
Mismatch Repair Deficiency and Somatic Mutations in Human Sinonasal Tumors

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Specimens
2.2. Sinonasal Cancer Cell Lines
2.3. Immunohistochemistry of Mismatch Repair (MMR) Proteins
2.4. Targeted Panel NGS
2.5. Microsatellite Instability (MSI) Analysis
2.6. Whole Exome Sequencing of Sinonasal Cancer Cell Lines
2.7. Statistical Analysis and Visualization
3. Results
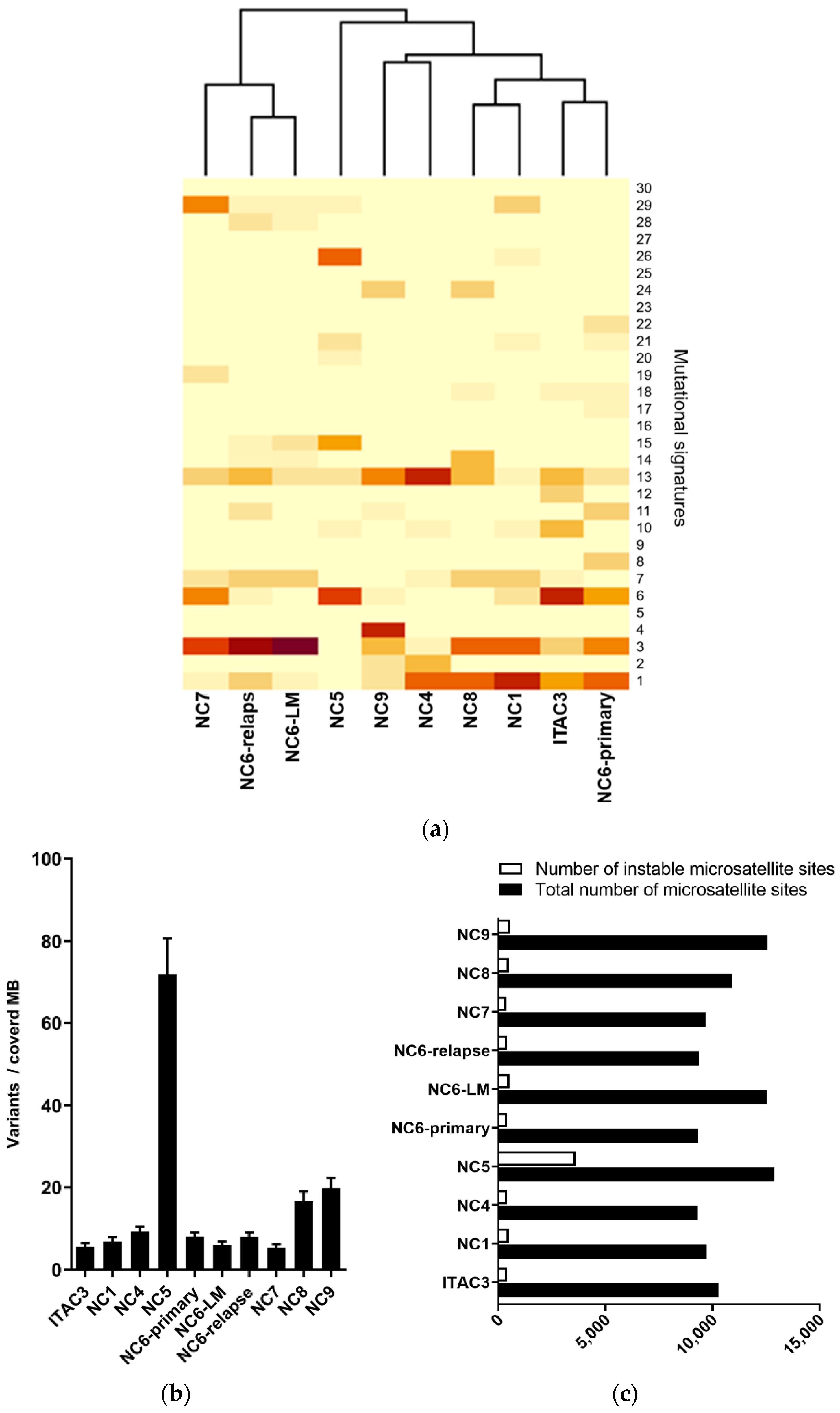
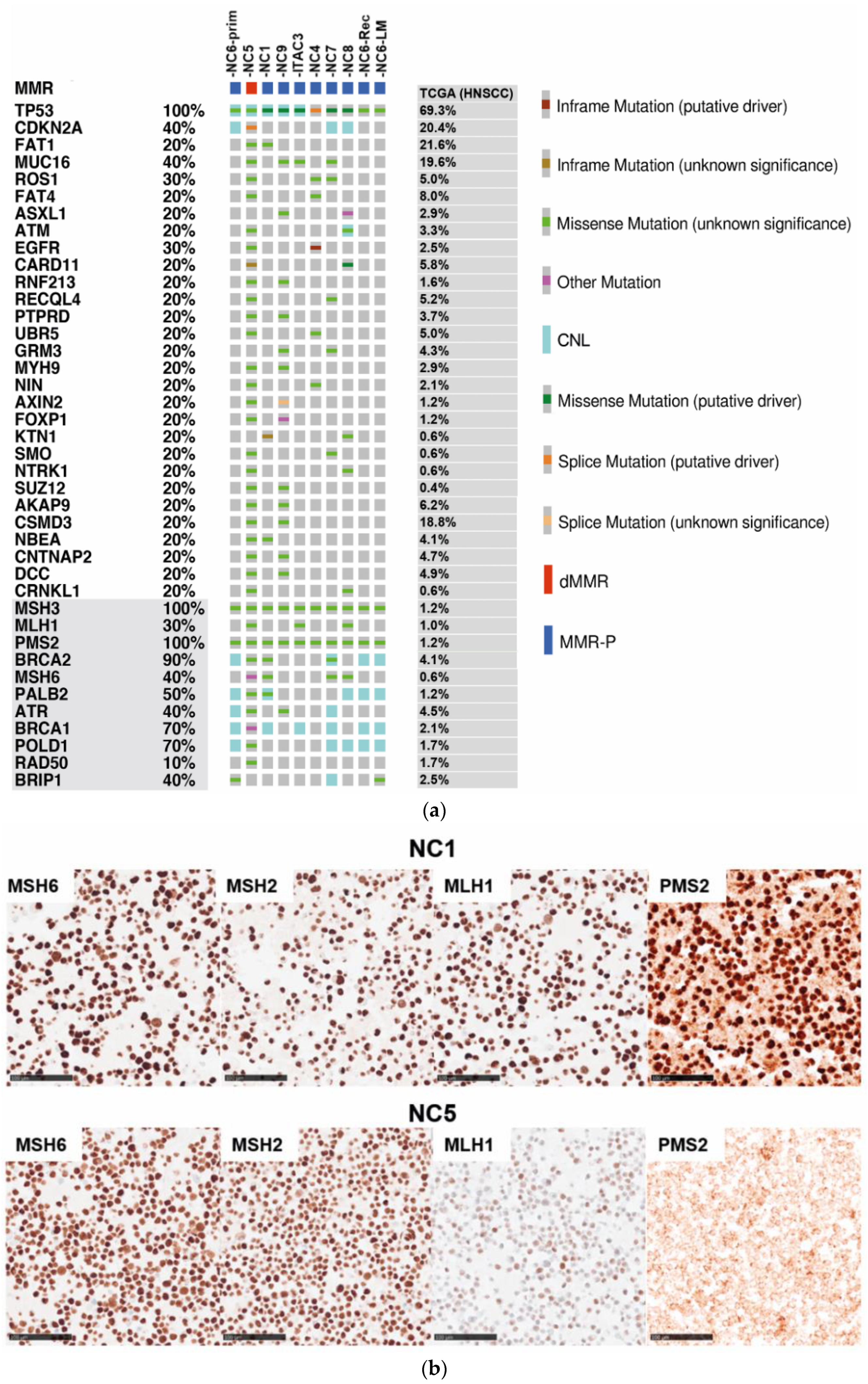
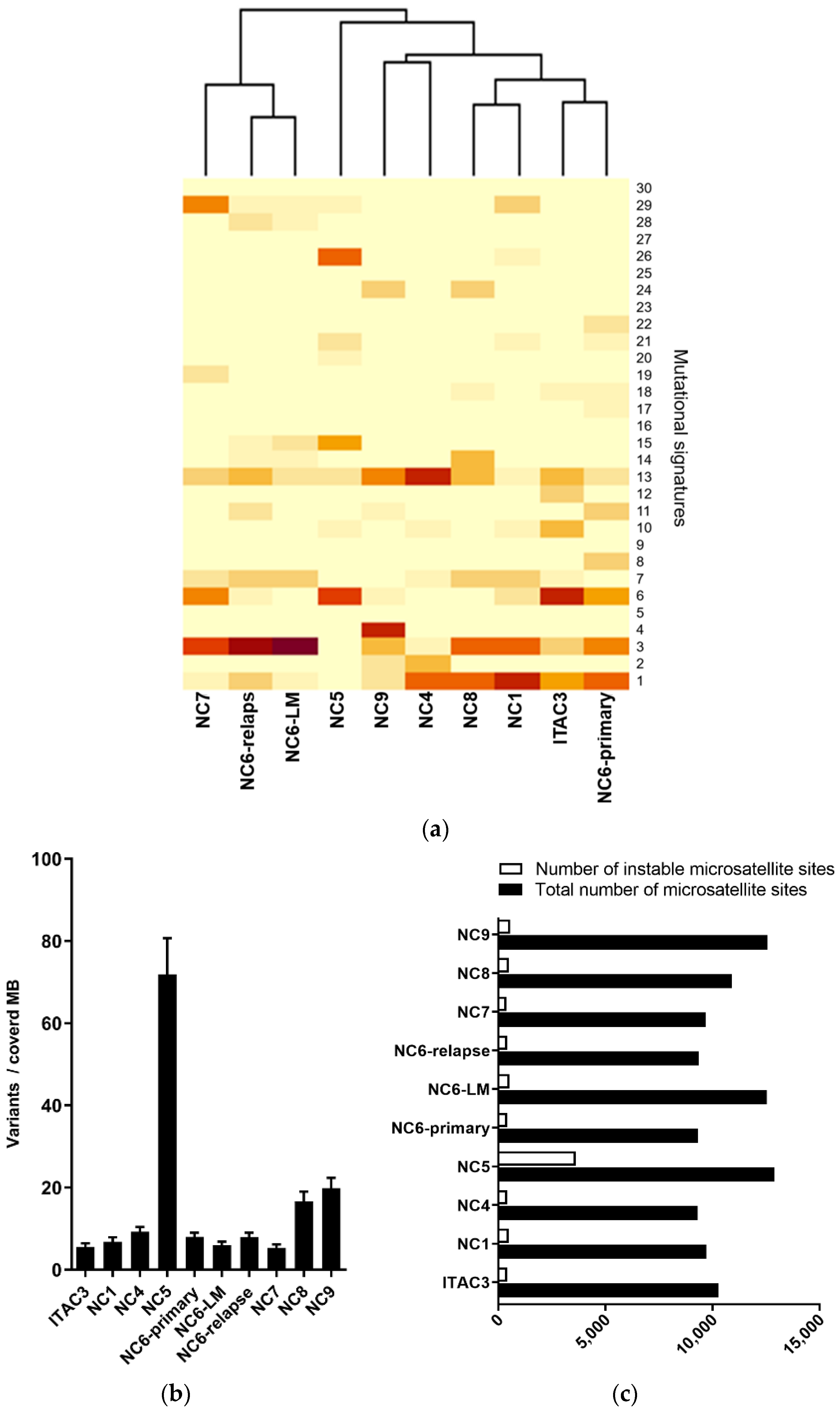
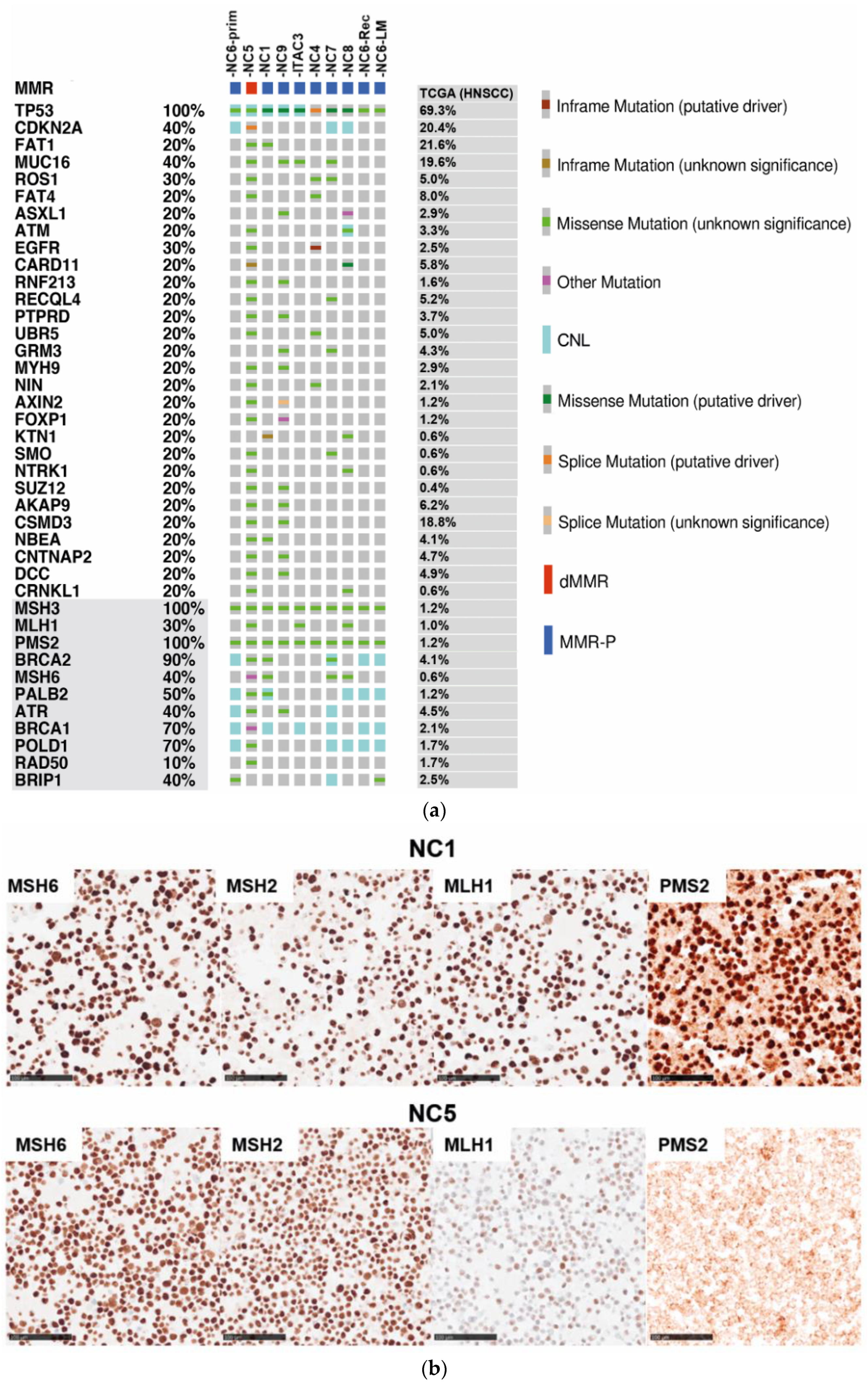
3.1. Genomic Profiling of Cell Lines Revealed a MMR Deficient SNSCC
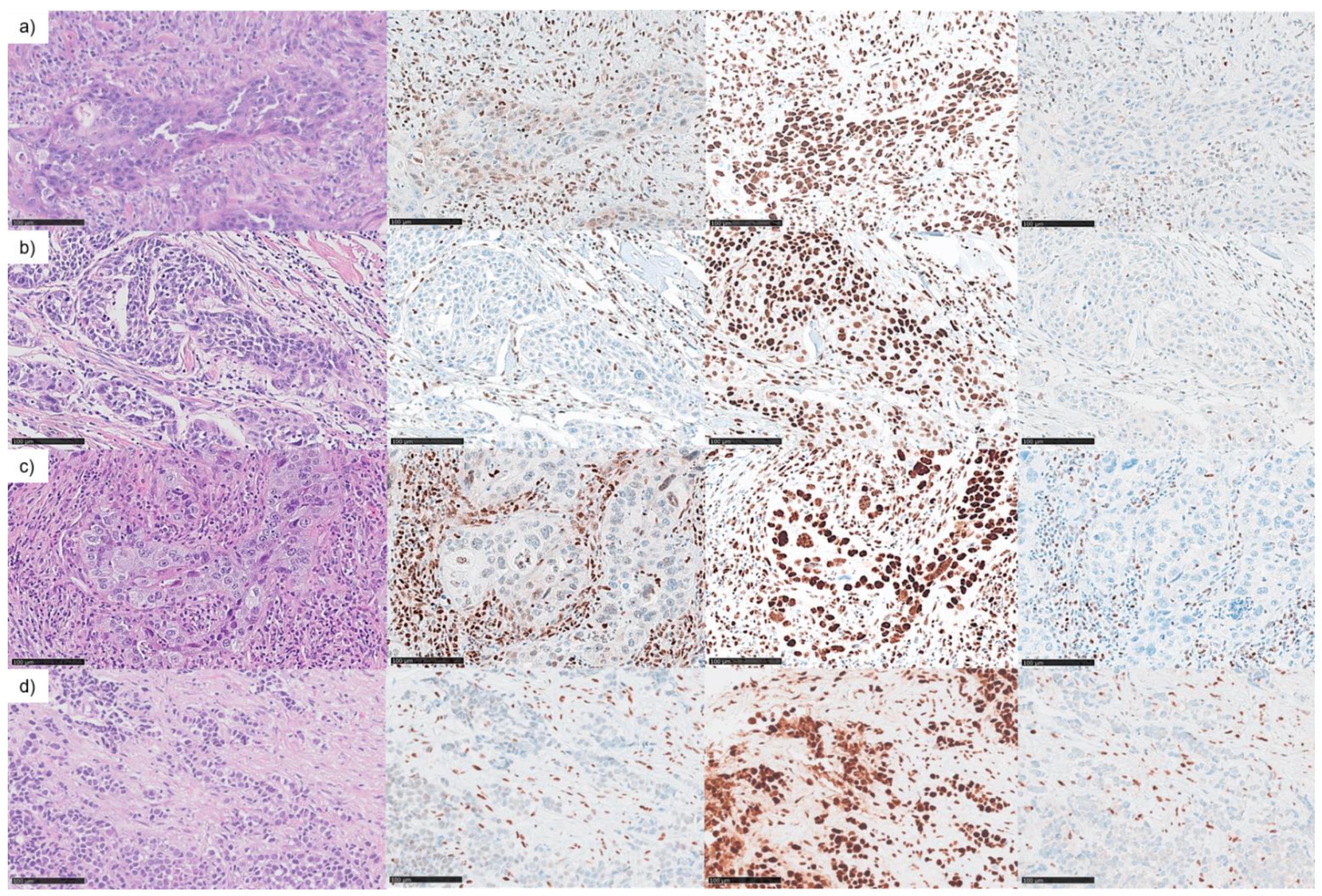
3.2. Mismatch Repair Deficiency Based on Somatic MHL1 Mutation
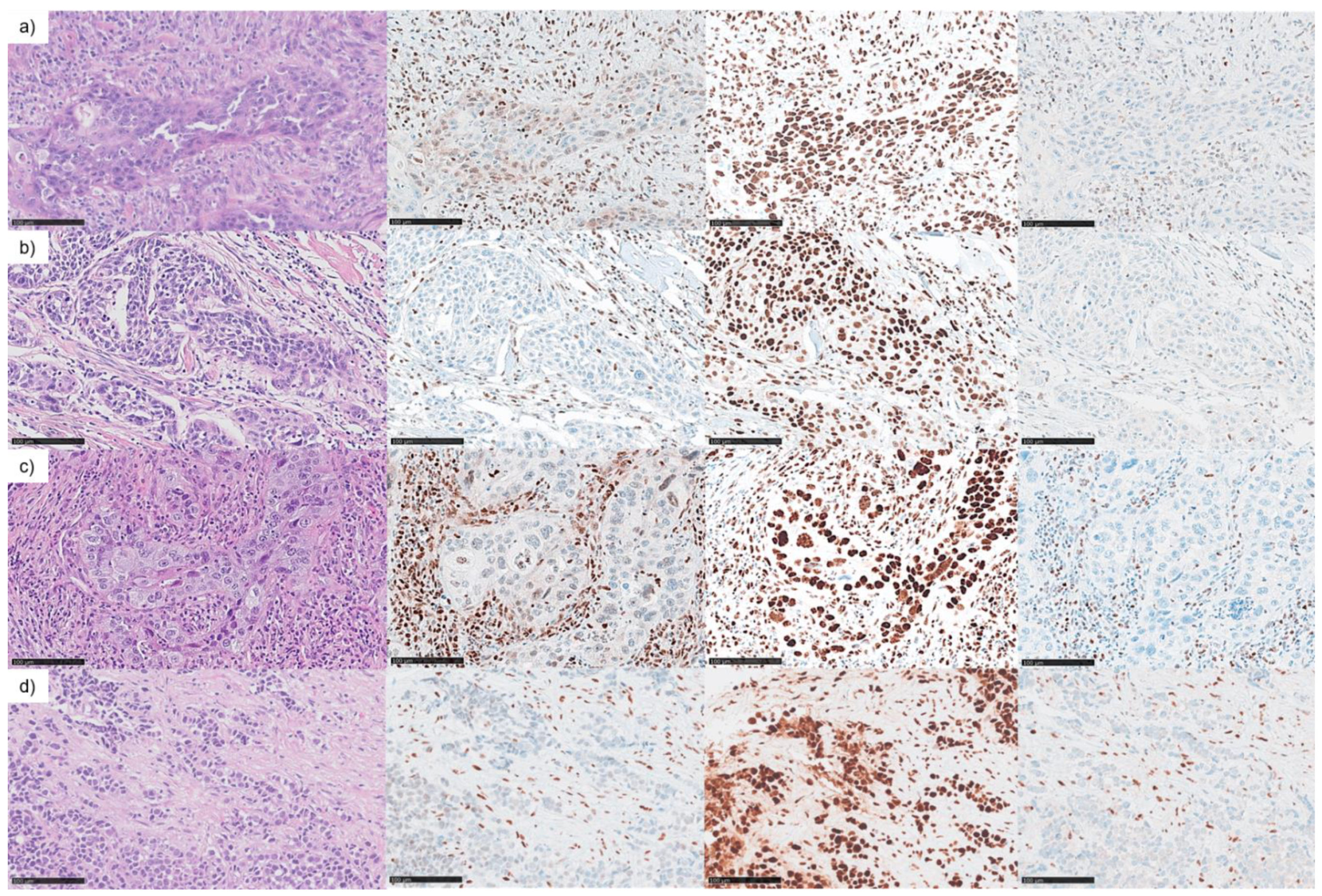
3.3. MMR/MSI Analysis Elucidated further dMMR/MSI-H SNSCC
3.4. Targeted Panel Analysis of Cancer Biomarkers in Sinonasal Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Youlden, D.R.; Cramb, S.M.; Peters, S.; Porceddu, S.V.; Møller, H.; Fritschi, L.; Baade, P.D. International comparisons of the incidence and mortality of sinonasal cancer. Cancer Epidemiol. 2013, 37, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Cantu, G.; Solero, C.L.; Mariani, L.; Lo Vullo, S.; Riccio, S.; Colombo, S.; Pompilio, M.; Perrone, F.; Formillo, P.; Quattrone, P. Intestinal type adenocarcinoma of the ethmoid sinus in wood and leather workers: A retrospective study of 153 cases. Head Neck 2011, 33, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, S.; Khan, M.N.; Patel, N.R.; Yeldandi, S.; Baredes, S.; Eloy, J.A. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4994 patients. Laryngoscope 2014, 124, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Llorente, J.L.; López, F.; Suárez, C.; Hermsen, M.A. Sinonasal carcinoma: Clinical, pathological, genetic and therapeutic advances. Nat. Rev. Clin. Oncol. 2014, 11, 460–472. [Google Scholar] [CrossRef]
- Chatelet, F.; Simon, F.; Bedarida, V.; Le Clerc, N.; Adle-Biassette, H.; Manivet, P.; Herman, P.; Verillaud, B. Surgical Management of Sinonasal Cancers: A Comprehensive Review. Cancers 2021, 13, 3995. [Google Scholar] [CrossRef]
- Castelnuovo, P.; Lambertoni, A.; Sileo, G.; Valentini, M.; Karligkiotis, A.; Battaglia, P.; Turri-Zanoni, M. Critical review of multidisciplinary approaches for managing sinonasal tumors with orbital involvement. Acta Otorhinolaryngol. Ital. 2021, 41, S76–S89. [Google Scholar] [CrossRef]
- Ferrari, M.; Taboni, S.; Carobbio, A.L.C.; Emanuelli, E.; Maroldi, R.; Bossi, P.; Nicolai, P. Sinonasal Squamous Cell Carcinoma, a Narrative Reappraisal of the Current Evidence. Cancers 2021, 13, 2835. [Google Scholar] [CrossRef]
- Turner, J.H.; Reh, D.D. Incidence and survival in patients with sinonasal cancer: A historical analysis of population-based data. Head Neck 2012, 34, 877–885. [Google Scholar] [CrossRef]
- Wieneke, J.A.; Thompson, L.D.; Wenig, B.M. Basaloid squamous cell carcinoma of the sinonasal tract. Cancer 1999, 85, 841–854. [Google Scholar] [CrossRef]
- Rytkönen, A.E.; Hirvikoski, P.P.; Salo, T.A. Lymphoepithelial carcinoma: Two case reports and a systematic review of oral and sinonasal cases. Head Neck Pathol. 2011, 5, 327–334. [Google Scholar] [CrossRef] [Green Version]
- West, R.B.; Kong, C.; Clarke, N.; Gilks, T.; Lipsick, J.S.; Cao, H.; Kwok, S.; Montgomery, K.D.; Varma, S.; Le, Q.T. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am. J. Surg. Pathol. 2011, 35, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.A.; Ogawa, T.; Stelow, E.B.; Moskaluk, C.A.; Koch, W.M.; Pai, S.I.; Westra, W.H. Human papillomavirus-related carcinoma with adenoid cystic-like features: A peculiar variant of head and neck cancer restricted to the sinonasal tract. Am. J. Surg. Pathol. 2013, 37, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, J.-F.; Hsieh, M.-S.; Li, W.-Y.; Chen, J.-Y.; Lin, S.-Y.; Liu, S.-H.; Pan, C.-C.; Kuo, Y.-J. Human papillomavirus-related carcinoma with adenoid cystic-like features: A series of five cases expanding the pathological spectrum. Histopathology 2017, 71, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.A.; Westra, W.H. NUT Midline Carcinomas of the Sinonasal Tract. Am. J. Surg. Pathol. 2012, 36, 1216–1221. [Google Scholar] [CrossRef] [Green Version]
- Haack, H.; Johnson, L.A.; Fry, C.J.; Crosby, K.; Polakiewicz, R.D.; Stelow, E.B.; Hong, S.M.; Schwartz, B.E.; Cameron, M.J.; Rubin, M.A.; et al. Diagnosis of NUT midline carcinoma using a NUT-specific monoclonal antibody. Am. J. Surg. Pathol. 2009, 33, 984–991. [Google Scholar] [CrossRef] [Green Version]
- López-Hernández, A.; Vivanco, B.; Franchi, A.; Bloemena, E.; Cabal, V.N.; Potes, S.; Riobello, C.; García-Inclán, C.; López, F.; Llorente, J.L.; et al. Genetic profiling of poorly differentiated sinonasal tumours. Sci. Rep. 2018, 8, 3998. [Google Scholar] [CrossRef] [Green Version]
- Agaimy, A.; Hartmann, A.; Antonescu, C.R.; Chiosea, S.I.; El-Mofty, S.K.; Geddert, H.; Iro, H.; Lewis, J.S., Jr.; Märkl, B.; Mills, S.E.; et al. SMARCB1 (INI-1)-deficient Sinonasal Carcinoma: A Series of 39 Cases Expanding the Morphologic and Clinicopathologic Spectrum of a Recently Described Entity. Am. J. Surg. Pathol. 2017, 41, 458–471. [Google Scholar] [CrossRef] [Green Version]
- Abdelmeguid, A.S.; Bell, D.; Hanna, E.Y. Neuroendocrine Carcinoma and Sinonasal Undifferentiated Carcinoma. Adv. Oto-Rhino-Laryngol. 2020, 84, 168–184. [Google Scholar] [CrossRef]
- Hongo, T.; Yamamoto, H.; Jiromaru, R.; Nozaki, Y.; Yasumatsu, R.; Hashimoto, K.; Yoneda, R.; Sugii, A.; Taguchi, K.; Masuda, M.; et al. Clinicopathologic Significance of EGFR Mutation and HPV Infection in Sinonasal Squamous Cell Carcinoma. Am. J. Surg. Pathol. 2021, 45, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Cabal, V.N.; Menendez, M.; Vivanco, B.; Potes-Ares, S.; Riobello, C.; Suarez-Fernandez, L.; Garcia-Marin, R.; Blanco-Lorenzo, V.; Lopez, F.; Alvarez-Marcos, C.; et al. EGFR mutation and HPV infection in sinonasal inverted papilloma and squamous cell carcinoma. Rhinology 2020, 58, 368–376. [Google Scholar] [CrossRef]
- Jiromaru, R.; Yamamoto, H.; Yasumatsu, R.; Hongo, T.; Nozaki, Y.; Hashimoto, K.; Taguchi, K.; Masuda, M.; Nakagawa, T.; Oda, Y. HPV-related Sinonasal Carcinoma: Clinicopathologic Features, Diagnostic Utility of p16 and Rb Immunohistochemistry, and EGFR Copy Number Alteration. Am. J. Surg. Pathol. 2020, 44, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Schlussel Markovic, E.; Marqueen, K.E.; Sindhu, K.K.; Lehrer, E.J.; Liu, J.; Miles, B.; Genden, E.; Sharma, S.; Gupta, V.; Westra, W.; et al. The prognostic significance of human papilloma virus in sinonasal squamous cell carcinoma. Laryngoscope Investig. Otolaryngol. 2020, 5, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Elgart, K.; Faden, D.L. Sinonasal Squamous Cell Carcinoma: Etiology, Pathogenesis, and the Role of Human Papilloma Virus. Curr. Otorhinolaryngol. Rep. 2020, 8, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; McHugh, J.B.; Goudsmit, C.M.; Weigelin, H.C.; Lim, M.S.; Elenitoba-Johnson, K.S.J.; Betz, B.L.; Carey, T.E.; Brown, N.A. Human papillomavirus (HPV) and somatic EGFR mutations are essential, mutually exclusive oncogenic mechanisms for inverted sinonasal papillomas and associated sinonasal squamous cell carcinomas. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 466–471. [Google Scholar] [CrossRef]
- Sasaki, E.; Nishikawa, D.; Hanai, N.; Hasegawa, Y.; Yatabe, Y. Sinonasal squamous cell carcinoma and EGFR mutations: A molecular footprint of a benign lesion. Histopathology 2018, 73, 953–962. [Google Scholar] [CrossRef]
- Udager, A.M.; Rolland, D.C.M.; McHugh, J.B.; Betz, B.L.; Murga-Zamalloa, C.; Carey, T.E.; Marentette, L.J.; Hermsen, M.A.; DuRoss, K.E.; Lim, M.S.; et al. High-Frequency Targetable EGFR Mutations in Sinonasal Squamous Cell Carcinomas Arising from Inverted Sinonasal Papilloma. Cancer Res. 2015, 75, 2600. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.A.; Plouffe, K.R.; Yilmaz, O.; Weindorf, S.C.; Betz, B.L.; Carey, T.E.; Seethala, R.R.; McHugh, J.B.; Tomlins, S.A.; Udager, A.M. TP53 mutations and CDKN2A mutations/deletions are highly recurrent molecular alterations in the malignant progression of sinonasal papillomas. Mod. Pathol. 2021, 34, 1133–1142. [Google Scholar] [CrossRef]
- Sánchez-Fernández, P.; Riobello, C.; Costales, M.; Vivanco, B.; Cabal, V.N.; García-Marín, R.; Suárez-Fernández, L.; López, F.; Cabanillas, R.; Hermsen, M.A.; et al. Next-generation sequencing for identification of actionable gene mutations in intestinal-type sinonasal adenocarcinoma. Sci. Rep. 2021, 11, 2247. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.-F.; Lou, P.-J. Immune checkpoint inhibitors for head and neck squamous cell carcinoma: Current landscape and future directions. Head Neck 2019, 41, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Vermaut, C.; Buisine, M.-P. Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors. Cancers 2021, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 38, 1–10. [Google Scholar] [CrossRef]
- Watt, S.A.; Purdie, K.J.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; McHugh, A.T.; Xue, D.J.; Dayal, J.H.; Proby, C.M.; Harwood, C.A.; et al. Novel CARD11 Mutations in Human Cutaneous Squamous Cell Carcinoma Lead to Aberrant NF-κB Regulation. Am. J. Pathol. 2015, 185, 2354–2363. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.F.; Huebbers, C.U.; Siefer, O.G.; Vent, J.; Engbert, I.; Eslick, G.D.; Valter, M.; Klussmann, J.P.; Preuss, S.F. Prevalence and risk factors for oral human papillomavirus infection in 129 women screened for cervical HPV infection. Oral Oncol. 2014, 50, 27–31. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; García Martínez, J.; García-Inclán, C.; Vivanco, B.; Costales, M.; Álvarez Marcos, C.; Llorente, J.L.; Hermsen, M.A. Establishment and genetic characterization of an immortal tumor cell line derived from intestinal-type sinonasal adenocarcinoma. Cell. Oncol. 2011, 34, 23–31. [Google Scholar] [CrossRef]
- Siemanowski, J.; Schömig-Markiefka, B.; Buhl, T.; Haak, A.; Siebolts, U.; Dietmaier, W.; Arens, N.; Pauly, N.; Ataseven, B.; Büttner, R.; et al. Managing Difficulties of Microsatellite Instability Testing in Endometrial Cancer-Limitations and Advantages of Four Different PCR-Based Approaches. Cancers 2021, 13, 1268. [Google Scholar] [CrossRef]
- Heydt, C.; Rehker, J.; Pappesch, R.; Buhl, T.; Ball, M.; Siebolts, U.; Haak, A.; Lohneis, P.; Büttner, R.; Hillmer, A.M.; et al. Analysis of tumor mutational burden: Correlation of five large gene panels with whole exome sequencing. Sci. Rep. 2020, 10, 11387. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Yang, X.; Guo, L.; Liu, B.; Lin, J.; Liang, H.; Sun, J.; Zhang, C.; Ye, K. MSIsensor-pro: Fast, Accurate, and Matched-normal-sample-free Detection of Microsatellite Instability. Genom. Proteom. Bioinform. 2020, 18, 65–71. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Vila-Casadesús, M.; Franch-Expósito, S.; Hernández-Illán, E.; Lozano, J.J.; Castellví-Bel, S. Mutational Signatures in Cancer (MuSiCa): A web application to implement mutational signatures analysis in cancer samples. BMC Bioinform. 2018, 19, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational signatures of ionizing radiation in second malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.E.; Adjei, N.N.; Khadraoui, W.; Altwerger, G. Defective mismatch repair associated mutational signatures, a prognostic and predictive biomarker in endometrial cancer. J. Clin. Oncol. 2021, 39, 5528. [Google Scholar] [CrossRef]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef]
- Li, L.; Wang, X.L.; Lei, Q.; Sun, C.Z.; Xi, Y.; Chen, R.; He, Y.W. Comprehensive immunogenomic landscape analysis of prognosis-related genes in head and neck cancer. Sci. Rep. 2020, 10, 6395. [Google Scholar] [CrossRef]
- Räschle, M.; Dufner, P.; Marra, G.; Jiricny, J. Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J. Biol. Chem. 2002, 277, 21810–21820. [Google Scholar] [CrossRef] [Green Version]
- Oliver, J.R.; Lieberman, S.M.; Tam, M.M.; Liu, C.Z.; Li, Z.; Hu, K.S.; Morris, L.G.T.; Givi, B. Human papillomavirus and survival of patients with sinonasal squamous cell carcinoma. Cancer 2020, 126, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Chang Sing Pang, K.J.W.; Mur, T.; Collins, L.; Rao, S.R.; Faden, D.L. Human Papillomavirus in Sinonasal Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. Cancers 2020, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.J.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018, 81, 45–51. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.J.; Soria, A.; Machiels, J.P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Sacco, A.G.; Chen, R.; Worden, F.P.; Wong, D.J.L.; Adkins, D.; Swiecicki, P.; Chai-Ho, W.; Oppelt, P.; Ghosh, D.; Bykowski, J.; et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: An open-label, multi-arm, non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2021, 22, 883–892. [Google Scholar] [CrossRef]
- Joost, P.; Veurink, N.; Holck, S.; Klarskov, L.; Bojesen, A.; Harbo, M.; Baldetorp, B.; Rambech, E.; Nilbert, M. Heterogenous mismatch-repair status in colorectal cancer. Diagn. Pathol. 2014, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lage, P.; Belga, S.; Filipe, B.; Francisco, I.; Rodrigues, P.; Fonseca, R.; Chaves, P.; Claro, I.; Albuquerque, C.; et al. Bethesda criteria for microsatellite instability testing: Impact on the detection of new cases of Lynch syndrome. Fam. Cancer 2012, 11, 571–578. [Google Scholar] [CrossRef]
- Zighelboim, I.; Powell, M.A.; Babb, S.A.; Whelan, A.J.; Schmidt, A.P.; Clendenning, M.; Senter, L.; Thibodeau, S.N.; de la Chapelle, A.; Goodfellow, P.J. Epitope-positive truncating MLH1 mutation and loss of PMS2: Implications for IHC-directed genetic testing for Lynch syndrome. Fam. Cancer 2009, 8, 501–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Sato, N.; Sugawara, T.; Takahashi, K.; Kito, M.; Makino, K.; Sato, T.; Shimizu, D.; Shirasawa, H.; Miura, H.; et al. Isolated Loss of PMS2 Immunohistochemical Expression is Frequently Caused by Heterogenous MLH1 Promoter Hypermethylation in Lynch Syndrome Screening for Endometrial Cancer Patients. Am. J. Surg. Pathol. 2016, 40, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uryu, H.; Oda, Y.; Shiratsuchi, H.; Oda, S.; Yamamoto, H.; Komune, S.; Tsuneyoshi, M. Microsatellite instability and proliferating activity in sinonasal carcinoma: Molecular genetic and immunohistochemical comparison with oral squamous cell carcinoma. Oncol. Rep. 2005, 14, 1133–1142. [Google Scholar] [CrossRef]
- Hongo, T.; Yamamoto, H.; Jiromaru, R.; Yasumatsu, R.; Kuga, R.; Nozaki, Y.; Hashimoto, K.; Matsuo, M.; Wakasaki, T.; Tamae, A.; et al. PD-L1 expression, tumor-infiltrating lymphocytes, mismatch repair deficiency, EGFR alteration and HPV infection in sinonasal squamous cell carcinoma. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2021, 34, 1966–1978. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.G.; Pérez-Escuredo, J.; López, F.; Suárez, C.; Alvarez-Marcos, C.; Llorente, J.L.; Hermsen, M.A. Microsatellite instability analysis of sinonasal carcinomas. Otolaryngol.-Head Neck Surg. Off. J. Am. Acad. Otolaryngol. Head Neck Surg. 2009, 140, 55–60. [Google Scholar] [CrossRef]
- Hermsen, M.A.; Llorente, J.L.; Pérez-Escuredo, J.; López, F.; Ylstra, B.; Alvarez-Marcos, C.; Suárez, C. Genome-wide analysis of genetic changes in intestinal-type sinonasal adenocarcinoma. Head Neck 2009, 31, 290–297. [Google Scholar] [CrossRef]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Wang, Y.; Turner, J.H. Proinflammatory mediators alter expression of nuclear factor kappa B-regulating deubiquitinases in sinonasal epithelial cells. Int. Forum Allergy Rhinol. 2015, 5, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, Z.; Fintelmann, F.J.; Sequist, L.V.; Jahagirdar, B. Response to Osimertinib in an EGFR Exon 20 Insertion-Positive Lung Adenocarcinoma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, e204–e206. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, T.M.; Kim, D.W.; Kim, S.; Kim, M.; Keam, B.; Ku, J.L.; Heo, D.S. Preclinical Modeling of Osimertinib for NSCLC With EGFR Exon 20 Insertion Mutations. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1556–1566. [Google Scholar] [CrossRef]
- Syed, Y.Y. Amivantamab: First Approval. Drugs 2021, 81, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; Vincent, S.; Baker, T.E.; Gould, A.E.; Li, S.; Wardwell, S.D.; Nadworny, S.; Ning, Y.; Zhang, S.; Huang, W.S.; et al. Mobocertinib (TAK-788): A Targeted Inhibitor of EGFR Exon 20 Insertion Mutants in Non-Small Cell Lung Cancer. Cancer Discov. 2021, 11, 1672–1687. [Google Scholar] [CrossRef]
- Gao, G.; Liao, W.; Ma, Q.; Zhang, B.; Chen, Y.; Wang, Y. KRAS G12D mutation predicts lower TMB and drives immune suppression in lung adenocarcinoma. Lung Cancer 2020, 149, 41–45. [Google Scholar] [CrossRef]
- Udager, A.M.; McHugh, J.B.; Betz, B.L.; Montone, K.T.; Livolsi, V.A.; Seethala, R.R.; Yakirevich, E.; Iwenofu, O.H.; Perez-Ordonez, B.; DuRoss, K.E.; et al. Activating KRAS mutations are characteristic of oncocytic sinonasal papilloma and associated sinonasal squamous cell carcinoma. J. Pathol. 2016, 239, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hieggelke, L.; Heydt, C.; Castiglione, R.; Rehker, J.; Merkelbach-Bruse, S.; Riobello, C.; Llorente, J.L.; Hermsen, M.A.; Buettner, R. Mismatch Repair Deficiency and Somatic Mutations in Human Sinonasal Tumors. Cancers 2021, 13, 6081. https://doi.org/10.3390/cancers13236081
Hieggelke L, Heydt C, Castiglione R, Rehker J, Merkelbach-Bruse S, Riobello C, Llorente JL, Hermsen MA, Buettner R. Mismatch Repair Deficiency and Somatic Mutations in Human Sinonasal Tumors. Cancers. 2021; 13(23):6081. https://doi.org/10.3390/cancers13236081
Chicago/Turabian StyleHieggelke, Lena, Carina Heydt, Roberta Castiglione, Jan Rehker, Sabine Merkelbach-Bruse, Cristina Riobello, José Luis Llorente, Mario A. Hermsen, and Reinhard Buettner. 2021. "Mismatch Repair Deficiency and Somatic Mutations in Human Sinonasal Tumors" Cancers 13, no. 23: 6081. https://doi.org/10.3390/cancers13236081
APA StyleHieggelke, L., Heydt, C., Castiglione, R., Rehker, J., Merkelbach-Bruse, S., Riobello, C., Llorente, J. L., Hermsen, M. A., & Buettner, R. (2021). Mismatch Repair Deficiency and Somatic Mutations in Human Sinonasal Tumors. Cancers, 13(23), 6081. https://doi.org/10.3390/cancers13236081