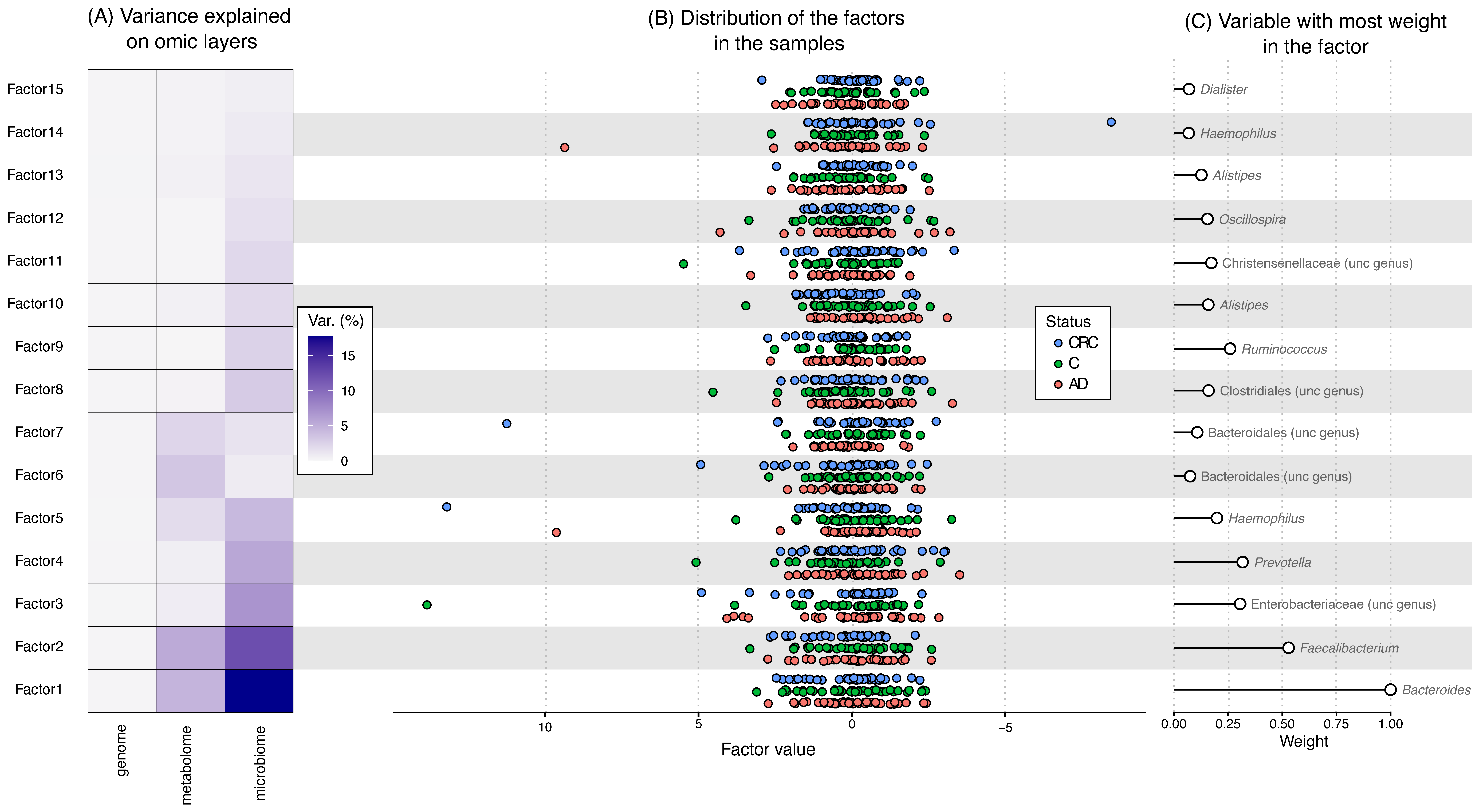

{kind=link}
{kind=link}
{kind=link}
| Gene | Analysis | SNP | p | OR (CI 95%) |
|---|---|---|---|---|
| LIPA | AD vs. C | rs2246833 | 0.019 | 2.9 (1.2–7.1) |
| LIPA | CRC vs. AD | rs885561 | 0.028 | 0.4 (0.2–0.9) |
| NCEH1 | AD vs. C | rs17756312 | 0.019 | 12.3 (1.5–100.7) |
| NCEH1 | CRC vs. C | rs522028 | 0.017 | 3.8 (1.3–11.2) |
| NCEH1 | CRC vs. AD | rs630736 | 0.049 | 0.3 (0.1–1.0) |
| NCEH1 | CRC + AD vs. C | rs522028 | 0.014 | 2.8 (1.2–6.2) |
| CES5A | AD vs. C | rs34266217 | 0.013 | 0.1 (0.0–0.6) |
| CES5A | CRC vs. C | rs34266217 | 0.016 | 0.2 (0.0–0.7) |
| CES5A | CRC vs. AD | rs11864688 | 0.003 | 0.1 (0.0–0.4) |
| CES5A | CRC + AD vs. C | rs34266217 | 0.004 | 0.2 (0.1–0.6) |
| SOAT1 | CRC vs. C | rs61824389 | 0.012 | 5.4 (1.5–20.2) |
| SOAT1 | CRC vs. AD | rs61824371 | 0.028 | 2.6 (1.1–6.0) |
| ABCA1 | AD vs. C | rs4149297 | 0.011 | 0.2 (0.0–0.7) |
| ABCA1 | CRC vs. C | rs2487060 | 0.029 | 9.4 (1.2–70.3) |
| ABCA1 | CRC vs. AD | rs2515617 | 0.007 | 0.3 (0.1–0.7) |
| ABCA1 | CRC + AD vs. C | rs4149297 | 0.011 | 0.3 (0.1–0.7) |
| SCARB1 | AD vs. C | rs1031605 | 0.012 | 15.8 (1.8–136.2) |
| SCARB1 | CRC vs. C | rs7485656 | 0.021 | 0.3 (0.1–0.8) |
| SCARB1 | CRC vs. AD | rs1031605 | 0.006 | 0.2 (0.0–0.6) |
| SCARB1 | CRC + AD vs. C | rs7485656 | 0.012 | 0.4 (0.2–0.8) |
| STARD3 | CRC + AD vs. C | rs11556624 | 0.04 | 0.1 (0.0–0.9) |
| NPC2 | AD vs. C | rs8008540 | 0.025 | 2.5 (1.1–5.6) |
| NPC2 | CRC + AD vs. C | rs10139122 | 0.026 | 0.4 (0.1–0.9) |
| OSBPL5 | AD vs. C | rs11025414 | 0.012 | 0.2 (0.1–0.7) |
| OSBPL5 | CRC vs. C | rs7122180 | 0.03 | 0.1 (0.0–0.8) |
| OSBPL5 | CRC vs. AD | rs11025414 | 0.003 | 5.5 (1.8–16.6) |
| OSBPL5 | CRC + AD vs. C | rs111415444 | 0.035 | 0.3 (0.1–0.9) |
| Gene | Analysis | SNP | p | OR (CI 95%) |
|---|---|---|---|---|
| LPCAT1 | AD vs. C | rs2962043 | 0.022 | 0.3 (0.1–0.8) |
| LPCAT1 | CRC vs. C | rs13170616 | 0.047 | 0.3 (0.1–1.0) |
| LPCAT1 | CRC vs. AD | rs55707452 | 0.02 | 14.4 (1.5–136.8) |
| LPCAT1 | CRC + AD vs. C | rs2962043 | 0.018 | 0.4 (0.2–0.8) |
| PLA2G12B | CRC vs. AD | rs77284811 | 0.02 | 0.1 (0.0–0.6) |
| PLA2G4C | AD vs. C | rs1985341 | 0.016 | 3.2 (1.2–8.2) |
| PLA2G4C | CRC vs. C | rs8110925 | 0.006 | 0.0 (0.0–0.4) |
| PLA2G4C | CRC + AD vs. C | rs8110925 | 0.005 | 0.1 (0.0–0.5) |
| PLB1 | AD vs. C | rs1528178 | 0.009 | 16.6 (2.0–138.4) |
| PLB1 | CRC vs. C | rs10176136 | 0.006 | 6.7 (1.7–25.9) |
| PLB1 | CRC vs. AD | rs7607457 | 0.001 | 7.6 (2.3–25.7) |
| PLB1 | CRC + AD vs. C | rs80005535 | 0.014 | 0.1 (0.0–0.6) |
| PLD1 | AD vs. C | rs9823312 | 0.022 | 4.2 (1.2–14.2) |
| PLD1 | CRC vs. C | rs416158 | 0.007 | 0.2 (0.1–0.6) |
| PLD1 | CRC vs. AD | rs41273601 | 0.008 | 4.1 (1.4–11.4) |
| PLD1 | CRC + AD vs. C | rs416158 | 0.005 | 0.3 (0.1–0.7) |
| PNPLA7 | CRC vs. C | rs12685481 | 0.033 | 0.2 (0.0–0.9) |
| PNPLA7 | CRC + AD vs. C | rs2488592 | 0.04 | 0.4 (0.2–1.0) |
| PLPP1 | CRC vs. C | rs111922583 | 0.019 | 13.9 (1.6–125.3) |
| PLPP1 | CRC + AD vs. C | rs4865950 | 0.026 | 0.0 (0.0–0.7) |
| PLPP3 | CRC vs. C | rs149181632 | 0.03 | 0.1 (0.0–0.8) |
| PLPP3 | CRC vs. AD | rs17429642 | 0.029 | 0.2 (0.0–0.8) |
| CERS4 | AD vs. C | rs7250035 | 0.017 | 4.2 (1.3–13.4) |
| CERS4 | CRC vs. C | rs11666971 | 0.01 | 4.5 (1.4–14.1) |
| CERS4 | CRC + AD vs. C | rs7250035 | 0.022 | 3.5 (1.2–10.1) |
| PIGK | AD vs. C | rs72683932 | 0.024 | 6.4 (1.3–31.7) |
| PIGK | CRC vs. C | rs12562725 | 0.014 | 0.2 (0.1–0.7) |
| PIGK | CRC + AD vs. C | rs72683932 | 0.008 | 6.5 (1.6–26.5) |
| PIGZ | AD vs. C | rs746037 | 0.006 | 3.5 (1.4–8.6) |
| PIGZ | CRC vs. C | rs76347663 | 0.032 | 7.9 (1.2–52.3) |
| PIGZ | CRC + AD vs. C | rs746037 | 0.008 | 2.7 (1.3–5.8) |
| SMPD3 | AD vs. C | rs9940621 | 0.048 | 0.3 (0.1–1.0) |
| SMPD3 | CRC vs. AD | rs7200932 | 0.006 | 0.3 (0.1–0.7) |
| Association with Trait | |||||||
|---|---|---|---|---|---|---|---|
| Traits | SNP | Gene | p | Effect (CI 95%) | Analysis | p | OR (CI 95%) |
| Bacteroidetes | rs2470641 | LOC105376940 | 1.7 × 10−6 | −0.086 (−0.119–0.053) | CRC vs. C | 0.049 | 0.3 (0.1–0.9) |
| Firmicutes | rs2662642 | SPAG16 | 1.1 × 10−7 | 0.072 (0.048–0.097) | CRC vs. AD | 0.034 | 2.2 (1.1–4.5) |
| Firmicutes | rs13280938 | LINC01605 | 1.6 × 10−6 | 0.14 (0.087–0.194) | CRC vs. AD | 0.034 | 6.8 (1.2–39.8) |
| Firmicutes | rs9538330 | - | 4.6 × 10−6 | −0.073 (−0.103–0.044) | AD vs. C | 0.025 | 3.5 (1.2–10.6) |
| Firmicutes | rs7166734 | LOC107983974 | 2.5 × 10−6 | −0.077 (−0.107–0.047) | AD vs. C | 0.0389 | 2.7 (1.1–6.9) |
| Shannon | rs73523611 | CRAT37 | 7.66 × 10−7 | 0.42 (0.26–0.57) | AD vs. C | 0.044 | 3.5 (1.0–11.9) |
| ChoE(18:1) | rs142370545 | - | 1.97 × 10−6 | −3.1 (−4.4–1.9) | CRC + AD vs. C | 0.049 | 0.1 (0.01–0.9) |
| ChoE(18:1) | rs691563 | SLC39A12 | 1.14 × 10−6 | 1.0 (0.6–1.3) | CRC vs. C | 0.010 | 5.5 (1.5–20.4) |
| ChoE(18:1) | rs691563 | SLC39A12 | 1.14 × 10−6 | 1.0 (0.6–1.3) | CRC + AD vs. C | 0.022 | 3.4 (1.2–10.2) |
| ChoE(18:1) | rs6085078 | PROKR2 | 1.73 × 10−6 | 0.69 (0.42–0.96) | CRC vs. C | 0.021 | 2.9 (1.2–7.2) |
| ChoE(18:1) | rs6085078 | PROKR2 | 1.73 × 10−6 | 0.69 (0.42–0.96) | CRC + AD vs. C | 0.037 | 2.0 (1.0–3.8) |
| ChoE(18:2) | rs62249239 | LOC100130207 | 1.17 × 10−6 | 1.7 (1.0–2.3) | CRC vs. AD | 0.036 | 9.8 (1.1–83.6) |
| ChoE(20:4) | rs2146594 | LOC102724520 | 4.36 × 10−6 | −1.1 (−1.6–0.8) | CRC vs. C | 0.007 | 0.2 (0.1–0.7) |
| ChoE(20:4) | rs56191847 | PCDH15 | 3.24 × 10−6 | −1.4 (−2.0–0.9) | CRC vs. C | 0.020 | 0.3 (0.1–0.8) |
| ChoE(20:4) | rs56191847 | PCDH15 | 3.24 × 10−6 | −1.4 (−2.0–0.9) | CRC + AD vs. C | 0.032 | 0.4 (0.2–0.9) |
| PE(16:0/18:1) | rs145994977 | - | 6.89 × 10−7 | −3.5 (−4.8–2.2) | CRC + AD vs. C | 0.035 | 0.1 (0.005–0.8) |
| PE(16:0/18:1) | rs17450393 | SNX2 | 1.33 × 10−6 | −3.1 (−4.3–1.9) | CRC + AD vs. C | 0.036 | 0.1 (0.01–0.8) |
| PE(16:0/18:1) | rs2048236 | EPM2A | 3.21 × 10−6 | −1.7 (−2.4–1.1) | AD vs. C | 0.027 | 0.09 (0.01–0.8) |
| PE(16:0/18:1) | rs2048236 | EPM2A | 3.21 × 10−6 | −1.7 (−2.4–1.1) | CRC vs. C | 0.030 | 0.1 (0.004–0.8) |
| PE(16:0/18:1) | rs2048236 | EPM2A | 3.21 × 10−6 | −1.7 (−2.4–1.1) | CRC + AD vs. C | 0.005 | 0.1 (0.01–0.4) |
| PE(16:0/18:1) | rs73153821 | CREB3L2 | 3.31 × 10−6 | −2.4 (−3.4–1.5) | CRC + AD vs. C | 0.022 | 0.1 (0.01–0.7) |
| SM(42:3) | rs17018424 | CCSER1 | 2.28 × 10−6 | −1.7 (−2.4–1.1) | CRC vs. AD | 0.012 | 0.3 (0.1–0.8) |
| SM(42:3) | rs186239 | - | 1.13 × 10−6 | 1.7 (1.1–2.4) | CRC vs. C | 0.028 | 5.1 (1.2–21.6) |
| SM(42:3) | rs186239 | - | 1.13 × 10−6 | 1.7 (1.1–2.4) | CRC vs. AD | 0.041 | 2.7 (1.0–7.2) |
| SM(42:3) | rs4745374 | CARNMT1 | 2.77 × 10−6 | 1.5 (0.9–2.2) | CRC vs. C | 0.014 | 3.4 (1.3–9.3) |
| SM(42:3) | rs4745374 | CARNMT1 | 2.77 × 10−6 | 1.5 (0.9–2.2) | CRC vs. AD | 0.015 | 3.1 (1.2–7.6) |
| SM(42:3) | rs11058736 | - | 1.53 × 10−6 | 2.4 (1.4–3.3) | CRC vs. C | 0.030 | 5.1 (1.2–21.9) |
| SM(42:3) | rs11058736 | - | 1.53 × 10−6 | 2.4 (1.4–3.3) | CRC vs. AD | 0.014 | 7 (1.5–33.1) |
| SM(42:3) | rs6085078 | PROKR2 | 2.32 × 10−6 | 1.3 (0.8–1.7) | CRC vs. C | 0.021 | 2.9 (1.2–7.2) |
| SM(42:3) | rs6085078 | PROKR2 | 2.32 × 10−6 | 1.3 (0.8–1.7) | CRC + AD vs. C | 0.037 | 2.0 (1.0–3.8) |
| SM(42:3) | rs118183318 | - | 1.75 × 10−6 | −2.5 (−3.5–1.5) | CRC vs. C | 0.012 | 0.03 (0.002–0.5) |
| SM(42:3) | rs118183318 | - | 1.75 × 10−6 | −2.5 (−3.5–1.5) | CRC vs. AD | 0.027 | 0.1 (0.01–0.7) |
| SM(42:3) | rs118183318 | - | 1.75 × 10−6 | −2.5 (−3.5–1.5) | CRC + AD vs. C | 0.032 | 0.3 (0.1–0.9) |
| TG(54:1) | rs117956865 | EPS15L1 | 2.73 × 10−8 | −3.1 (−4.1–2.1) | CRC vs. AD | 0.039 | 12.1 (1.1–127.7) |
| Model | Measurement | AD vs. C | CRC vs. C | AD vs. CRC | CRC + AD vs. C |
|---|---|---|---|---|---|
| Model 1 | AUC | 0.91 (0.84–0.94) | 0.87 (0.79–0.91) | 0.76 (0.66–0.81) | 0.91 (0.86–0.94) |
| Specificity | 0.81 (0.67–1) | 0.92 (0.81–1) | 0.8 (0.66–0.91) | 0.94 (0.69–1) | |
| Sensitivity | 0.94 (0.64–1) | 0.83 (0.69–0.94) | 0.72 (0.58–0.86) | 0.79 (0.65–0.97) | |
| Model 1 + f-Hb | AUC | 1 | 1 | 0.77 (0.67–0.82) | 0.81 (0.73–0.85) |
| Specificity | 1 | 1 | 0.66 (0.49–0.8) | 0.72 (0.58–0.86) | |
| Sensitivity | 1 | 1 | 0.89 (0.78–0.97) | 0.9 (0.83–0.97) | |
| Model 2 | AUC | 0.85 (0.76–0.89) | 0.85 (0.76–0.89) | 0.67 (0.55–0.73) | 0.84 (0.76–0.88) |
| Specificity | 0.82 (0.61–0.92) | 0.82 (0.61–0.97) | 0.7 (0.32–0.92) | 0.79 (0.61–0.95) | |
| Sensitivity | 0.86 (0.7–0.97) | 0.82 (0.6–0.98) | 0.7 (0.35–0.97) | 0.84 (0.65–0.96) | |
| Model 2 + f-Hb | AUC | 0.92 (0.86–0.95) | 0.95 (0.9–0.98) | 0.75 (0.64–0.81) | 0.94 (0.89–0.96) |
| Specificity | 0.82 (0.68–0.92) | 0.95 (0.82–1) | 0.8 (0.65–0.98) | 0.84 (0.68–1) | |
| Sensitivity | 0.97 (0.89–1) | 0.9 (0.78–1) | 0.7 (0.43–0.86) | 0.94 (0.7–1) | |
| Model 1 + Model 2 | AUC | 0.97 (0.94–0.99) | 1 | 1 | 0.96 (0.93–0.98) |
| Specificity | 0.97 (0.83–1) | 1 | 1 | 1 (0.89–1) | |
| Sensitivity | 0.89 (0.78–1) | 1 | 1 | 0.86 (0.77–0.97) | |
| Model 1 + Model 2 + f-Hb | AUC | 1 | 1 | 1 | 0.93 (0.87–0.96) |
| Specificity | 1 | 1 | 1 | 0.89 (0.78–0.97) | |
| Sensitivity | 1 | 1 | 1 | 0.97 (0.93–1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Etxebarria, K.; Clos-Garcia, M.; Telleria, O.; Nafría, B.; Alonso, C.; Iruarrizaga-Lejarreta, M.; Franke, A.; Crespo, A.; Iglesias, A.; Cubiella, J.; et al. Interplay between Genome, Metabolome and Microbiome in Colorectal Cancer. Cancers 2021, 13, 6216. https://doi.org/10.3390/cancers13246216
Garcia-Etxebarria K, Clos-Garcia M, Telleria O, Nafría B, Alonso C, Iruarrizaga-Lejarreta M, Franke A, Crespo A, Iglesias A, Cubiella J, et al. Interplay between Genome, Metabolome and Microbiome in Colorectal Cancer. Cancers. 2021; 13(24):6216. https://doi.org/10.3390/cancers13246216
Chicago/Turabian StyleGarcia-Etxebarria, Koldo, Marc Clos-Garcia, Oiana Telleria, Beatriz Nafría, Cristina Alonso, Marta Iruarrizaga-Lejarreta, Andre Franke, Anais Crespo, Agueda Iglesias, Joaquín Cubiella, and et al. 2021. "Interplay between Genome, Metabolome and Microbiome in Colorectal Cancer" Cancers 13, no. 24: 6216. https://doi.org/10.3390/cancers13246216
APA StyleGarcia-Etxebarria, K., Clos-Garcia, M., Telleria, O., Nafría, B., Alonso, C., Iruarrizaga-Lejarreta, M., Franke, A., Crespo, A., Iglesias, A., Cubiella, J., Bujanda, L., & Falcón-Pérez, J. M. (2021). Interplay between Genome, Metabolome and Microbiome in Colorectal Cancer. Cancers, 13(24), 6216. https://doi.org/10.3390/cancers13246216