
Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy


Abstract
:Simple Summary
Abstract
1. Cutaneous Melanoma and the Immune System
1.1. The Tumorigenesis of Cutaneous Melanoma
1.2. The Immune System against Cancer
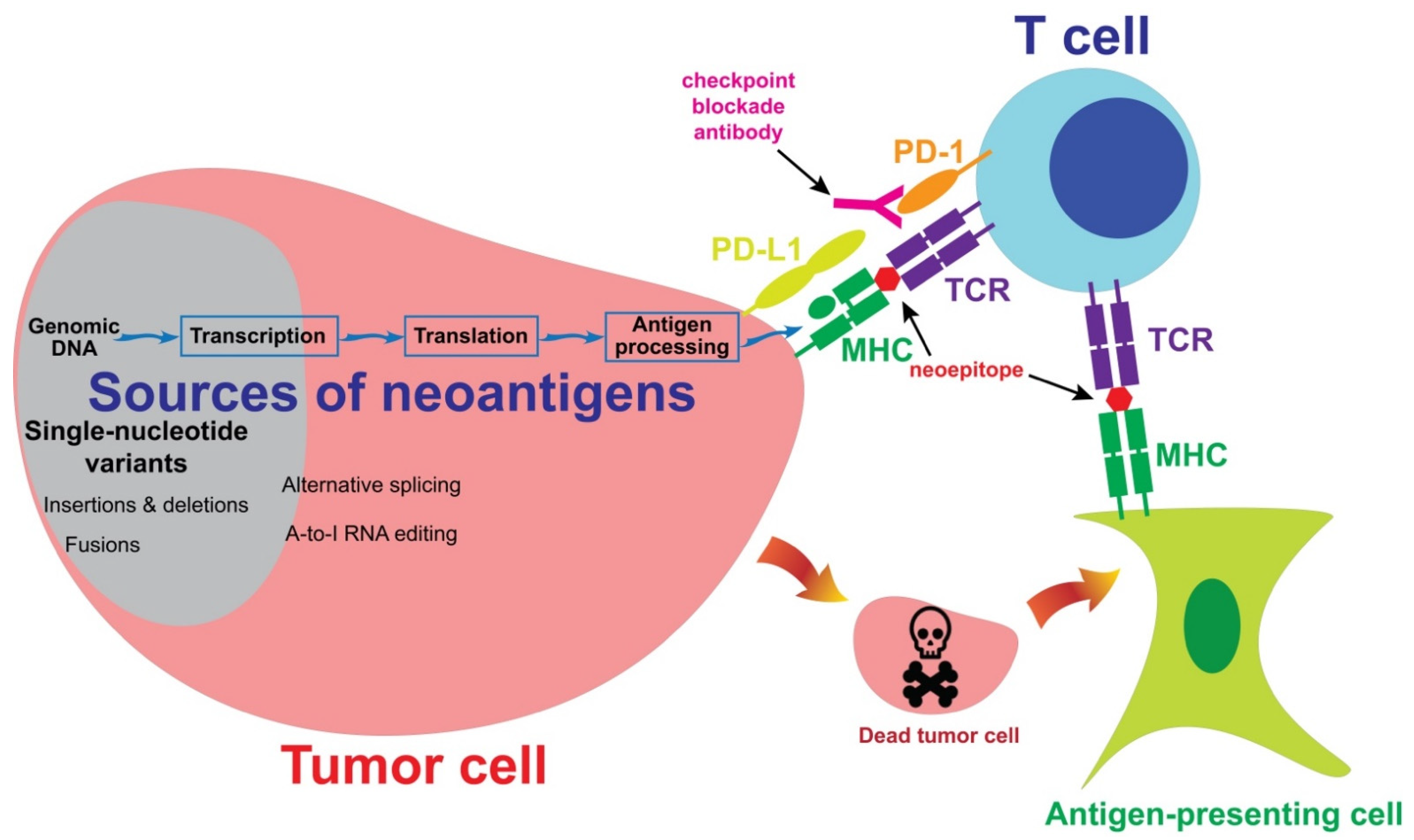
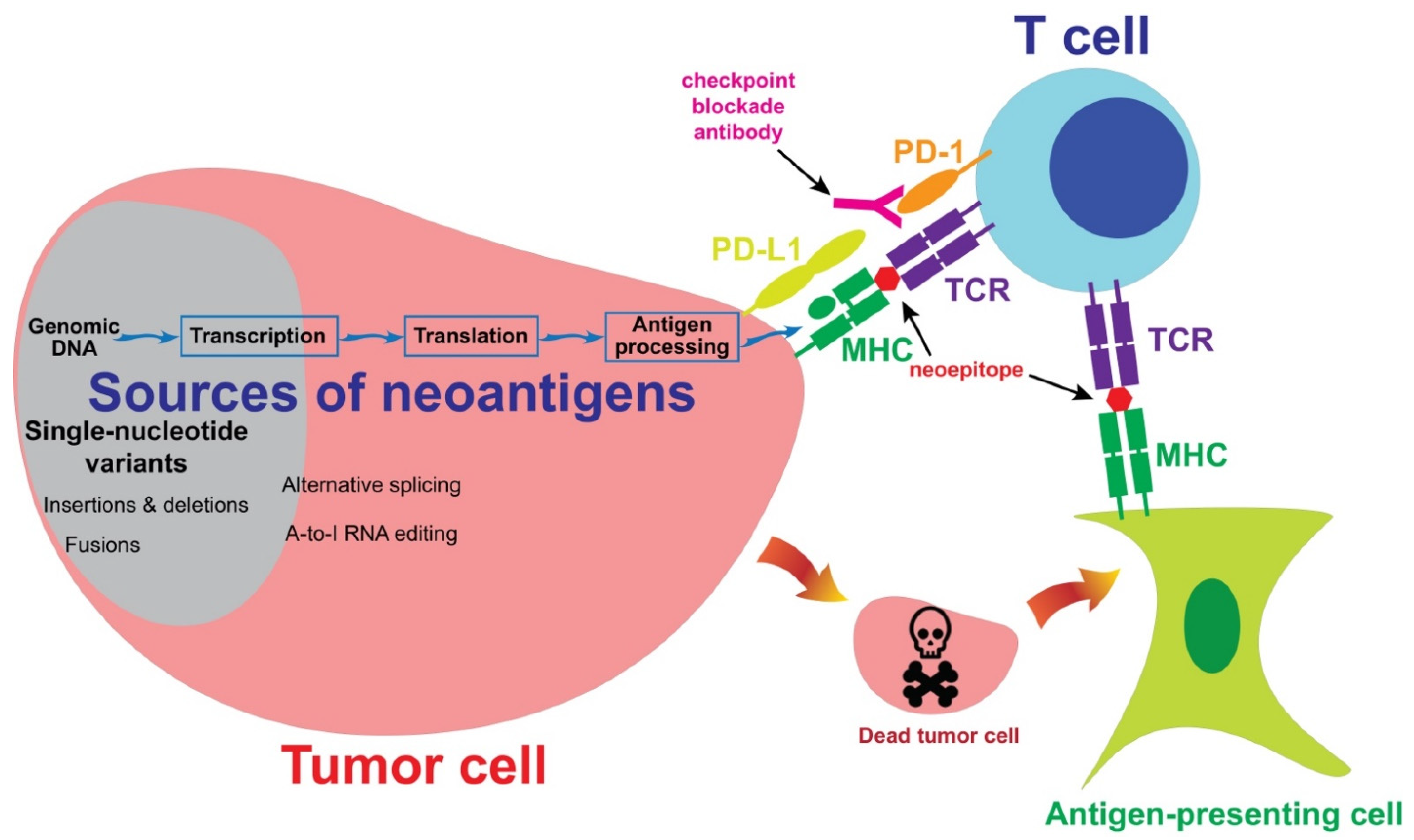
1.3. Antigen Process and Presenting Pathways
1.4. Co-Inhibitory Molecules
1.5. Overview of Tumor Antigens
1.5.1. Tissue-Specific Differentiation Antigens
1.5.2. Cancer Germline Antigens
1.5.3. Viral Antigens
1.5.4. Neoantigens
2. Sources of Neoantigens
2.1. Single-Nucleotide Variants
2.2. Insertion, Deletion and Fusion
2.3. Post-Transcriptional and Post-Translational Alternations
3. Approaches to the Identification of Neoantigens
3.1. cDNA Library Screening
3.2. Neoepitope Prediction
3.3. Tandem Minigene and Long-Peptide Libraries
3.4. HLA Peptidomics
4. Clinical Trials for Melanoma Immunotherapy
4.1. Monotherapy
4.1.1. Cytokines
4.1.2. Checkpoint Blockade Antibodies
4.1.3. Other Therapeutic Agents
4.2. Combination Therapy
4.3. Adjuvant Therapy
4.4. Neoadjuvant Therapy

{kind=link}
{kind=link}
| Drug | Trial/ID | Dosage | Primary Outcome/ Estimated Completion Date |
|---|---|---|---|
| Pembrolizumab | NCT02434354 | 200 mg Pembro followed by surgery, then adjuvant Pembro therapy every 3 weeks for 1 year | July 2022 |
| Ipilimumab + nivolumab | OpACIN NCT02977052 | Arm A: 3 mg/kg Ipi + 1 mg/kg Nivo every 3 weeks for 2 cycles prior to surgery. Arm B: 1 mg/kg Ipi + 3 mg/kg Nivo every 3 weeks for 2 cycles prior to surgery Arm C: 3 mg/kg Ipi every 3 weeks for 2 cycles, followed immediately by 3 mg/kg Nivo every 2 weeks for 2 cycles prior to surgery | June 2025 |
| Dabrafenib+ trametinib | NCT01972347 | 150 mg Dab + 2 mg Tram for 12 weeks followed by surgery, then 40 weeks of adjuvant Dab/Tram | May 2022 |
| Ipilimumab | NCT00972933 | Two doses of 10 mg/kg of Ipi followed by surgery, then two doses of adjuvant Ipi | Median PFS: 11 months [125] |
| Nivolumab vs. Ipilimumab + nivolumab vs. Nivolumab + relatlimab | NCT02519322 | Arm A: 3 mg/kg Nivo every 2 weeks for 4 cycles prior to surgery, then adjuvant Nivo every 2 weeks for 13 cycles Arm B: 1 mg/kg Nivo and 3 mg/kg Ipi every 3 weeks for 3 cycles prior to surgery, then adjuvant Nivo every 2 weeks for 13 cycles Arm C: 480 mg Nivo + 160 mg relatlimab every 4 weeks for 2 cycles prior to surgery, then adjuvant Nivo + relatlimab every 4 weeks for 10 cycles | December 2022 |
5. The Role of Neoantigens in Immunotherapy
5.1. Animal Models
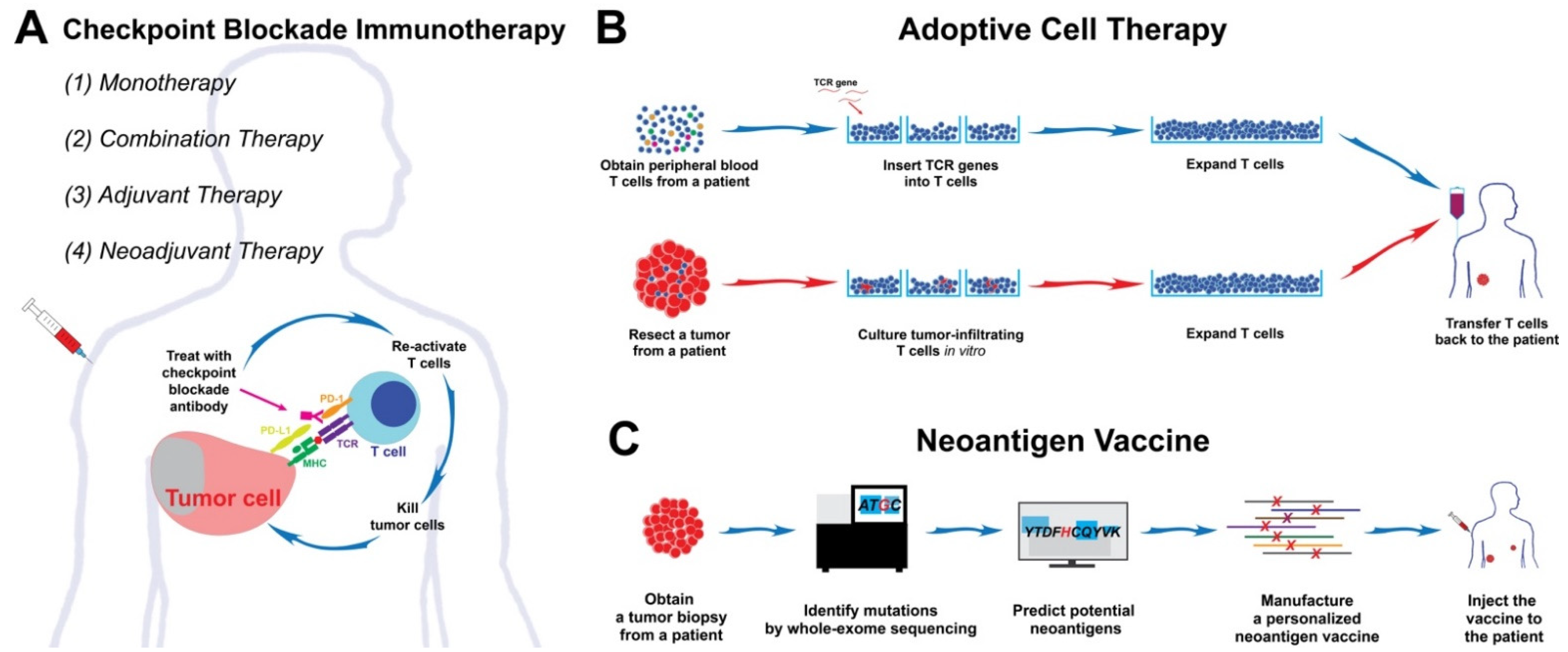
5.2. Checkpoint Blockade Immunotherapy
5.3. Adoptive Cell Therapy (ACT)
5.4. Neoantigen Vaccine
5.5. The Dysfunction and Re-Activation of Neoantigen-Specific T Cells
5.6. Neoantigen-Specific T Cells: A Minor Population in TILs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Mo, X.; Preston, S.; Zaidi, M.R. Macroenvironment-gene-microenvironment interactions in ultraviolet radiation-induced melanomagenesis. Adv. Cancer Res. 2019, 144, 1–54. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Slominski, A.; Kim, T.K.; Brozyna, A.A.; Janjetovic, Z.; Brooks, D.L.; Schwab, L.P.; Skobowiat, C.; Jozwicki, W.; Seagroves, T.N. The role of melanogenesis in regulation of melanoma behavior: Melanogenesis leads to stimulation of HIF-1alpha expression and HIF-dependent attendant pathways. Arch. Biochem. Biophys. 2014, 563, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.; Zbytek, B.; Slominski, R. Inhibitors of melanogenesis increase toxicity of cyclophosphamide and lymphocytes against melanoma cells. Int. J. Cancer. 2009, 124, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Buart, S.; Terry, S.; Noman, M.Z.; Lanoy, E.; Boutros, C.; Fogel, P.; Dessen, P.; Meurice, G.; Gaston-Mathe, Y.; Vielh, P.; et al. Transcriptional response to hypoxic stress in melanoma and prognostic potential of GBE1 and BNIP3. Oncotarget 2017, 8, 108786–108801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The role of melanin pigment in melanoma. Exp. Dermatol. 2015, 24, 258–259. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Carlson, J.A. Melanoma resistance: A bright future for academicians and a challenge for patient advocates. Mayo Clin. Proc. 2014, 89, 429–433. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Oak, A.S.W.; Slominski, R.M.; Brozyna, A.A.; Slominski, A.T. Current Molecular Markers of Melanoma and Treatment Targets. Int. J. Mol. Sci. 2020, 21, 3535. [Google Scholar] [CrossRef]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef] [PubMed]
- He, S.J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Willsmore, Z.N.; Harris, R.J.; Crescioli, S.; Hussein, K.; Kakkassery, H.; Thapa, D.; Cheung, A.; Chauhan, J.; Bax, H.J.; Chenoweth, A.; et al. B Cells in Patients With Melanoma: Implications for Treatment With Checkpoint Inhibitor Antibodies. Front. Immunol. 2020, 11, 622442. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016, 1, eaai8593. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8(+) T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, M.L.; Cruz, A.M.; Crossland, G.E.; Gaglia, G.; Ritch, C.C.; Blatt, S.E.; Bhutkar, A.; Canner, D.; Kienka, T.; Tavana, S.Z.; et al. Antigen dominance hierarchies shape TCF1(+) progenitor CD8 T cell phenotypes in tumors. Cell 2021, 184, 4996–5014.e26. [Google Scholar] [CrossRef] [PubMed]
- Brightman, S.E.; Naradikian, M.S.; Miller, A.M.; Schoenberger, S.P. Harnessing neoantigen specific CD4 T cells for cancer immunotherapy. J. Leukoc. Biol. 2020, 107, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Poncette, L.; Bluhm, J.; Blankenstein, T. The role of CD4 T cells in rejection of solid tumors. Curr. Opin. Immunol. 2021, 74, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lu, S.; Rospo, G.; Lamba, S.; Rousseau, B.; Fanelli, S.; Stenech, D.; Le, D.T.; Hays, J.; Totaro, M.G.; et al. CD4 T Cell-Dependent Rejection of Beta-2 Microglobulin Null Mismatch Repair-Deficient Tumors. Cancer Discov. 2021, 11, 1844–1859. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Oddone, F.; Kudriaeva, A.A.; Lacal, P.M.; Belogurov, A.A., Jr.; Graziani, G.; Marini, S. At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention. Cancers 2021, 13, 4852. [Google Scholar] [CrossRef]
- Tubio-Santamaria, N.; Ebstein, F.; Heidel, F.H.; Kruger, E. Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells 2021, 10, 1577. [Google Scholar] [CrossRef]
- Harton, J.; Jin, L.; Hahn, A.; Drake, J. Immunological Functions of the Membrane Proximal Region of MHC Class II Molecules. F1000Res 2016, 5, 368. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Ruiz-Cabello, F.; Aptsiauri, N. Rejection versus escape: The tumor MHC dilemma. Cancer Immunol. Immunother. 2017, 66, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, M.; Mezquita, L.; De Nerville, G.G.; Tihy, I.; Malenica, I.; Chouaib, S.; Mami-Chouaib, F. Recent Advances in Lung Cancer Immunotherapy: Input of T-Cell Epitopes Associated With Impaired Peptide Processing. Front. Immunol. 2019, 10, 1505. [Google Scholar] [CrossRef] [Green Version]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Salama, A.D.; Chitnis, T.; Imitola, J.; Ansari, M.J.; Akiba, H.; Tushima, F.; Azuma, M.; Yagita, H.; Sayegh, M.H.; Khoury, S.J. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2003, 198, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, F.; Iwai, H.; Otsuki, N.; Abe, M.; Hirose, S.; Yamazaki, T.; Akiba, H.; Yagita, H.; Takahashi, Y.; Omura, K.; et al. Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur. J. Immunol. 2003, 33, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfel, T.; Van Pel, A.; Brichard, V.; Schneider, J.; Seliger, B.; Meyer zum Buschenfelde, K.H.; Boon, T. Two tyrosinase nonapeptides recognized on HLA-A2 melanomas by autologous cytolytic T lymphocytes. Eur. J. Immunol. 1994, 24, 759–764. [Google Scholar] [CrossRef]
- Engelhard, V.H.; Bullock, T.N.; Colella, T.A.; Sheasley, S.L.; Mullins, D.W. Antigens derived from melanocyte differentiation proteins: Self-tolerance, autoimmunity, and use for cancer immunotherapy. Immunol. Rev. 2002, 188, 136–146. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Van Der Bruggen, P.; Zhang, Y.; Chaux, P.; Stroobant, V.; Panichelli, C.; Schultz, E.S.; Chapiro, J.; Van Den Eynde, B.J.; Brasseur, F.; Boon, T. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol. Rev. 2002, 188, 51–64. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Cell transfer immunotherapy for metastatic solid cancer--what clinicians need to know. Nat. Rev. Clin. Oncol. 2011, 8, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Hatta, M.N.A.; Mohamad Hanif, E.A.; Chin, S.F.; Neoh, H.M. Pathogens and Carcinogenesis: A Review. Biology 2021, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef]
- Doran, S.L.; Stevanovic, S.; Adhikary, S.; Gartner, J.J.; Jia, L.; Kwong, M.L.M.; Faquin, W.C.; Hewitt, S.M.; Sherry, R.M.; Yang, J.C.; et al. T-Cell Receptor Gene Therapy for Human Papillomavirus-Associated Epithelial Cancers: A First-in-Human, Phase I/II Study. J. Clin. Oncol. 2019, 37, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.B.; Norberg, S.M.; Sinkoe, A.L.; Adhikary, S.; Meyer, T.J.; Lack, J.B.; Warner, A.C.; Schweitzer, C.; Doran, S.L.; Korrapati, S.; et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat. Med. 2021, 27, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2018, 37, 173–200. [Google Scholar] [CrossRef]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Wolfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wolfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; zum Buschenfelde, K.H.M.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996, 183, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [Green Version]
- Robinson, T.J.; Freedman, J.A.; Al Abo, M.; Deveaux, A.E.; LaCroix, B.; Patierno, B.M.; George, D.J.; Patierno, S.R. Alternative RNA Splicing as a Potential Major Source of Untapped Molecular Targets in Precision Oncology and Cancer Disparities. Clin. Cancer Res. 2019, 25, 2963–2968. [Google Scholar] [CrossRef] [Green Version]
- Guilloux, Y.; Lucas, S.; Brichard, V.G.; Van Pel, A.; Viret, C.; De Plaen, E.; Brasseur, F.; Lethe, B.; Jotereau, F.; Boon, T. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J. Exp. Med. 1996, 183, 1173–1183. [Google Scholar] [CrossRef]
- Labarriere, N.; Diez, E.; Pandolfino, M.C.; Viret, C.; Guilloux, Y.; Le Guiner, S.; Fonteneau, J.F.; Dreno, B.; Jotereau, F. Optimal T cell activation by melanoma cells depends on a minimal level of antigen transcription. J. Immunol. 1997, 158, 1238–1245. [Google Scholar]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.; Chiu, Y.; Tsou, C.C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018, 9, 3919. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Lehmann, F.; Lethe, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [Green Version]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.F.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [Green Version]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Pasetto, A.; Gros, A.; Robbins, P.F.; Deniger, D.C.; Prickett, T.D.; Matus-Nicodemos, R.; Douek, D.C.; Howie, B.; Robins, H.; Parkhurst, M.R.; et al. Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol. Res. 2016, 4, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Hu, Z.; Leet, D.E.; Allesoe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef]
- Kalaora, S.; Wolf, Y.; Feferman, T.; Barnea, E.; Greenstein, E.; Reshef, D.; Tirosh, I.; Reuben, A.; Patkar, S.; Levy, R.; et al. Combined Analysis of Antigen Presentation and T-cell Recognition Reveals Restricted Immune Responses in Melanoma. Cancer Discov. 2018, 8, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann. Surg. 1998, 228, 307–319. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Lebbe, C.; Weber, J.S.; Maio, M.; Neyns, B.; Harmankaya, K.; Hamid, O.; O’Day, S.J.; Konto, C.; Cykowski, L.; McHenry, M.B.; et al. Survival follow-up and ipilimumab retreatment of patients with advanced melanoma who received ipilimumab in prior phase II studies. Ann. Oncol. 2014, 25, 2277–2284. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Del Vecchio, M.; Robert, C.; Mackiewicz, A.; Chiarion-Sileni, V.; Arance, A.; Lebbe, C.; Bastholt, L.; Hamid, O.; Rutkowski, P.; et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017, 18, 611–622. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Hamid, O.; Molinero, L.; Bolen, C.R.; Sosman, J.A.; Munoz-Couselo, E.; Kluger, H.M.; McDermott, D.F.; Powderly, J.D.; Sarkar, I.; Ballinger, M.; et al. Safety, Clinical Activity, and Biological Correlates of Response in Patients with Metastatic Melanoma: Results from a Phase I Trial of Atezolizumab. Clin. Cancer Res. 2019, 25, 6061–6072. [Google Scholar] [CrossRef] [Green Version]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.J.; Taylor, M.H.; Wyrwicz, L.; Lee, K.W.; et al. Avelumab in patients with previously treated metastatic melanoma: Phase 1b results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer 2019, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Hrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.; Ross, M.; Puzanov, I.; Milhem, M.; Collichio, F.; Delman, K.A.; Amatruda, T.; Zager, J.S.; Cranmer, L.; Hsueh, E.; et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann. Surg. Oncol. 2016, 23, 4169–4177. [Google Scholar] [CrossRef] [Green Version]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Byers, B.A.; Temple-Oberle, C.F.; Hurdle, V.; McKinnon, J.G. Treatment of in-transit melanoma with intra-lesional interleukin-2: A systematic review. J. Surg. Oncol. 2014, 110, 770–775. [Google Scholar] [CrossRef]
- Radny, P.; Caroli, U.M.; Bauer, J.; Paul, T.; Schlegel, C.; Eigentler, T.K.; Weide, B.; Schwarz, M.; Garbe, C. Phase II trial of intralesional therapy with interleukin-2 in soft-tissue melanoma metastases. Br. J. Cancer 2003, 89, 1620–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.H.; Jessup, J.M.; Felix, E.L.; Weese, J.L.; Herberman, R.B. Intralesional treatment of recurrent metastatic cutaneous malignant melanoma: A randomized prospective study of intralesional Bacillus Calmette-Guerin versus intralesional dinitrochlorobenzene. Cancer 1978, 41, 2456–2463. [Google Scholar] [CrossRef]
- Thompson, J.F.; Hersey, P.; Wachter, E. Chemoablation of metastatic melanoma using intralesional Rose Bengal. Melanoma Res. 2008, 18, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Ascierto, P.A.; Schadendorf, D.; Grob, J.J.; Ribas, A.; Kiecker, F.; Dutriaux, C.; Demidov, L.V.; Lebbe, C.; Rutkowski, P.; et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib monotherapy: Analysis from phase 2 and 3 clinical trials. Eur. J. Cancer 2020, 125, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Ribas, A.; Robert, C.; Schachter, J.; Nyakas, M.; Daud, A.; Arance, A.; Carlino, M.S.; O’Day, S.J.; Long, G.V.; et al. Association of BRAF V600E/K Mutation Status and Prior BRAF/MEK Inhibition With Pembrolizumab Outcomes in Advanced Melanoma: Pooled Analysis of 3 Clinical Trials. JAMA Oncol. 2020, 6, 1256–1264. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Atkins, M.B.; Tarhini, A.; Rael, M.; Gupte-Singh, K.; O’Brien, E.; Ritchings, C.; Rao, S.; McDermott, D.F. Comparative efficacy of combination immunotherapy and targeted therapy in the treatment of BRAF-mutant advanced melanoma: A matching-adjusted indirect comparison. Immunotherapy 2019, 11, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Maio, M.; Lewis, K.; Demidov, L.; Mandala, M.; Bondarenko, I.; Ascierto, P.A.; Herbert, C.; Mackiewicz, A.; Rutkowski, P.; Guminski, A.; et al. Adjuvant vemurafenib in resected, BRAF(V600) mutation-positive melanoma (BRIM8): A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 510–520. [Google Scholar] [CrossRef]
- Amaria, R.N.; Menzies, A.M.; Burton, E.M.; Scolyer, R.A.; Tetzlaff, M.T.; Antdbacka, R.; Ariyan, C.; Bassett, R.; Carter, B.; Daud, A.; et al. Neoadjuvant systemic therapy in melanoma: Recommendations of the International Neoadjuvant Melanoma Consortium. Lancet Oncol. 2019, 20, e378–e389. [Google Scholar] [CrossRef]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpaa, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef]
- Rozeman, E.A.; Hoefsmit, E.P.; Reijers, I.L.M.; Saw, R.P.M.; Versluis, J.M.; Krijgsman, O.; Dimitriadis, P.; Sikorska, K.; van de Wiel, B.A.; Eriksson, H.; et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat. Med. 2021, 27, 256–263. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Zahoor, H.; Lin, Y.; Malhotra, U.; Sander, C.; Butterfield, L.H.; Kirkwood, J.M. Baseline circulating IL-17 predicts toxicity while TGF-beta1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J. Immunother. Cancer 2015, 3, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Lower, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Solit, D.B.; Chan, T.A.; Kurzrock, R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) >/=10: A decision centered on empowering patients and their physicians. Ann. Oncol. 2020, 31, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, F.; Sonnemann, H.; Jackson, K.R.; Talukder, A.H.; Katailiha, A.S.; Lizee, G. Evolution of CD8(+) T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer. Cells 2021, 10, 2379. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef]
- Forget, M.A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin. Cancer Res. 2018, 24, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Saibil, S.D.; Sotov, V.; Le, M.X.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.M.; et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besser, M.J.; Itzhaki, O.; Ben-Betzalel, G.; Zippel, D.B.; Zikich, D.; Kubi, A.; Brezinger, K.; Nissani, A.; Levi, M.; Zeltzer, L.A.; et al. Comprehensive single institute experience with melanoma TIL: Long term clinical results, toxicity profile, and prognostic factors of response. Mol. Carcinog. 2020, 59, 736–744. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yao, X.; Li, Y.F.; El-Gamil, M.; Dudley, M.E.; Yang, J.C.; Almeida, J.R.; Douek, D.C.; Samuels, Y.; Rosenberg, S.A.; et al. Mutated PPP1R3B Is Recognized by T Cells Used To Treat a Melanoma Patient Who Experienced a Durable Complete Tumor Regression. J. Immunol. 2013, 190, 6034–6042. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Saberian, C.; Amaria, R.N.; Najjar, A.M.; Radvanyi, L.G.; Haymaker, C.L.; Forget, M.A.; Bassett, R.L.; Faria, S.C.; Glitza, I.C.; Alvarez, E.; et al. Randomized phase II trial of lymphodepletion plus adoptive cell transfer of tumor-infiltrating lymphocytes, with or without dendritic cell vaccination, in patients with metastatic melanoma. J. Immunother. Cancer 2021, 9, e002449. [Google Scholar] [CrossRef]
- Bigot, J.; Lalanne, A.I.; Lucibello, F.; Gueguen, P.; Houy, A.; Dayot, S.; Ganier, O.; Gilet, J.; Tosello, J.; Nemati, F.; et al. Splicing Patterns in SF3B1-Mutated Uveal Melanoma Generate Shared Immunogenic Tumor-Specific Neoepitopes. Cancer Discov. 2021, 11, 1938–1951. [Google Scholar] [CrossRef]
- Peri, A.; Greenstein, E.; Alon, M.; Pai, J.A.; Dingjan, T.; Reich-Zeliger, S.; Barnea, E.; Barbolin, C.; Levy, R.; Arnedo-Pac, C.; et al. Combined presentation and immunogenicity analysis reveals a recurrent RAS.Q61K neoantigen in melanoma. J. Clin. Invest. 2021, 131, e129466. [Google Scholar] [CrossRef] [PubMed]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gurusamy, D.; et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef]
- Caushi, J.X.; Zhang, J.; Ji, Z.; Vaghasia, A.; Zhang, B.; Hsiue, E.H.; Mog, B.J.; Hou, W.; Justesen, S.; Blosser, R.; et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 2021, 596, 126–132. [Google Scholar] [CrossRef]
- Braunlein, E.; Lupoli, G.; Fuchsl, F.; Abualrous, E.T.; de Andrade Kratzig, N.; Gosmann, D.; Wietbrock, L.; Lange, S.; Engleitner, T.; Lan, H.; et al. Functional analysis of peripheral and intratumoral neoantigen-specific TCRs identified in a patient with melanoma. J. Immunother. Cancer 2021, 9, e002754. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Zheng, Z.; Lowery, F.J.; Gartner, J.J.; Prickett, T.D.; Robbins, P.F.; Rosenberg, S.A. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing. J. Immunother. Cancer 2021, 9, e002595. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
| Drug (Manufacturer) | Trial ID | Mechanism of Action | Dosage | Primary Outcome |
|---|---|---|---|---|
| Interferon (IFN-α-2b) (Merck) | ECOG 1684 | Immune stimulator | IFN-α-2b 20 MU/m2 intravenously, followed by 10 MU/m2 subcutaneously | Median OS: 3.8 years (IFN-α-2b) vs. 2.8 years (observation) |
| Aldesleukin (Clinigen) | T-cell stimulator | 600,000 or 720,000 IU/kg | Median OS: 11.4 months | |
| Ipilimumab (Bristol-Myers Squibb) | CA 184-002 (NCT00094653) | CTLA-4 checkpoint inhibitor | Ipi 3 mg/kg + gp100 | Median OS: 10.0 months (Ipi + gp100) vs. 6.4 months (gp100 alone) |
| CA 184-169 (NCT01515189) | Ipi 10 mg/kg vs. Ipi 3 mg/kg | Median OS: 15.7 months (10 m/kg) vs. 11.5 months (3 mg/kg) | ||
| Pembrolizumab (Merck) | KEYNOTE-002 (NCT01704287) | PD-1 checkpoint inhibitor | Pembro 2 mg/kg vs. 10 mg/kg vs. chemo | 6 month PFS: 34% (Pembro 2 mg/kg), 38% (Pembro 10 mg/kg), 16% (chemo) |
| KEYNOTE-006 (NCT01866319) | Pembro 10 mg/kg vs. Ipi 3 mg/kg | Median OS: 32.7 months (Pembro) vs. 15.9 months (Ipi) | ||
| Nivolumab (Bristol-Myers Squibb) | Checkmate-066 (NCT01721772) | PD-1 checkpoint inhibitor | Nivo 3 mg/kg vs dacarbazine 1000 mg/m2 | 3 yr OS 51.2% (Nivo) vs. 21.6% (Dab) |
| Checkmate-067 (NCT01844505) | Nivo 3 mg/kg vs. Ipi 3 mg/kg | 5 yr OS 44% (Nivo) vs. 26% (Ipi) | ||
| Talimogene laherparepve (T-VEC) (Amgen) | OPTiM (NCT00769704) | Oncolytic virus | Up to 4 mL of 10⁸ pfu/mL per intratumoral injection vs. GM-CSF 125 μg/m2 | Median OS: 23.3 months (T-VEC) vs. 18.9 months (GM-CSF) |
| Vemurafenib (Genentech) | BRIM-3 (NCT01006980) | BRAF inhibitor | Oral Vem 960 mg vs. dacarbazine 1000 mg/m2 | Median OS: 13.6 months (Vem) vs. 9.7 months (dacarbazine) |
| Dabrafenib (Novartis) | BREAK-2 (NCT01153763) | BRAF inhibitor | Oral Dab 150 mg | OS at 3, 4, 5 years: 30%, 23%, 20% |
| BREAK-3 (NCT01227889) | OS at 3, 4, 5 years: 31%, 27%, 24% |
| Drug (Manufacturer) | Trial ID | Mechanism of Action | Dosage | Primary Outcome |
|---|---|---|---|---|
| Dabrafenib + trametinib (Novartis) | COMBI-d (NCT01584648)COMBI-v (NCT01597908) | BRAF inhibitor + MEK inhibitor | Dab 150 mg + Tram 2 mg | OS at 2 & 3 years: 52%, 44% |
| Vemurafenib + cobimetinib (Genentech) | CO-BRIM (NCT01689519) | BRAF inhibitor + MEK inhibitor | Vem 960 mg + Cobi 60 mg | Median PFS: 9.9 months |
| Encorafenib + binimetinib (Pfizer) | COLUMBUS (NCT01909453) | BRAF inhibitor + MEK inhibitor | 450 mg encorafenib + 45 mg binimetinib | Median PFS: 14.9 months |
| Atezolizumab + vemurafenib + cobimetinib (Genentech) | IMspire 150 (NCT02908672) | PD-L1 checkpoint inhibitor + BRAF inhibitor + MEK inhibitor | cycle 1: Vem 960 mg for 21 days + Cob 60 mg, followed by Vem 720 mg cycle 2: atezolizumab 840 mg, Vem 720 mg, Cob 60 mg | Median PFS: 15.1 months |
| Nivolumab + ipilimumab (Bristol-Myers Squibb) | Checkmate-067 (NCT01844505) Checkmate-069 (NCT01927419) | PD-1 checkpoint inhibitor + CTLA-4 checkpoint inhibitor | Nivo 1 mg/kg + Ipi 3 mg/kg, followed by Nivo 3 mg/kg | ORR: 58% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, L.; Tarduno, A.; Lu, Y.-C. Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers 2021, 13, 6061. https://doi.org/10.3390/cancers13236061
Davis L, Tarduno A, Lu Y-C. Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers. 2021; 13(23):6061. https://doi.org/10.3390/cancers13236061
Chicago/Turabian StyleDavis, Lindy, Ashley Tarduno, and Yong-Chen Lu. 2021. "Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy" Cancers 13, no. 23: 6061. https://doi.org/10.3390/cancers13236061
APA StyleDavis, L., Tarduno, A., & Lu, Y.-C. (2021). Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers, 13(23), 6061. https://doi.org/10.3390/cancers13236061