
RNA Sequencing of Primary Cutaneous and Breast-Implant Associated Anaplastic Large Cell Lymphomas Reveals Infrequent Fusion Transcripts and Upregulation of PI3K/AKT Signaling via Neurotrophin Pathway Genes

 , , ,
, , ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Laser Microdissection and RNA Extraction
2.3. RNA Sequencing
2.4. RNA-Sequencing Data Analysis and Fusion Detection
2.5. Confirmation of Novel Fusions by Real-Time Polymerase Chain Reaction Analysis
2.6. Differential Gene Expression Analysis
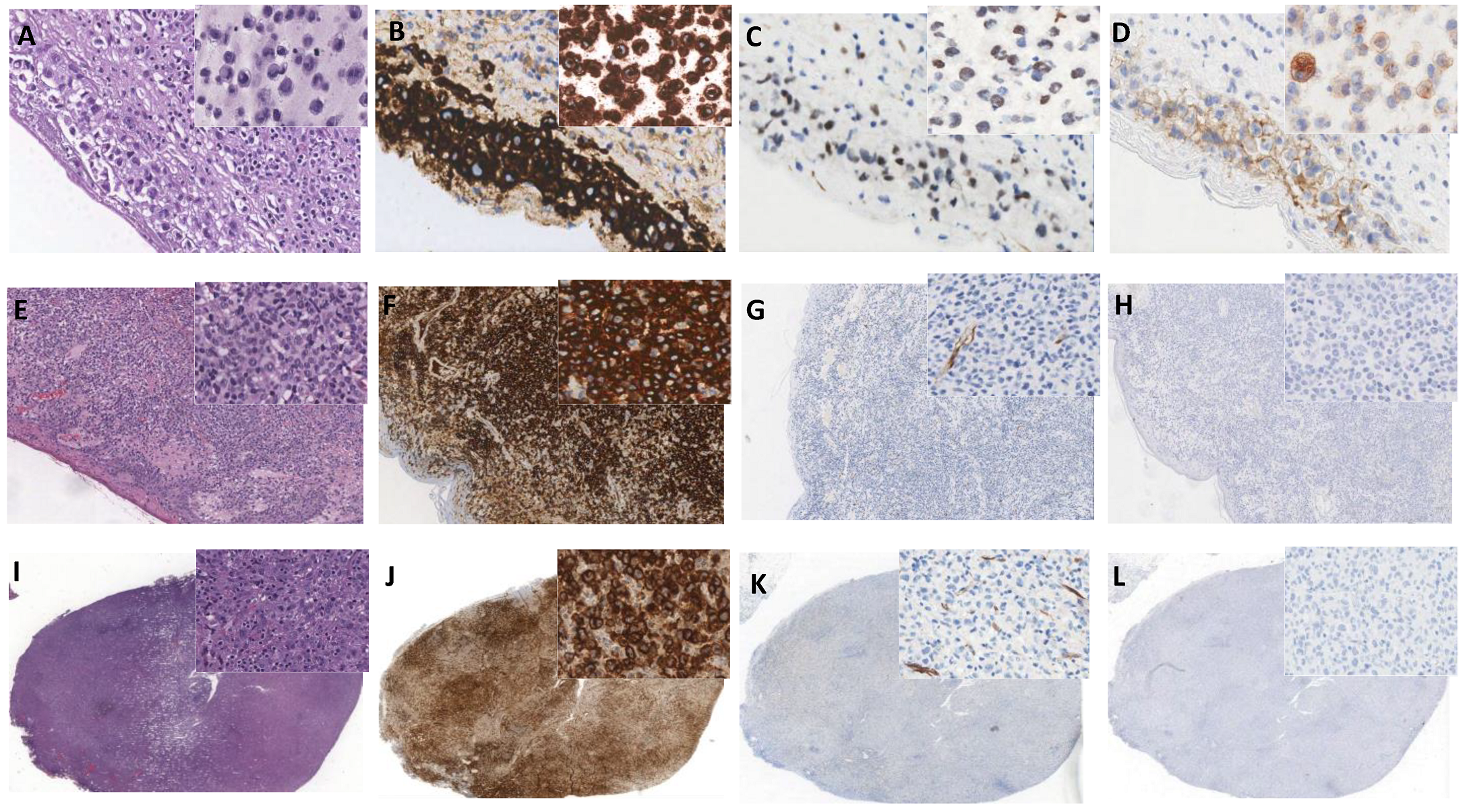
2.7. Immunohistochemistry
2.8. Cytogenetic Analysis
2.9. Targeted Next-Generation Sequencing
3. Results
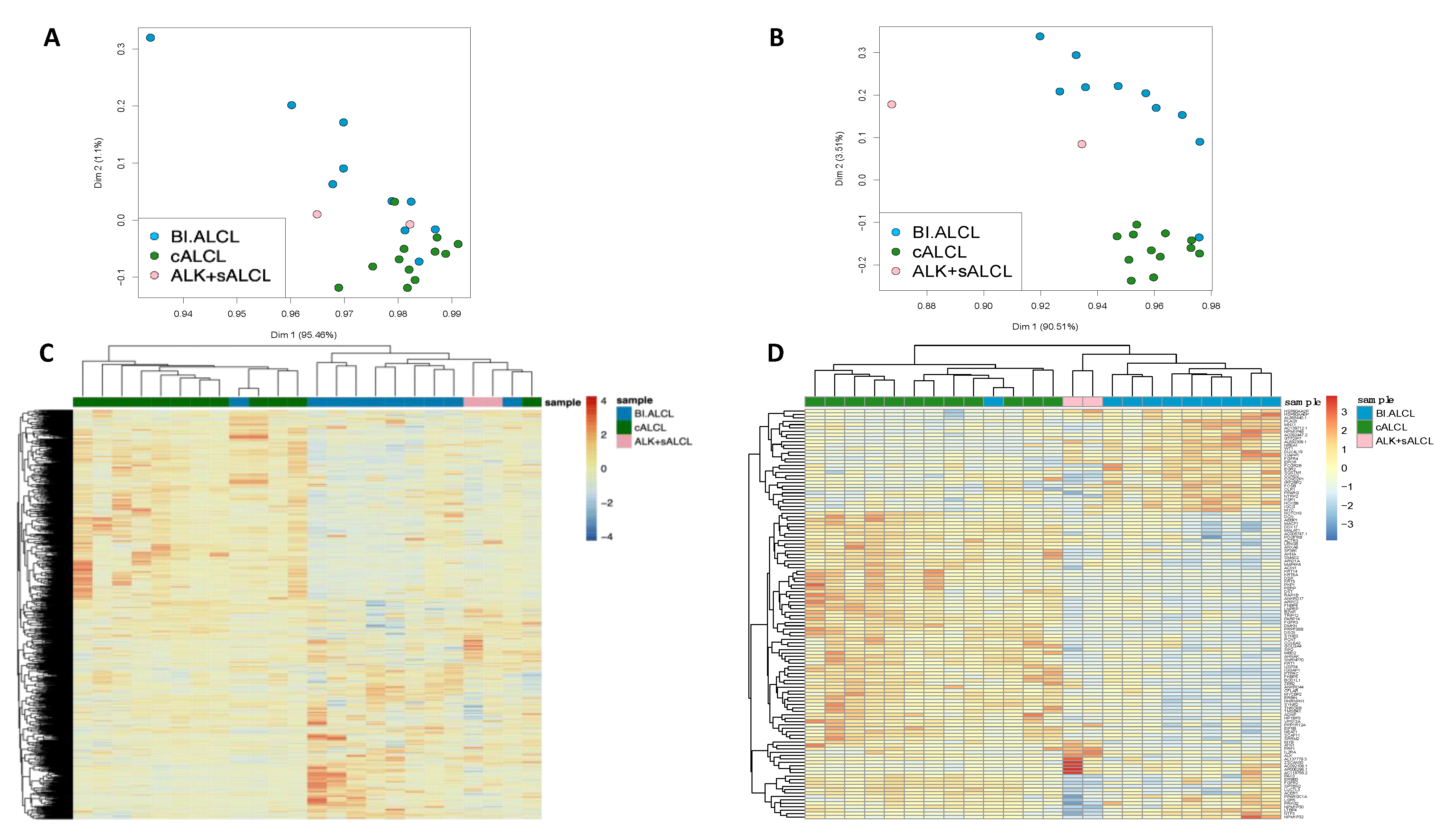
3.1. Fusion Transcripts in cALCL and BI-ALCL Are Uncommon and May Suggest Genomic Instability in BI-ALCL
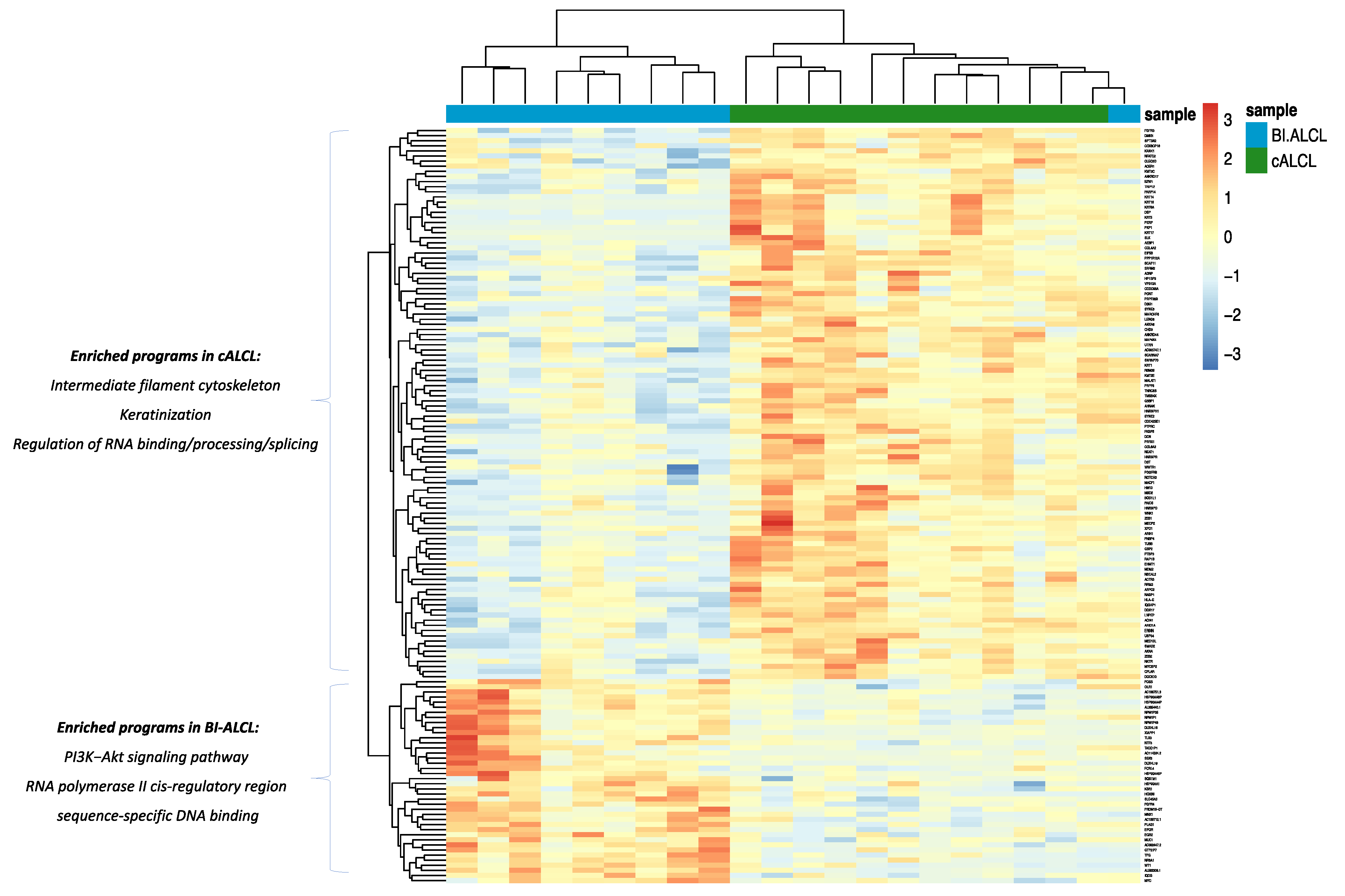
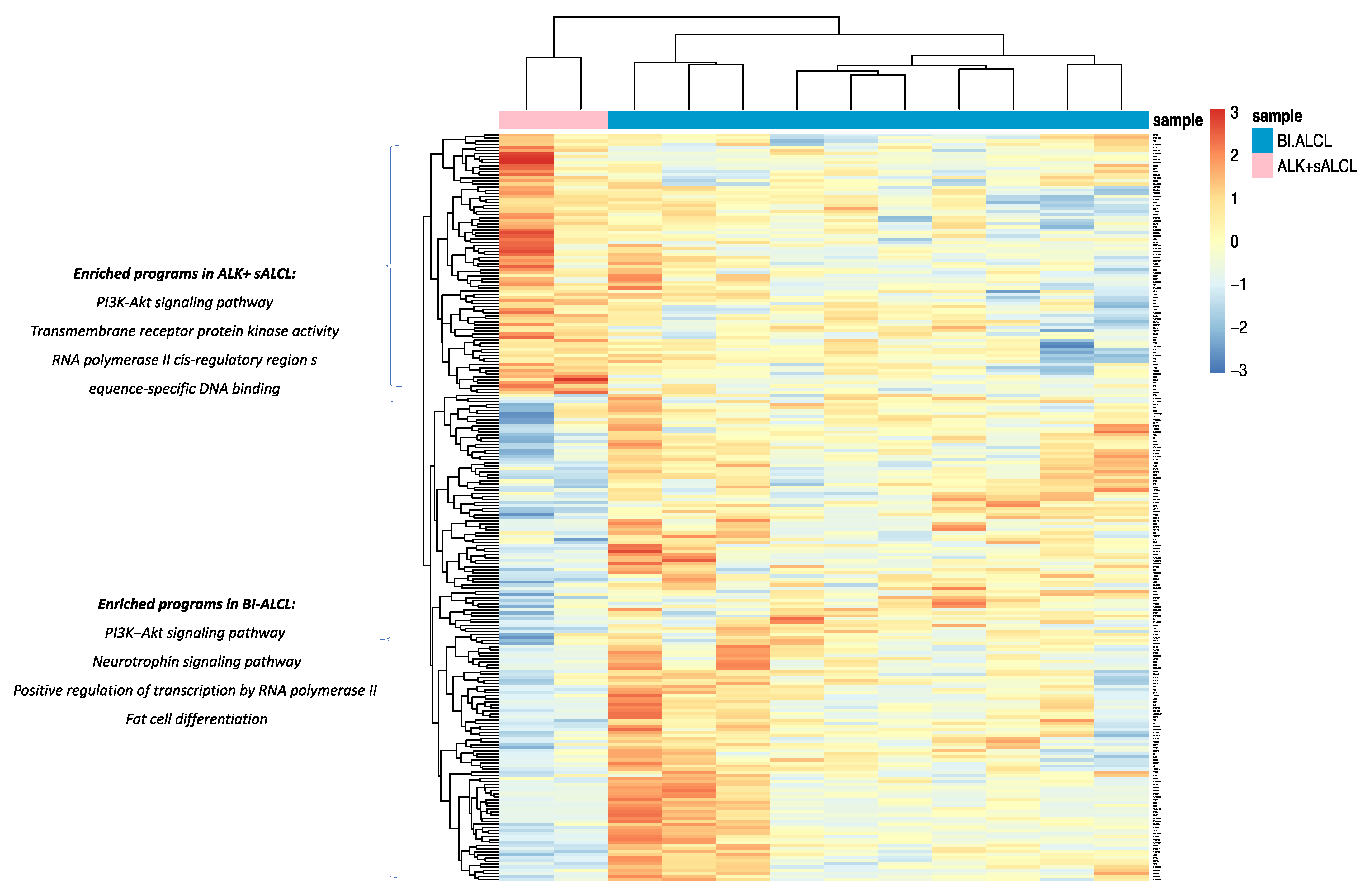
3.2. cALCL and BI-ALCL Show Upregulation of the PI3K/Akt Pathway and Differences in Transcription and Spicing Processes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. Mature T- and NK-cell neoplasms. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017; pp. 345–421. [Google Scholar]
- Amin, H.M.; Medeiros, L.J.; Ma, Y.; Feretzaki, M.; Das, P.; Leventaki, V.; Rassidakis, G.Z.; O’Connor, S.L.; McDonnell, T.J.; Lai, R. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene 2003, 22, 5399–5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mereu, E.; Pellegrino, E.; Scarfò, I.; Inghirami, G.; Piva, R. The heterogeneous landscape of ALK negative ALCL. Oncotarget 2017, 8, 18525–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrentraut, S.; Nagel, S.; Scherr, M.E.; Schneider, B.; Quentmeier, H.; Geffers, R.; Kaufmann, M.; Meyer, C.; Prochorec-Sobieszek, M.; Ketterling, R.P.; et al. t(8;9)(p22;p24)/PCM1-JAK2 activates SOCS2 and SOCS3 via STAT5. PLoS ONE 2013, 8, e53767. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef] [Green Version]
- Kadin, M.E.; Deva, A.; Xu, H.; Morgan, J.; Khare, P.; MacLeod, R.A.; Van Natta, B.W.; Adams, W.P., Jr.; Brody, G.S.; Epstein, A.L. Biomarkers Provide Clues to Early Events in the Pathogenesis of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthetic Surg. J. 2016, 36, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Di Napoli, A.; De Cecco, L.; Piccaluga, P.P.; Navari, M.; Cancila, V.; Cippitelli, C.; Pepe, G.; Lopez, G.; Monardo, F.; Bianchi, A.; et al. Transcriptional analysis distinguishes breast implant-associated anaplastic large cell lymphoma from other peripheral T-cell lymphomas. Mod. Pathol. 2019, 32, 216–230. [Google Scholar] [CrossRef]
- Oishi, N.; Hundal, T.; Phillips, J.L.; Dasari, S.; Hu, G.; Viswanatha, D.S.; He, R.; Mai, M.; Jacobs, H.K.; Ahmed, N.H.; et al. Molecular profiling reveals a hypoxia signature in breast implant-associated anaplastic large cell lymphoma. Haematologica 2020, 106, 1714–1724. [Google Scholar] [CrossRef]
- Van Kester, M.S.; Tensen, C.P.; Vermeer, M.H.; Dijkman, R.; Mulder, A.A.; Szuhai, K.; Willemze, R.; van Doorn, R. Cutaneous Anaplastic Large Cell Lymphoma and Peripheral T-Cell Lymphoma NOS Show Distinct Chromosomal Alterations and Differential Expression of Chemokine Receptors and Apoptosis Regulators. J. Investig. Dermatol. 2010, 130, 563–575. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.L.; Law, M.; Remstein, E.D.; Macon, W.R.; Erickson, L.A.; Grogg, K.L.; Kurtin, P.J.; Dogan, A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia 2008, 23, 574–580. [Google Scholar] [CrossRef]
- Feldman, A.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Özsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Wada, D.A.; Law, M.E.; Hsi, E.D.; DiCaudo, D.J.; Ma, L.; Lim, M.; De Souza, A.; Comfere, N.I.; Weenig, R.H.; Macon, W.R.; et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: A multicenter study of 204 skin biopsies. Mod. Pathol. 2010, 24, 596–605. [Google Scholar] [CrossRef]
- Castellar, E.R.P.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, X.; He, Y.H. Detection of chromosomal translocations of DUSP22 and TP63 in ALK-negative anaplastic large cell lymphoma by fluorescence in situ hybridization and related clinical relevance. Zhonghua Bing Li Xue Za Zhi 2019, 48, 791–795. [Google Scholar] [CrossRef]
- Vasmatzis, G.; Johnson, S.H.; Knudson, R.A.; Ketterling, R.P.; Braggio, E.; Fonseca, R.; Viswanatha, D.S.; Law, M.E.; Kip, N.S.; Özsan, N.; et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood 2012, 120, 2280–2289. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Feldman, A.L. Anaplastic large cell lymphomas: ALK positive, ALK negative, and primary cutaneous. Adv. Anat. Pathol. 2015, 22, 29–49. [Google Scholar] [CrossRef]
- Schrader, A.M.R.; Chung, Y.-Y.; Jansen, P.M.; Szuhai, K.; Torres, A.N.B.; Tensen, C.P.; Willemze, R. No TP63 rearrangements in a selected group of primary cutaneous CD30+ lymphoproliferative disorders with aggressive clinical course. Blood 2016, 128, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Delas, A.; Gaulard, P.; Haioun, C.; Moreau, A.; Xerri, L.; Traverse-Glehen, A.; Rousset, T.; Quintin-Roue, I.; Petrella, T.; et al. Breast implant-associated anaplastic large cell lymphoma: Two distinct clinicopathological variants with different outcomes. Ann. Oncol. 2016, 27, 306–314. [Google Scholar] [CrossRef]
- Oishi, N.; Brody, G.S.; Ketterling, R.P.; Viswanatha, D.S.; He, R.; Dasari, S.; Mai, M.; Benson, H.K.; Sattler, C.A.; Boddicker, R.L.; et al. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood 2018, 132, 544–547. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Rassidakis, G.Z.; Thomaides, A.; Atwell, C.; Ford, R.; Jones, D.; Claret, F.-X.; Medeiros, L.J. JunB expression is a common feature of CD30+ lymphomas and lymphomatoid papulosis. Mod. Pathol. 2005, 18, 1365–1370. [Google Scholar] [CrossRef]
- Los-de Vries, G.T.; de Boer, M.; van Dijk, E.; Stathi, P.; Hijmering, N.J.; Roemer, M.G.M.; Mendeville, M.; Miedema, D.M.; de Boer, J.P.; Rakhorst, H.A.; et al. Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 2020, 136, 2927–2932. [Google Scholar] [CrossRef]
- Tabanelli, V.; Corsini, C.; Fiori, S.; Agostinelli, C.; Calleri, A.; Orecchioni, S.; Melle, F.; Motta, G.; Rotili, A.; Di Napoli, A.; et al. Recurrent PDL1 expression and PDL1 (CD274) copy number alterations in breast implant—Associated anaplastic large cell lymphomas. Hum. Pathol. 2019, 90, 60–69. [Google Scholar] [CrossRef]
- Blombery, P.; Thompson, E.; Ryland, G.L.; Joyce, R.; Byrne, D.J.; Khoo, C.; Lade, S.; Hertzberg, M.; Hapgood, G.; Marlton, P.; et al. Frequent activating STAT3 mutations and novel recurrent genomic abnormalities detected in breast implant-associated anaplastic large cell lymphoma. Oncotarget 2018, 9, 36126–36136. [Google Scholar] [CrossRef] [Green Version]
- Blombery, P.; Thompson, E.R.; Jones, K.; Arnau, G.M.; Lade, S.; Markham, J.F.; Li, J.; Deva, A.; Johnstone, R.W.; Khot, A.; et al. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast implant-associated anaplastic large cell lymphoma anaplastic large cell lymphoma. Haematologica 2016, 101, e387–e390. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Nicolae, A.; Laurent, C.; Le Bras, F.; Haioun, C.; Fataccioli, V.; Amara, N.; Adélaïde, J.; Guille, A.; Schiano, J.-M.; et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood 2019, 135, 360–370. [Google Scholar] [CrossRef]
- Letourneau, A.; Maerevoet, M.; Milowich, D.; DeWind, R.; Bisig, B.; Missiaglia, E.; De Leval, L. Dual JAK1 and STAT3 mutations in a breast implant-associated anaplastic large cell lymphoma. Virchows Arch. 2018, 473, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Di Napoli, A.; Jain, P.; Duranti, E.; Margolskee, E.; Arancio, W.; Facchetti, F.; Alobeid, B.; Di Pompeo, F.S.; Mansukhani, M.; Bhagat, G. Targeted next generation sequencing of breast implant-associated anaplastic large cell lymphoma reveals mutations in JAK/STAT signalling pathway genes, TP53 and DNMT3A. Br. J. Haematol. 2018, 180, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.-L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Liu, K.; Juan, T.; Fang, F.; Newman, M.; Hoeck, W. FusionMap: Detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics 2011, 27, 1922–1928. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Reactome Tool. Available online: https://reactome.org/PathwayBrowser/#TOOL=AT (accessed on 24 August 2021).
- ChimerDB 4.0 Database. Available online: https://www.kobic.re.kr/chimerdb_mirror (accessed on 24 August 2021).
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Laharanne, E.; Vergier, B.; Jouary, T.; Beylot-Barry, M.; Merlio, J.P. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: A study of 54 cases. J. Investig. Dermatol. 2010, 130, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.D. Chromosomal translocations: Revisited yet again. Blood 2008, 112, 2183–2189. [Google Scholar] [CrossRef]
- Vose, J.; Armitage, J.; Weisenburger, D. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Oishi, N.; Miranda, R.N.; Feldman, A.L. Genetics of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthetic Surg. J. 2019, 39, S14–S20. [Google Scholar] [CrossRef]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G.W. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [Green Version]
- George, E.V.; Pharm, J.; Houston, C.; Al-Quran, S.; Brian, G.; Dong, H.; Hai, W.; Reeves, W.; Yang, L.-J. Breast implant-associated ALK-negative anaplastic large cell lymphoma: A case report and discussion of possible pathogenesis. Int. J. Clin. Exp. Pathol. 2013, 6, 1631–1642. [Google Scholar]
- Lechner, M.; Megiel, C.; Church, C.H.; Angell, T.; Russell, S.M.; Sevell, R.B.; Jang, J.K.; Brody, G.S.; Epstein, A.L. Survival Signals and Targets for Therapy in Breast Implant–Associated ALK− Anaplastic Large Cell Lymphoma. Clin. Cancer Res. 2012, 18, 4549–4559. [Google Scholar] [CrossRef] [Green Version]
- Alobeid, B.; Sevilla, D.W.; El-Tamer, M.B.; Murty, V.V.; Savage, D.G.; Bhagat, G. Aggressive presentation of breast implant-associated ALK-1 negative anaplastic large cell lymphoma with bilateral axillary lymph node involvement. Leuk. Lymphoma 2009, 50, 831–833. [Google Scholar] [CrossRef]
- Dhanasekaran, S.M.; Balbin, O.A.; Chen, G.; Nadal, E.; Kalyana-Sundaram, S.; Pan, J.; Veeneman, B.A.; Cao, X.; Malik, R.S.; Vats, P.; et al. Transcriptome meta-analysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Lobello, C.; Tichy, B.; Bystry, V.; Radova, L.; Filip, D.; Mraz, M.; Montes-Mojarro, I.A.; Prokoph, N.; Larose, H.; Liang, H.C.; et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia 2021, 35, 1500–1505. [Google Scholar] [CrossRef]
- Laimer, D.; Dolznig, H.; Kollmann, K.; Vesely, P.W.; Schlederer, M.; Merkel, O.; Schiefer, A.-I.; Hassler, M.R.; Heider, S.; Amenitsch, L.; et al. PDGFR blockade is a rational and effective therapy for NPM-ALK–driven lymphomas. Nat. Med. 2012, 18, 1699–1704. [Google Scholar] [CrossRef] [Green Version]
- Piva, R.; Agnelli, L.; Pellegrino, E.; Todoerti, K.; Grosso, V.; Tamagno, I.; Fornari, A.; Martinoglio, B.; Medico, E.; Zamò, A.; et al. Gene Expression Profiling Uncovers Molecular Classifiers for the Recognition of Anaplastic Large-Cell Lymphoma Within Peripheral T-Cell Neoplasms. J. Clin. Oncol. 2010, 28, 1583–1590. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Joshi, S.; Davare, M.A.; Druker, B.J.; Tognon, C.E. Revisiting NTRKs as an emerging oncogene in hematological malignancies. Leukemia 2019, 33, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Fuda, N.J.; Ardehali, M.B.; Lis, J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nat. Cell Biol. 2009, 461, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Nicoll, M.; Ingham, R.J. AP-1 family transcription factors: A diverse family of proteins that regulate varied cellular activities in classical hodgkin lymphoma and ALK+ ALCL. Exp. Hematol. Oncol. 2021, 10, 4. [Google Scholar] [CrossRef]
- Parenti, R.; Salvatorelli, L.; Musumeci, G.; Parenti, C.; Giorlandino, A.; Motta, F.; Magro, G. Wilms’ tumor 1 (WT1) protein expression in human developing tissues. Acta Histochem. 2015, 117, 386–396. [Google Scholar] [CrossRef]
- Miwa, H.; Beran, M.; Saunders, G.F. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar]
- Drakos, E.; Rassidakis, G.Z.; Tsioli, P.; Lai, R.; Jones, D.; Medeiros, L.J. Differential Expression of WT1 Gene Product in Non-Hodgkin Lymphomas. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 132–137. [Google Scholar] [CrossRef]
- Boyle, K.; Hadaschik, D.; Virtue, S.; Cawthorn, W.; Ridley, S.; O’Rahilly, S.; Siddle, K. The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ. 2019, 16, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, A.; Pepe, G.; Giarnieri, E.; Cippitelli, C.; Bonifacino, A.; Mattei, M.; Martelli, M.; Falasca, C.; Cox, M.C.; Santino, I.; et al. Cytological diagnostic features of late breast implant seromas: From reactive to anaplastic large cell lymphoma. PLoS ONE 2017, 12, e0181097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, T.; Raymond, M.; Bhullar, P.; Ghaffari, E.; Symonds, A.L.J.; Meier, U.C.; Giovannoni, G.; Li, S.; Wang, P. Early Growth Response Gene-2 Controls IL-17 Expression and Th17 Differentiation by Negatively Regulating Batf. J. Immunol. 2013, 190, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Symonds, A.; Martin, J.E.; Kioussis, D.; Wraith, D.C.; Li, S.; Wang, P. Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J. Exp. Med. 2008, 205, 2295–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safford, M.; Collins, S.; Lutz, M.A.; Allen, A.; Huang, C.T.; Kowalski, J.; Blackford, A.; Horton, M.R.; Drake, C.; Schwartz, R.H.; et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol. 2005, 6, 472–480. [Google Scholar] [CrossRef]
- Di Napoli, A.; Greco, D.; Scafetta, G.; Ascenzi, F.; Gulino, A.; Aurisicchio, L.; Di Pompeo, F.S.; Bonifacino, A.; Giarnieri, E.; Morgan, J.; et al. IL-10, IL-13, Eotaxin and IL-10/IL-6 ratio distinguish breast implant-associated anaplastic large-cell lymphoma from all types of benign late seromas. Cancer Immunol. Immunother. 2021, 70, 1379–1392. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward 5′-3′ | Reverse 5′-3′ | Probe 5′-3′ | Amplicon Size (bp) |
|---|---|---|---|---|
| NPM1-TYK2 | GGAAAAGACTCAAAACCATC | CCAGGATCACTCAGCTTG | CCTGGTTCATGGTAATGTGTGTGGCCGGAA | 130 |
| MYO18A-GIT1 | CAAGGCAGCTTCTGATGATGG | AGTATTCCGGGTTAACAGGCAG | TGGTGACAGAGCGCAGTGCCGTGCCCTT | 242 |
| GORS1-NF1 | TTTCTACCAAGCTGGGACTTCC | CCCAATAACACCCCCTAGGATG | GGATCAACCTGAGGAAGCGGCGGGACTCGC | 229 |
| Sample ID | Fusion Transcript | Genomic Location | Frame |
|---|---|---|---|
| ALK+ sALCL #1 | NPM1/ALK | t(2;5)(p23;q35) | In-frame |
| ALK+ sALCL #2 | NPM1/ALK | t(2;5)(p23;q35) | In-frame |
| cALCL #4 | NPM1/TYK2 | t(5;19)(q35;p13) | In-frame |
| BI-ALCL #3 | MYO18A/GIT1 | chr17(q11.2) | In-frame |
| EPS15/GNG12 | chr1(p32.3-p31.3) | In-frame | |
| NF1/GOSR1 | chr17(q11.2) | In-frame | |
| BI-ALCL #7 | ARNT/GOLPH3L | chr1(q21.3) | In-frame |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Napoli, A.; Vacca, D.; Bertolazzi, G.; Lopez, G.; Piane, M.; Germani, A.; Rogges, E.; Pepe, G.; Santanelli Di Pompeo, F.; Salgarello, M.; et al. RNA Sequencing of Primary Cutaneous and Breast-Implant Associated Anaplastic Large Cell Lymphomas Reveals Infrequent Fusion Transcripts and Upregulation of PI3K/AKT Signaling via Neurotrophin Pathway Genes. Cancers 2021, 13, 6174. https://doi.org/10.3390/cancers13246174
Di Napoli A, Vacca D, Bertolazzi G, Lopez G, Piane M, Germani A, Rogges E, Pepe G, Santanelli Di Pompeo F, Salgarello M, et al. RNA Sequencing of Primary Cutaneous and Breast-Implant Associated Anaplastic Large Cell Lymphomas Reveals Infrequent Fusion Transcripts and Upregulation of PI3K/AKT Signaling via Neurotrophin Pathway Genes. Cancers. 2021; 13(24):6174. https://doi.org/10.3390/cancers13246174
Chicago/Turabian StyleDi Napoli, Arianna, Davide Vacca, Giorgio Bertolazzi, Gianluca Lopez, Maria Piane, Aldo Germani, Evelina Rogges, Giuseppina Pepe, Fabio Santanelli Di Pompeo, Marzia Salgarello, and et al. 2021. "RNA Sequencing of Primary Cutaneous and Breast-Implant Associated Anaplastic Large Cell Lymphomas Reveals Infrequent Fusion Transcripts and Upregulation of PI3K/AKT Signaling via Neurotrophin Pathway Genes" Cancers 13, no. 24: 6174. https://doi.org/10.3390/cancers13246174
APA StyleDi Napoli, A., Vacca, D., Bertolazzi, G., Lopez, G., Piane, M., Germani, A., Rogges, E., Pepe, G., Santanelli Di Pompeo, F., Salgarello, M., Jobanputra, V., Hsiao, S., Wrzeszczynski, K. O., Berti, E., & Bhagat, G. (2021). RNA Sequencing of Primary Cutaneous and Breast-Implant Associated Anaplastic Large Cell Lymphomas Reveals Infrequent Fusion Transcripts and Upregulation of PI3K/AKT Signaling via Neurotrophin Pathway Genes. Cancers, 13(24), 6174. https://doi.org/10.3390/cancers13246174