Simple Summary
Tumor mutation burden (TMB) has shown promise as a biomarker for immune checkpoint blockade therapy in some cancers, but not consistently in gliomas. The goal of our study was to systematically investigate the association between TMB, expressed neoantigens, and the tumor immune microenvironment in IDH-mutant and IDH-wildtype gliomas, which are two types of biologically distinct gliomas. We demonstrated that TMB positively correlated with expressed neoantigens, but inversely correlated with immune score in IDH-wildtype tumors but showed no correlation in IDH-mutant tumors. The antigen processing and presenting (APP) score may have potential as a clinical biomarker to predict immune therapy response in gliomas. Lastly, 19% of patients had pathogenic or likely pathogenic germline mutations, primarily in DNA damage repair genes.
Abstract
Background: A consistent correlation between tumor mutation burden (TMB) and tumor immune microenvironment has not been observed in gliomas as in other cancers. Methods: Driver germline and somatic mutations, TMB, neoantigen, and immune cell signatures were analyzed using whole exome sequencing (WES) and transcriptome sequencing of tumor and WES of matched germline DNA in a cohort of 66 glioma samples (44 IDH-mutant and 22 IDH-wildtype). Results: Fourteen samples revealed a hypermutator phenotype (HMP). Eight pathogenic (P) or likely pathogenic (LP) germline variants were detected in 9 (19%) patients. Six of these 8 genes were DNA damage repair genes. P/LP germline variants were found in 22% of IDH-mutant gliomas and 12.5% of IDH-wildtype gliomas (p = 0.7). TMB was correlated with expressed neoantigen but showed an inverse correlation with immune score (R = −0.46, p = 0.03) in IDH-wildtype tumors and no correlation in IDH-mutant tumors. The Antigen Processing and Presentation (APP) score correlated with immune score and was surprisingly higher in NHMP versus HMP samples in IDH-wildtype gliomas, but higher in HMP versus NHMP in IDH-mutant gliomas. Conclusion: TMB was inversely correlated with immune score in IDH-wildtype gliomas and showed no correlation in IDH-mutant tumors. APP was correlated with immune score and may be further investigated as a biomarker for response to immunotherapy in gliomas. Studies of germline variants in a larger glioma cohort are warranted.
1. Introduction
Gliomas are the most common primary malignant brain tumor and remain a fatal disease [1]. They are challenging to treat, largely due to the high level of intra- and inter-tumoral heterogeneity and a genomic landscape that constantly evolves due to selective pressure in response to therapies [2]. In addition, the immunosuppressive tumor microenvironment (TME) counteracts the efficacy of therapies, particularly immunotherapies [2].
In the past decade, immunotherapy such as immune checkpoint blockade has emerged as an effective therapeutic approach for several types of cancers, such as melanoma and lung cancer [3]. However, response to immunotherapy varies in patients with the same type of cancer, demonstrating the importance of identifying predictive biomarkers [3]. Tumor mutation burden (TMB), which is often proportional to the neoantigen burden, has emerged as a promising predictive biomarker of immune response in melanoma and lung cancer [4]. These efforts are highlighted in the KEYNOTE-158 study, which led to the recent US Food and Drug Administration (FDA) approval of using pembrolizumab, an anti-PD1 immune checkpoint inhibitor, in solid tumors with a TMB above 10 mutations per mega base (Mb) (defined as having a hypermutator phenotype (HMP) [5]. However, this correlation between TMB and response to immunotherapy has not been consistently observed in gliomas [6,7].
A recently published seminal study by Touat et al. comprehensively analyzed the molecular determinants of TMB in over 10,000 glioma samples [6]. Two major pathways to hypermutation were elucidated: a de novo pathway associated with constitutional defects in mismatch repair (MMR) genes, and an acquired resistance driven by MMR deficiency following temozolomide (TMZ) treatment. While MMR deficient tumors are more likely to accumulate TMB, they were found to have a lack of T cell infiltrates and a low rate of response to anti-PD1 therapy. This study provided evidence that TMZ can drive the accumulation of mutations without promoting a response to immunotherapy. While detailed characterization of the phenotypic and molecular features of hypermutated gliomas has been performed, a systematic analysis of the associations between TMB, expressed neoantigens, and tumor microenvironment has not been previously performed and may provide a better understanding of the discordance between a high TMB and poor response to immunotherapy in gliomas.
In addition, the mechanisms underlying this discordance may not be the same in biologically distinct subsets of gliomas. Isocitrate dehydrogenase (IDH)-mutant gliomas have a distinct tumor biology compared to IDH-wildtype gliomas at genetic and epigenetic levels [8]. Moreover, IDH mutation status has been considered a favorable predictive biomarker for clinical outcomes [9]. The discovery of mutations in IDH genes has led to a better understanding of glioma biology as well as a major change in diagnostic criteria and standards of care.
In this study, we performed a comprehensive genomic analysis including whole exome sequencing (WES) and transcriptomic analysis of primary and recurrent tumor samples in both IDH-mutant and IDH-wildtype gliomas. Furthermore, we examined germline cancer predisposition genes (CPGs) by conducting WES of matched blood samples. The focus of our study was to analyze the correlations between TMB, expressed neoantigens, immune score of the tumor microenvironment, and antigen processing and presentation (APP) function in IDH-wildtype and -mutant gliomas separately. Our data shows promise for further investigating APP score as a clinical biomarker for determining immune response in glioma patients.
2. Results
2.1. Sample Characteristics
A total of 66 tumor samples and matched blood samples collected from 48 glioma patients from January 2016 to March 2020 were analyzed. As summarized in Table 1 and further expanded on in Table S1, the sample cohort included both IDH-mutant (n = 44) and IDH-wildtype (n = 22) tumors, as well as samples collected from primary (n = 13) and recurrent disease stages (n = 53), ranging from the 1st to more than 5th recurrence, which represent different stages of the disease (Table 1, Table S1). The samples used in this study also exhibited different histology and tumor World Health Organization (WHO) grades.

Table 1.
Sample Characteristics by IDH mutation status.
2.2. Pathogenic Germline Mutations
Among 48 patients, nine (19%) were found to carry heterozygous pathogenic (P) or likely pathogenic (LP) germline alterations in eight cancer predisposition genes (CPGs): TP53, MUTYH, BLM, RET, ERCC6, MITF, BRIP1, and MSH2 (Table 2). Importantly, six of them, except for RET and MITF, are involved in the DNA damage repair (DDR) pathway, indicating the importance of genomic instability in glioma genesis. Among these nine patients, seven had IDH-mutant gliomas and two had IDH-wildtype gliomas. No correlation was found between P/LP variants and the IDH somatic mutation status (P/LP germline variants in 21.9% of patients with IDH-mutant gliomas versus 12.5% of patients with IDH-wildtype gliomas, two-tailed Fisher’s exact test, p = 0.7). Analysis of the TMB revealed HMP in two patients with IDH-mutant gliomas at the time of disease recurrence, each carrying P/LP germline variants in MUTYH and ERCC6, respectively and in one patient with de novo IDH-wildtype tumor (NCI0392) carrying a pathogenic variant in MSH2. Therefore, three of 11 (27.3%) patients with HMP tumors and three of 37 (7.9%) patients with NHMP tumors had P/LP germline mutations in DDR genes. However, we found no association between the hypermutation phenotype and the presence of P/LP germline variants in the DDR pathway (two-tailed Fisher’s exact, p = 0.12). Taken together, mutations in DDR-related genes are common among P/LP germline variants. However, the association of DDR germline variants with HMP development needs to be further studied in a larger cohort.
Table 2.
Pathogenic or likely pathogenic germline mutations detected in 9 patients.
2.3. Mutational Landscape
Among the 66 samples, tumor DNA was available for 65 samples. A total of 28,630 high confidence somatic mutations were detected by WES analysis. Using our in-house tiering system, 353 pathogenic or hotspot mutations (tier 1) were detected [11]. The top somatic mutations in the tier 1 list are summarized in Figure 1A. The common genetic alterations include IDH1 (58%), TP53 (58%), ATRX (47%), IDH2 (11%), CIC (13%), SETD2 (13%), PIK3CA (11%), PIK3R1(11%), PTEN (8%), and RB1(8%), which are consistent with previously reported genomic alterations in gliomas [12]. To further understand the potential of these high confidence somatic mutations to generate tumor antigens, we examined the percentage of mutant genes that are expressed. We looked for the exact variant reads from RNAseq of the corresponding tumor using a set of filters (VAF ≥ 0.1, total RNA coverage ≥ 10, variant coverage ≥ 2) to identify the expressed somatic mutations from all high confidence somatic mutations. Tier 1 mutations were more likely to be expressed compared to all high confidence somatic mutations (52.4% vs. 30.1%, p < 0.0001; Fisher’s exact test, two-tailed) (Figure 1B).
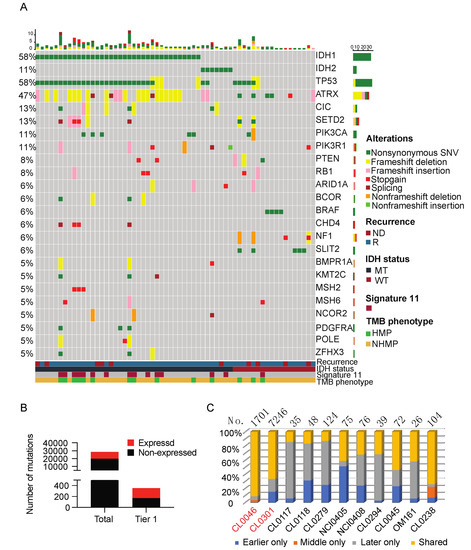
Figure 1.
Somatic mutations detected in the sample cohort. (A) An integrated analysis of the sample cohort (66 samples) depicts the top tier 1 mutations. The samples are grouped by recurrence status, IDH mutation status, presence of mutational signature 11, and TMB phenotype. Complete information of all genetic alterations can be found in the database (https://clinomics.ccr.cancer.gov/clinomics/public/login accessed date: 20 November 2021) (B) High confidence somatic variants count analysis shows that tier 1 high confident somatic mutations contain a higher percentage of expressed mutations than the total high confident somatic mutations. (C) Matched recurrent glioma samples share expressed somatic mutations. Total number of expressed somatic mutations is labeled for each patient. Patients labeled in red carry HMP tumors, and patients labeled in black carry NHMP tumors. NHMP, TMB less than 10 mutations per Mb. HMP, TMB more than 10 mutations per Mb. ND, newly diagnosed tumor. R, recurrent tumor.
A comparison of the genetic alterations in recurrent IDH-mutant gliomas with those in the matched newly diagnosed tumors demonstrated a significant number of acquired mutations that are specific to the recurrent tumors [13]. To examine the genetic alterations that evolve through disease progression, we analyzed the high confidence somatic mutations in the samples collected at early recurrences to their matched samples collected at later recurrences. Patients CL0046 and CL0301, who developed HMP, had the highest number of shared mutations in the matched samples (Figure 1C). This suggests that recurrent HMP gliomas harbor mutations that persist, indicating the existence of a resistant clone. Patient CL0238, previously reported by our group to harbor a pathogenic fusion gene BCR-ABL [14], was diagnosed with a NHMP glioma that also had a high percentage of shared mutations, indicating that the fusion event of BCR-ABL occurred early and that it is an oncogenic driver leading to rapid progression of disease without significant clonal divergence.
To better understand the mutational profiles of gliomas, we calculated the TMB in our sample cohort using WES data of tumor samples and their matched blood samples. The TMB in all 65 samples ranged from 0.6 to 254 mutations per Mb. We then compared TMB in newly diagnosed (ND) and recurrent tumors in both IDH-wildtype and IDH-mutant gliomas. TMB values of recurrent samples were significantly higher than that of ND samples for IDH-mutant tumors (median: 1.17 versus 2.63, p = 0.007). However, no statistically significant difference in TMB values was found between recurrent and ND samples for IDH-wildtype tumors (Figure 2A). Using 10 mutations per Mb as the cutoff, 14 tumor samples were defined as HMP and 51 samples were NHMP. Among the 14 HMP samples, 11 samples were IDH-mutant and three were IDH-wildtype, and among the 51 NHMP samples, 32 were IDH-mutant and 19 were IDH-wildtype (Table S2). There was no difference in hypermutation phenotype incidence between IDH-mutant and IDH-wildtype gliomas (26% and 13.6%, respectively, two-tailed Fisher’s exact test, p = 0.35).
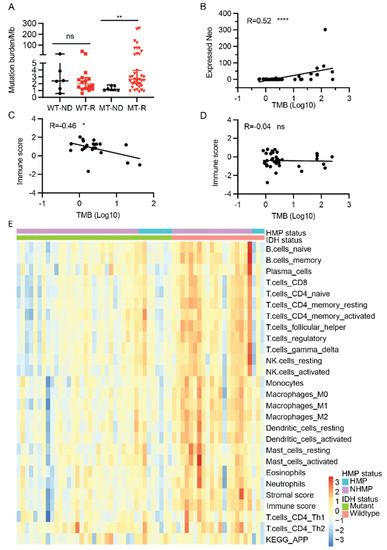
Figure 2.
Neoantigen profile and immune signatures (n = 60). (A) Tumor mutation burden at initial diagnosis and recurrence in IDH-mutant (MT) and IDH-wildtype (WT) gliomas. (B) A significant correlation between expressed neoantigens and tumor mutation burden in all samples (R = 0.52, p < 0.0001. (C) An inverse correlation between TMB and immune score in IDH-wildtype glioma samples (R = −0.46, p < 0.05). (D) No correlation between TMB and immune score in IDH-mutant samples (R = 0.04, p > 0.05). (E) Heatmap of immune signatures in gliomas. Samples are grouped by their IDH mutation status and HMP status. Expressed neo, expressed neoantigen. ns: not statistically significant, *, p < 0.05; **, p < 0.01; ****, p < 0.0001. WT-ND, IDH-wildtype, newly diagnosed tumor. WT-R, IDH-wildtype, recurrent tumor. MT-ND, IDH-mutant, newly diagnosed tumor. MT-R, IDH-mutant, recurrent tumor. Wilcoxon rank sum test, ns: not statistically significant; *, p < 0.05.
The TMZ-induced mutational signature (G:C > A:T), defined as signature 11, is often observed in post-TMZ recurrent gliomas and relevant to clinical management of glioma patient [6]. Touat et al. demonstrated that over 98% of post-treatment HMP gliomas showed signature 11 and that exposing MMR-deficient cells to TMZ induces HMP with signature 11, suggesting that HMP and signature 11 represent MMR deficiency and TMZ resistance [6]. In order to examine the prevalence of signature 11 in all tumors exposed to TMZ, we analyzed the 52 samples that were collected at disease recurrence in our cohort. Among all recurrent samples, 43 were from tumors that had prior exposure to TMZ or TMZ + radiotherapy (Table 1), and 42 of them had DNA samples. Among all samples, 15 of them demonstrated signature 11. Interestingly, 35.7% (15 of 42) samples exposed to TMZ developed signature 11, and 93.3% (14 of 15) were IDH-mutant gliomas. Of the other 27 samples without signature 11, 16 were IDH-mutant and 11 were IDH-wildtype. With the exposure to TMZ, 45.2% (14 out of 30) IDH-mutant tumors and 8.3% (1 out of 12) IDH-wildtype tumors developed signature 11, suggesting that the IDH-mutant tumors were more likely to harbor signature 11 following TMZ exposure (two-tailed Fisher’s exact test, p = 0.02).
2.4. TMB, Neoantigens, and Immune Signatures
Tumor neoantigens play a vital role in anti-tumor immunity. To better understand the immune landscape of gliomas, neoantigens from tumor samples in our cohort were predicted from mutations detected by WES of tumor DNA. In total, we found 1963 neoantigens (derived from 1325, 4.6% of all high confidence somatic variants) predicted to have a high binding affinity to human leukocyte antigen I (HLA-I) (IC50 < 500 nanomolar (nM)) and a lower HLA-I binding affinity (IC50 > 500 nM) to the corresponding wildtype peptides. Since immune cells must recognize neoantigens that are expressed and presented by HLA molecules on the tumor cell surface, we filtered out 619 expressed neoantigens from the predicted neoantigens by using a cut off total RNA read coverage ≥ 10, matched variant RNA read coverage ≥ 2 and VAF ≥ 0.1 (Table S3) (31.5%, 619/1963). As fusion genes are also a source of neoantigens, fusion gene-derived neoantigens were also included in the neoantigen calculation. In our samples, 20 high-confidence fusion gene-derived neoantigens were detected (IC50 < 500 nM). While the predicted neoantigens are directly derived from somatic mutations and are expected to correlate with TMB, we confirmed that the expressed neoantigens also have a statistically significant correlation with TMB in all samples (Pearson R = 0.52, p < 0.0001) (Figure 2B).
Although a correlation between TMB and the tumor immune response has been reported in other cancers, there is a discordance in gliomas [15]. Given the overall strong correlation between TMB and the expressed neoantigens, we next examined the correlation between TMB and tumor immune scores. In our cohort, TMB showed an inverse correlation with immune score in IDH-wildtype samples (R = −0.46, p = 0.03) (Figure 2C), and no correlation in IDH-mutant gliomas (Figure 2D), suggesting that the IDH mutation has an impact on the correlation of TMB and immune score.
To characterize the tumor immune microenvironment of HMP and NHMP in IDH-mutant and IDH-wildtype tumors, we performed ssGSEA using the transcriptomic data that was available for 60 samples in our sample cohort. Immune cell specific gene sets were used to calculate enrichment scores for infiltrating immune cell types and describe overall “immune signature score” in each sample [16]. Most of the immune cell infiltration scores for CD8 T cells, CD4 T cells, subtypes of dendritic cells, and macrophages were higher in IDH-wildtype samples compared to IDH-mutant samples (Figure 2E and Figure S1), which is similar to previous findings in primary gliomas from The Cancer Genome Atlas (TCGA) dataset [17]. It was also notable that several subsets of T cells and NK cells had a higher score in NHMP compared to HMP in IDH-wildtype tumors. However, no significant difference was observed between HMP and NHMP in IDH-mutant tumors (Figure S1). Overall, the immune signature clustered better by IDH mutation status, IDH-wildtype versus IDH-mutant, than by TMB, HMP versus NHMP (Figure 2E).
In order to better understand the immune signatures of the tumor microenvironment, we examined the infiltrating immune cell subtypes inferred by CIBERSORT scores [16]. As shown in Figure S2, regardless of IDH status or TMB, all glioma groups showed similarly high percentage of immune cells classified as M2 macrophages, but no significant difference between groups (one-way ANOVA test, p = 0.78) (Figure S3A). Monocytes and activated mast cells also had relatively high percentages (total average 13.9% and 12.7%, respectively) of infiltration compared to other immune cells such as CD8 T cells (total average 3.5%). Furthermore, there was no significant difference in CD8 T cell infiltration between HMP and NHMP samples, irrespective of IDH mutation status (One-way ANOVA test, p = 0.28) (Figure S3B). These data are consistent with previous findings that M2 macrophages are the dominant immune cell in the glioma microenvironment, whereas CD8 T cells are a minority [15]. In addition, the similar proportions of these immune cells across all groups are unlikely to explain the different correlations of TMB and immune scores in IDH-mutant and IDH-wildtype gliomas.
2.5. Antigen Processing and Presentation
Effective immune responses against tumors largely depend on immune cells recognizing antigens presented on the tumor surface. HLA-I loss and defects in the antigen processing machinery were reported to be common in various cancers, including gliomas [18,19,20,21]. To assess the ability of antigen presentation in gliomas, we first explored the expression of the major histocompatibility complex class I. As shown in Figure 3A, no significant difference in the expression levels of HLA-A, B, or C was found between HMP and NHMP samples in either IDH-mutant or IDH-wildtype tumors. These results suggest that HLA expression is unlikely to be the cause of the different correlation of TMB and immune scores in IDH-mutant and IDH-wildtype gliomas.
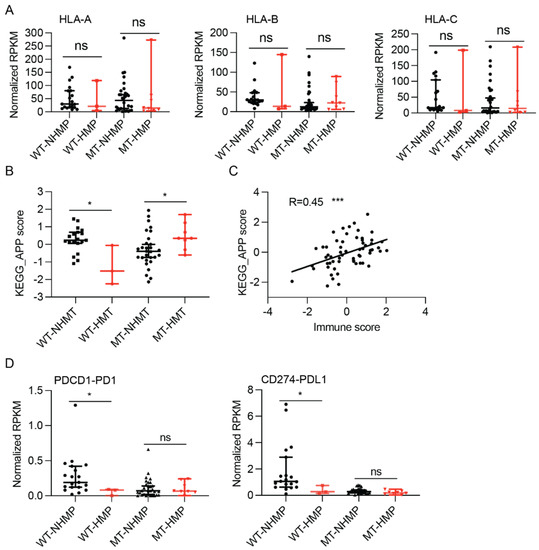
Figure 3.
Antigen processing and presentation (APP) and immunosuppressive gene expression in HMP and NHMP glioma samples. (A) No difference in expression of type I HLAs between HMP and NHMP samples is detected in either IDH-wildtype or IDH-mutant tumors. (B) APP score is higher in NHMP than HMP for IDH-wildtype glioma samples (p < 0.05), but higher in HMP than NHMP for IDH-mutant glioma samples (p < 0.05). (C) KEGG_APP score correlates with immune score (R = 0.45, p < 0.0001). (D) RNA expression level of PDCD1 and CD274 in HMP and NHMP in both IDH-mutant and wildtype gliomas. APP, antigen -processing and -presentation. RPKM, reads per kilobase of transcript per million reads mapped. Wilcoxon rank sum test, ns: not statistically significant. *, p < 0.05. ***, p < 0.001.
To further understand the discordance between neoantigen burden and immune infiltrate function in the tumor microenvironment, the KEGG Antigen Processing and Presentation (APP) score between HMP and NHMP samples was compared in both IDH-mutant and -wildtype gliomas. As shown in Figure 3B, the KEGG APP score was significantly higher in NHMP samples compared to HMP samples in IDH-wildtype tumor (median 0.2385 versus −1.518, p = 0.014). In contrast, a significantly higher KEGG APP score was found in HMP samples versus NHMP samples in IDH-mutant gliomas (median 0.35 versus −0.39, p = 0.03). To better understand the effect of APP score on the tumor microenvironment, the correlation between APP score and immune score was analyzed. As shown in Figure 3C, the APP score had a statistically significant correlation with immune score in gliomas (R = 0.45, p = 0.0003). These data indicate that APP function is different between HMP and NHMP samples with different IDH mutation status but correlates with immune score in our sample cohort.
2.6. Immunosuppressive Gene Expression in Gliomas
To understand the role of immunosuppressive factors in the tumor microenvironment of HMP and NHMP gliomas, we analyzed the expression of well-known immunosuppressive genes. The expression levels of most examined immunosuppressive genes did not show significant differences between HMP and NHMP samples in IDH-mutant gliomas, except for TGFB1, which had a trend of higher expression in NHMP IDH-mutant samples (median 4.34 versus 2.9, p = 0.054) (Figure S4). In IDH-wildtype gliomas, the immunosuppressive genes that showed a statistically significant difference in expression between NHMP and HMP samples were PD1 and PDL1 (median PD1: 0.19 versus 0.08, p = 0.04; PDL1: 1.06 versus 0.27, p = 0.04) (Figure 3D). These data suggest a potential therapeutic role of targeting TGFB1 and PD1/PDL1 in IDH-mutant and IDH-wildtype gliomas, respectively.
3. Discussion
TMB has been used as a predictive biomarker of response to immune checkpoint blockade therapy in several cancers, including melanoma and lung cancer [4]. However, a correlation between TMB and response to immunotherapy has not been observed in gliomas consistently [6]. In this study, we focused on a systematic assessment of the TMB, expressed neoantigens, and the tumor immune microenvironment in both IDH-wildtype and IDH-mutant gliomas, which have distinct tumor biology. Compared to IDH-wildtype glioma, IDH-mutant gliomas were more likely to accumulate mutation burden during their disease progression and more likely to harbor signature 11 following the exposure to TMZ. Most importantly, while TMB had a positive correlation with expressed neoantigens, it showed an inverse correlation with immune scores in IDH-wildtype gliomas and no correlation in IDH-mutant gliomas. In addition, we found a significantly higher APP score in NHMP compared to HMP samples in IDH-wildtype gliomas, but a higher APP score in HMP compared to NHMP in IDH-mutant gliomas. Together with the strong correlation between APP score and immune score, the data suggests that APP score could be further investigated as a biomarker for predicting response to immunotherapy, and that the impact of TMB on the immune signature depends on the IDH mutation status. Finally, we also analyzed germline alterations of CPGs, particularly P/LP genes, and explored the correlation with other tumor driver genes, such as IDH and TP53. Our results provide evidence for further evaluating P/LP germline variants in a larger glioma cohort and a potential value in screening patients prior to receiving treatment.
3.1. Germline Variants of P/LP CPGs in Gliomas
Despite the fact that we only analyzed a small cohort of glioma patients, 19% of them carried a germline monoallelic P/LP variant in CPG. The prevalence in our cohort is higher than what is reported in the literature in both pediatric and adult cancer patients [22,23]. To understand the spectrum of CPGs, particularly the P/LP mutations of CPGs in IDH-mutant and IDH-wildtype gliomas, we collected and analyzed germline genomic information in all cases. Although there was no statistically significant association between IDH mutation status and the occurrence of P/LP mutations, interesting observations between CPGs and somatic variants in the tumors were made. First, a germline TP53 mutation (p.R209Q) was detected in a patient with grade 3 astrocytoma (OM161). In addition to TP53 mutation and loss of heterozygosity (LOH), a frame-shift deletion of ATRX and somatic IDH1 mutation, which is considered a tumor driver gene in gliomas, were also detected in the tumor. IDH mutation was also detected in two patients with grade 4 astrocytoma (CL0095 and CL0332) who carried a monoallelic germline mutation of MUTYH (p.G396D), which is a common mutation in MUTYH-associated polyposis (MAP) with an autosomal recessive inheritance [24]. Another IDH-mutant grade 4 astrocytoma patient (CL0101) was found to have a monoallelic pathogenic nonsense BLM mutation (p.Q548X). Biallelic BLM mutation usually occurs in Bloom syndrome, which features abnormal DNA repair and high levels of chromosome breaks and rearrangements [25]. Evidently, IDH mutations frequently occurred in patients carrying P/LP germline mutations in our patient cohort. These observations raise a question about the role of another cancer driver gene such as IDH mutation in the presence of germline drivers such as TP53 mutation. Thus, it would be interesting to review the P/LP germline mutations of CPGs in a large cohort of IDH-mutant tumors. However, based on our available data, it may not be possible to determine with certainty which P/LP variants are incidental and therefore, less likely to contribute to the primary tumor diagnosis. For instance, while the TP53 variant reported in patient OM161 is likely causal of the patient’s astrocytoma, it would be less likely for a monoallelic MUTYH in patients CL0095 or CL00332 to contribute to their respective tumor diagnoses.
3.2. TMB, Immune Signatures, and IDH Mutation Status
There has been increasing evidence that TMB does not consistently correlate with immune response in gliomas [6,15] Our analysis revealed that TMB and immune scores are correlated differently in IDH-mutant and -wildtype gliomas. As summarized in Figure 4, in IDH-wildtype tumors, NHMP tumors have better APP function and immune scores than HMP tumors. In contrast, HMP tumors have higher APP function than NHMP, but not a better immune score, in IDH-mutant gliomas. Furthermore, APP score strongly correlates with immune score in all gliomas. While one would expect a higher TMB to result in a higher number of expressed neoantigens, in turn increasing the immune response, our findings revealed the opposite in the case of IDH-wildtype gliomas. A similar finding was described by Gromeier et al., who reported that IDH-wildtype samples with a lower TMB had higher immune inflammation, which was explained by the mechanism of neoantigen depletion/immunoediting [26]. Of note, we also found that the CD8 T cells showed a non-significant trend of being suppressed alongside APP suppression in the IDH-wildtype HMP gliomas (Figure S1), potentially dampening the immune response. Interestingly, while APP scores were elevated in the HMP subgroup in IDH-mutant gliomas, no significant correlation between the immune signature and TMB in this subset of patients was revealed. Therefore, it is possible that despite the high APP score in IDH-mutant HMP gliomas, 2-hydroxygluarate (2-HG) induces T cell suppression in some capacity. The production of this oncometabolite is a unique feature of IDH-mutant gliomas and has been previously shown to impair T cell activation and reduce T cell migration to the tumor site. Importantly, our ssGSEA data supported this because comparison of the CD8 T cell score in IDH-wildtype and -mutant gliomas revealed a significant suppression of these immune cells in the latter (Figure S5), consistent with findings from other studies [27].
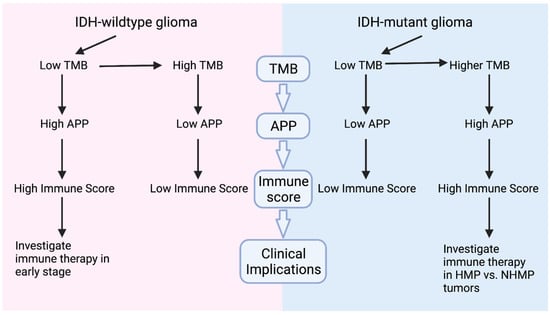
Figure 4.
Graphic summary of study findings. In IDH-wildtype tumors, NHMP tumors have higher APP and immune scores than HMP, suggesting a need to investigate potential benefit for immunotherapy in early stage of the disease. However, in IDH-mutant gliomas, HMP tumors have higher APP scores than NHMP. Despite a lack of correlation between TMB and immune scores, investigation of immune therapy in HMP versus HNMP glioma is warranted and ongoing.
3.3. Clinical Implications, Prospectives and Limitations
While conferring tumorigenesis, P/LP germline mutations may also provide important applications to aid patient management. For example, previous studies have shown that patients who are MSH6 mutation carriers should avoid treatment with alkylating agents such as TMZ [28]. In our patient cohort, we detected a MSH2 germline mutation in an IDH-wildtype glioblastoma patient (NCI0392), who was diagnosed with Lynch syndrome and had a de novo HMP brain tumor. In this case, alkylating agents such as TMZ should have been avoided if more treatment options were available to the patient. A patient with an IDH-mutant grade 4 astrocytoma (CL0301) was found to have a germline mutation in ERCC6, an important gene in the DNA double-stranded breaks (DSBs) repair pathway. This patient received more than 24 cycles of TMZ after the initial diagnosis of a lower grade astrocytoma, and both tumors from later recurrences were found to be HMP, harboring 2200 high confidence somatic mutations, which indicated a likely pathogenic function of this mutation and a potential role in HMP development when treated with TMZ. Although the association between the hypermutation phenotype and the presence of P/LP germline variants in the DDR pathway was not found to be statistically significant, investigation in a larger patient cohort is needed. Screening of those germline mutations of CPGs may provide insights to assist the clinical management of cancer patients.
In addition, further studying potential clinical biomarkers is vital for selecting patients who will benefit from immunotherapy. For an expressed neoantigen to elicit an immune response, a high APP score and HLA expression level are necessary. Currently, we do not completely understand why IDH-wildtype HMP gliomas show a decreased APP score. It is possible that critical genes are mutated at the time of development of HMP in IDH-WT tumors that disrupts APP and thus cause resistance to immunotherapy. This potential resistance mechanism can be further explored in a longitudinal study where matched tumor samples are collected and analyzed. Our findings of the correlation between APP function and immune score support testing the use of immunotherapy at an early stage of the disease for IDH-wildtype glioma patients when the TMB is low and APP function is high in a larger cohort study. The anti-PD1/PDL1 therapies may be valuable because of the increased expression level of PD1/PDL1 when TMB is relatively lower in the IDH-wildtype tumors. Interestingly, in the case of IDH-mutant gliomas, an opposite trend is seen, wherein a high APP score is seen in HMP gliomas, suggesting a potential value in considering IDH-mutant HMP gliomas for immunotherapy rather than their NHMP counterparts (Figure 4). While the findings of our study expand the knowledge of TMB, expressed neoantigens, and the tumor immune microenvironment and provided insights for clinical investigations, certain limitations are present. The conclusions are drawn from bioinformatic analyses of a sample cohort from a single institution. Further validation using in vitro and in vivo glioma models as well as larger cohort studies are thus warranted. Due to the retrospective nature of the study, the percentage of IDH-mutant glioma may not be representative of the incidence in the entire malignant glioma population. Nevertheless, our ongoing clinical trial (NCT 03718767) will provide prospectively collected data to further elucidate the correlation between TMB, expressed neoantigens, and tumor immune signatures.
4. Conclusions
TMB was inversely correlated with immune score in IDH-wildtype and showed no correlation in IDH-mutant gliomas. APP was correlated with immune score and may be further investigated as a biomarker for response to immunotherapy in gliomas. Studies of germline variants in a larger glioma cohort are warranted.
5. Materials and Methods
5.1. Patients and Samples
Adult patients with primary malignant brain tumors, who were evaluated at the Neuro-Oncology Branch, Center for Cancer Research, National Cancer Institute (NCI), were enrolled in NCI 16-C-0151 (NCT02851706), NCI 19-C-0006 (NCT03718767), and NCI 10-C-0086 (NCT01109394). The protocols were approved by the Institutional Review Board of the National Institutes of Health. Written consents were obtained from all patients. Both matched whole blood and brain tumor samples were collected and analyzed using the ClinOmics platform, a clinical next-generation sequencing program at NCI [29]. Tumor samples were only collected for sequencing if sufficient tissue for clinical diagnosis was available. The schema of overall experimental approach for this study is summarized in Figure S6.
5.2. mRNA Sequencing (RNAseq)
Tumor RNA was extracted from Formalin Fixed Paraffin-Embedded (FFPE) tumor sections by the Rneasy FFPE kit (Qiagen, Germantown, MD, USA). RNA libraries were prepared by using Illumina TruSeq RNA Access Library Preparation Kit according to the manufacturer’s protocol (TruSeq RNA Exome kits; Illumina, San Diego, CA, USA). The sequencing was performed on Illumina NextSeq500 (Illumina) according to the manufacturer’s protocols. Samples were sequenced at a depth of 40 million reads per sample. All the RNAseq data was processed by using an RNAseq data analysis pipeline, where reads were mapped to the ENSEMBL human genome GRCh37 build 71 using STAR. Single-sample Gene Set Enrichment Analysis (ssGSEA) was used for the generation of immune cell infiltration scores, immune scores, and antigen processing and presentation scores based on the previously published gene sets [16,30]. CIBERSORT was used to analyze the proportions of immune cells [29].
5.3. Whole Exome Sequencing
Tumor DNA was extracted from FFPE samples. Genomic DNA, which was used as germline exome sequencing, was extracted from the peripheral blood cells of individual patients. The exome was enriched by using SureSelect Clinical Research Exome Kits according to the manufacturer’s instructions (Agilent, Santa Clara, CA, USA). The prepared samples were sequenced on Illumina NextSeq500 (Illumina). Reportable germline mutations, which is defined as actionable genomic alterations to be targeted by the FDA approved drugs or clinical trials, were filtered out by in-house criteria [29]. TMB was defined as the number of somatic mutations in the coding region per Mb, which contain single nucleotide variants (SNVs), small insertions and deletions (INDELs) (usually less than 20 bases). TMB was calculated as indicated in the previous report [31].
5.4. Identification of Somatic Mutation
The bcl files of exome sequencing were converted to FASTQ files by using the bcl2fastq tool in CASAVA (Illumina). The sequences were then mapped to the human reference genome GRCH37 by using a customized NCI ClinOmics Bioinformatic Pipeline v3.2. MuTect and Strelka were used for somatic single nucleotide variant (SNV) and small indel calling respectively. The Genome Analysis Toolkit (GATK) and HaplotypeCaller (HAPLOC) for germline SNV and indel callings as previously described. High confidence somatic mutations were called by using the cutoffs: (1) tumor total coverage ≥20×, (2) normal total coverage ≥20× and (3) variant allele frequency (VAF) ≥0.10. Using these parameters, our assay has a high sensitivity of 100% and a positive predictive value (PPV) of 90% for the exome sequencing.
5.5. Neoantigen Prediction from Mutations and Fusions, and Expressed Neoantigen Computation
The high confidence of somatic mutations was used for the neoantigen prediction according to the previous report [32]. The amino acid change and the transcript peptide sequence were annotated by seq2HLA v2.2, HLAminer_v1.3.1, in-house developed script consensusHLA.pl, consencusSomaticsVCF, pl, VEP v.86, pvacseqtools 1.3.5. NeoFuse v1.1.1 was used for the prediction of fusion neoantigens. NeoFuse internally runs OptiType for genotyping of class-1 HLA and Arriba for predicting of fusion peptides and MHC flurry for binding affinity prediction. The neoantigen candidates with a mutant HLA type I binding score (IC50) lower than 500 nM, and a corresponding wild type binding IC50 of greater than 500 nM were selected as predicted neoantigens. The expressed high confidence neoantigens from somatic mutations were called based on the high confidence neoantigens from somatic mutations by further using the cutoffs: (1) total RNA read coverage ≥ 10, (2) matched variant RNA read coverage ≥ 2, (3) VAF ≥ 0.1. The total expressed neoantigen load was calculated by adding the high confidence expressed neoantigen mutation and high confidence neoantigen from fusion.
5.6. Statistical Analysis
Wilcoxon rank sum test was used for differential analyses between two subgroups. One-way ANOVA test was used in the comparison of more than two groups. Categorical variables were compared using Fisher’s exact test. All statistical analyses were performed by using GraphPad Prism software (Version 8, GraphPad Software, Inc., San Diego, CA, USA). p value < 0.05 was considered significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
5.7. Data Availability
All Data has been deposited in dbGaP and RNAseq and Somatic Data is available on an online database (https://clinomics.ccr.cancer.gov/clinomics/public/login, accessed date: 20 November 2021).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cancers13236092/s1, Figure S1. ssGSEA scores of immune cells, stromal score and immune score between HMP and NHMP samples with IDH status. MT, IDH-mutant, WT, IDH-wildtype. Figure S2. Different immune cell proportions are analyzed in HMP and NHMP samples by CIBERSORT. Figure S3. Comparison of percentages of M2 macrophages and CD8 T cells in HMP and NHMP samples. Figure S4. Immunosuppressive gene expressions are analyzed between HMP and NHMP samples. Figure S5. CD8 T cell scores in IDH-wildtype are higher than in IDH-mutant glioma. Figure S6. Schema of the experimental approach. Table S1. Sample cohort information. Table S2. HMP tumors. Table S3. High confidence expressed neoantigens from mutations.
Author Contributions
Conceptualization, G.Y., Y.P., J.K. and J.W.; Methodology, G.Y., Y.P., C.K., J.K. and J.W.; Software, V.G., A.A., H.-C.C., X.W. and S.S.; Formal Analysis, G.Y., Y.P., C.K., V.G., A.A., H.-C.C., J.K. and J.W.; Investigation, all authors; Resources, R.N.K., T.S.A., M.R.G., K.A., J.K. and J.W.; Data Curation, G.Y., Y.P., M.M., C.K., V.G., A.A., A.R., J.S.W., H.-C.C., X.W., S.S. and L.X.; Writing—Original Draft Preparation, G.Y., Y.P., M.M. and J.W.; Writing—Review & Editing, M.M., C.K., J.S.W., A.R., O.K. and J.K.; Supervision, J.K. and J.W.; Funding Acquisition, J.K. and J.W. All authors have read and agreed to the published version of the manuscript.
Funding
Supported by the NIH Lasker Clinical Research Scholars Program and NCI Intramural Research Program (1ZIABC011841 and 1ZIABC011840).
Institutional Review Board Statement
The study protocols for sample collection were reviewed and approved by the NCI Institutional Review Board. (Protocol code and date of approval, NCT02851706 2 August 2016, NCT03718767 24 October 2018, and NCT01109394 23 April 2010).
Informed Consent Statement
Informed consent was waived for data analysis report, due to all samples used in this analysis are deidentified.
Data Availability Statement
All Data has been deposited in dbGaP and RNAseq and Somatic Data is available on an online database (https://clinomics.ccr.cancer.gov/clinomics/public/login, accessed date: 20 November 2021).
Acknowledgments
This research was supported by the NIH Intramural Research Program and Lasker Clinical Research Scholar Program. We thank all the patients for participating in and supporting the clinical studies, which ultimately help other patients with brain tumors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Prasad, V.; Addeo, A. The FDA approval of pembrolizumab for patients with TMB >10 mut/Mb: Was it a wise decision? No. Ann. Oncol. 2020, 31, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.J.N. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Ranjan, A.; Pang, Y.; Yu, G.; Kim, O.; Khan, J.; Wu, J. Tumor mutational burden and immunotherapy in gliomas. Trends Cancer 2021, 7, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Turkalp, Z.; Karamchandani, J.; Das, S. IDH mutation in glioma: New insights and promises for the future. JAMA Neurol. 2014, 71, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Roper, N.; Brown, A.L.; Wei, J.S.; Pack, S.; Trindade, C.; Kim, C.; Restifo, O.; Gao, S.; Sindiri, S.; Mehrabadi, F.; et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep. Med. 2020, 1, 100007. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; Yu, G.; Butler, M.; Sindiri, S.; Song, Y.K.; Wei, J.S.; Wen, X.; Chou, H.C.; Quezado, M.; Pack, S.; et al. Report of Canonical BCR-ABL1 Fusion in Glioblastoma. JCO Precis. Oncol. 2021, 5, PO.20.00519. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Long, W.; Ma, Q.; Xiao, K.; Li, Y.; Xiao, Q.; Peng, G.; Yuan, J.; Liu, Q. Identification of a Tumor Microenvironment-Related Eight-Gene Signature for Predicting Prognosis in Lower-Grade Gliomas. Front. Genet. 2019, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehling, M.; Simon, P.; Mittelbronn, M.; Meyermann, R.; Ferrone, S.; Weller, M.; Wiendl, H. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: Does it reflect a potential immune escape mechanism? Acta Neuropathol. 2007, 114, 111–119. [Google Scholar] [CrossRef]
- Facoetti, A.; Nano, R.; Zelini, P.; Morbini, P.; Benericetti, E.; Ceroni, M.; Campoli, M.; Ferrone, S. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin. Cancer Res. 2005, 11, 8304–8311. [Google Scholar] [CrossRef] [Green Version]
- Yeung, J.T.; Hamilton, R.L.; Ohnishi, K.; Ikeura, M.; Potter, D.M.; Nikiforova, M.N.; Ferrone, S.; Jakacki, R.I.; Pollack, I.F.; Okada, H. LOH in the HLA class I region at 6p21 is associated with shorter survival in newly diagnosed adult glioblastoma. Clin. Cancer Res. 2013, 19, 1816–1826. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 2021, 11, 1–20. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Pitroski, C.E.; Cossio, S.L.; Koehler-Santos, P.; Graudenz, M.; Prolla, J.C.; Ashton-Prolla, P. Frequency of the common germline MUTYH mutations p.G396D and p.Y179C in patients diagnosed with colorectal cancer in Southern Brazil. Int. J. Colorectal Dis. 2011, 26, 841–846. [Google Scholar] [CrossRef]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef]
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; He, Y.; Desjardins, A.; et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat. Commun. 2021, 12, 352. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.H.; Mansour, S.; Pritchard-Jones, K.; Kumar, D.; MacSweeney, F.; Rahman, N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat. Clin. Pract. Oncol. 2007, 4, 130–134. [Google Scholar] [CrossRef]
- Chang, W.; Brohl, A.S.; Patidar, R.; Sindiri, S.; Shern, J.F.; Wei, J.S.; Song, Y.K.; Yohe, M.E.; Gryder, B.; Zhang, S.; et al. Multidimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clin. Cancer Res. 2016, 22, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).