
Going with the Flow: Modeling the Tumor Microenvironment Using Microfluidic Technology


Abstract
:Simple Summary
Abstract
1. Introduction
“The field of cancer research has largely been guided by a reductionist focus on cancer cells and the genes within them—a focus that has produced an extraordinary body of knowledge. Looking forward in time, we believe that important new inroads will come from regarding tumors as complex tissues in which mutant cancer cells have conscripted and subverted normal cell types to serve as active collaborators in their neoplastic agenda. The interactions between the genetically altered malignant cells and these supporting coconspirators will prove critical to understanding cancer pathogenesis and to the development of novel, effective therapies”.Hanahan and Weinberg, Cell 2000 [1]
2. Cancer Immunotherapy
3. Modeling the Tumor Microenvironment
“All models are wrong, but some are useful”.George E.P. Box (British statistician)
3.1. Tumor Heterogeneity and Composition of the TME
3.2. In Vivo Models
3.3. 2D versus 3D Culture
3.3.1. 2D Culture
3.3.2. 3D Culture
3.3.3. Comparison between 2D and 3D
3.3.4. Evaluating Tumor–Immune Interactions in 2D Culture Systems
3.3.5. Evaluating Tumor–Immune Interactions in 3D Culture Systems
4. Microfluidic Technology
5. Modeling Cancer in Microfluidic Chips
“A key consideration in the development of new microfluidic methods in academic research should be whether the use of microfluidics introduces truly enabling functionality compared to current methods. When a potential application passes this test, the chances of contributing useful technology to the field are substantially higher”.Sackmann et al. Nature 2014 [134]
5.1. Tumor Growth
5.2. Tumor Migration and Extravasation
5.3. Angiogenesis
5.4. Cancer Metastasis
5.5. Modeling the TME
5.6. Immune Cell Migration/Recruitment
5.7. T Lymphocyte Activation
5.8. Therapy Assessment
5.9. Disease and Therapeutic Response Monitoring
5.10. Drug Screening
6. Challenges, Opportunities, and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. Targeted therapy for cancer using PARP inhibitors. Curr. Opin. Pharmacol. 2008, 8, 363–369. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Callejo, D.; Gonzalez-Rincon, J.; Sanchez, A.; Provencio, M.; Sanchez-Beato, M. Action and resistance of monoclonal CD20 antibodies therapy in B-cell Non-Hodgkin Lymphomas. Cancer Treat. Rev. 2015, 41, 680–689. [Google Scholar] [CrossRef]
- Cameron, F.; Sanford, M. Ibrutinib: First global approval. Drugs 2014, 74, 263–271. [Google Scholar] [CrossRef]
- Markham, A. Idelalisib: First global approval. Drugs 2014, 74, 1701–1707. [Google Scholar] [CrossRef]
- Dienstmann, R.; Jang, I.S.; Bot, B.; Friend, S.; Guinney, J. Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov. 2015, 5, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Invest. Dermatol. 2021, 141, 23–31. [Google Scholar] [CrossRef]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Tissot, T.; Ujvari, B.; Solary, E.; Lassus, P.; Roche, B.; Thomas, F. Do cell-autonomous and non-cell-autonomous effects drive the structure of tumor ecosystems? Biochim. Biophys. Acta 2016, 1865, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor Microenvironment-Derived NRG1 Promotes Antiandrogen Resistance in Prostate Cancer. Cancer Cell 2020, 38, 279–296.e279. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Breakstone, R. Colon cancer and immunotherapy-can we go beyond microsatellite instability? Transl. Gastroenterol. Hepatol. 2021, 6, 12. [Google Scholar] [CrossRef]
- Gogas, H.; Dreno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: Primary results from the randomized phase III IMspire170 study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Uno, T.; Takeda, K.; Kojima, Y.; Yoshizawa, H.; Akiba, H.; Mittler, R.S.; Gejyo, F.; Okumura, K.; Yagita, H.; Smyth, M.J. Eradication of established tumors in mice by a combination antibody-based therapy. Nat. Med. 2006, 12, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tokheim, C.; Gu, S.S.; Wang, B.; Tang, Q.; Li, Y.; Traugh, N.; Zeng, Z.; Zhang, Y.; Li, Z.; et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Kooreman, N.G.; Kim, Y.; de Almeida, P.E.; Termglinchan, V.; Diecke, S.; Shao, N.Y.; Wei, T.T.; Yi, H.; Dey, D.; Nelakanti, R.; et al. Autologous iPSC-Based Vaccines Elicit Anti-tumor Responses In Vivo. Cell Stem Cell 2018, 22, 501–513.e507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Kracker, S.; Yasuda, T.; Casola, S.; Vanneman, M.; Homig-Holzel, C.; Wang, Z.; Derudder, E.; Li, S.; Chakraborty, T.; et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell 2012, 148, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e836. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Lin, L.J.; Moon, H.; Pham, A.T.; Wang, X.; Liu, S.; Ji, S.; Rezek, V.; Shimizu, S.; Ruiz, M.; et al. Propagating Humanized BLT Mice for the Study of Human Immunology and Immunotherapy. Stem Cells Dev. 2016, 25, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.N.; Lacroix, R.; et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019, 178, 585–599.e515. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gestermann, N.; Saugy, D.; Martignier, C.; Tille, L.; Fuertes Marraco, S.A.; Zettl, M.; Tirapu, I.; Speiser, D.E.; Verdeil, G. LAG-3 and PD-1+LAG-3 inhibition promote anti-tumor immune responses in human autologous melanoma/T cell co-cultures. Oncoimmunology 2020, 9, 1736792. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2019, 176, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ootani, A.; Kuo, C. An Air-Liquid Interface Culture System for 3D Organoid Culture of Diverse Primary Gastrointestinal Tissues. Methods Mol. Biol. 2016, 1422, 33–40. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e1512. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e122. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, M.J.; Sood, A.; Sevinsky, C.; Pris, A.D.; Zavodszky, M.I.; Ginty, F. Emerging understanding of multiscale tumor heterogeneity. Front. Oncol. 2014, 4, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e847. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Hurwitz, A.A.; Foster, B.A.; Kwon, E.D.; Truong, T.; Choi, E.M.; Greenberg, N.M.; Burg, M.B.; Allison, J.P. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000, 60, 2444–2448. [Google Scholar] [PubMed]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Mseka, T.; Bamburg, J.R.; Cramer, L.P. ADF/cofilin family proteins control formation of oriented actin-filament bundles in the cell body to trigger fibroblast polarization. J. Cell Sci. 2007, 120, 4332–4344. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)evolution: Unveiling Tumor-Stroma Interactions. Trends Cancer 2021, 7, 249–264. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Li, E.; Weiner, L.M. 3D Culture Systems for Exploring Cancer Immunology. Cancers 2020, 13, 56. [Google Scholar] [CrossRef]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.A.; Kazolias, D.; Duignan, D.B. Use of cryopreserved human hepatocytes in sandwich culture to measure hepatobiliary transport. Drug Metab. Dispos. 2006, 34, 1658–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.M.; Barton, H.A.; Lai, Y.; Bi, Y.A.; Kimoto, E.; Kempshall, S.; Tate, S.C.; El-Kattan, A.; Houston, J.B.; Galetin, A.; et al. Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 2012, 40, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, Y.K.; Yang, M.T.; Desai, R.A.; Yu, X.; Liu, Z.; Chen, C.S. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat. Methods 2010, 7, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Burdett, E.; Kasper, F.K.; Mikos, A.G.; Ludwig, J.A. Engineering tumors: A tissue engineering perspective in cancer biology. Tissue Eng. Part B Rev. 2010, 16, 351–359. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Nii, T.; Kuwahara, T.; Makino, K.; Tabata, Y. A Co-Culture System of Three-Dimensional Tumor-Associated Macrophages and Three-Dimensional Cancer-Associated Fibroblasts Combined with Biomolecule Release for Cancer Cell Migration. Tissue Eng. Part A 2020, 26, 1272–1282. [Google Scholar] [CrossRef]
- Molina, E.R.; Chim, L.K.; Salazar, M.C.; Koons, G.L.; Menegaz, B.A.; Ruiz-Velasco, A.; Lamhamedi-Cherradi, S.E.; Vetter, A.M.; Satish, T.; Cuglievan, B.; et al. 3D Tissue-Engineered Tumor Model for Ewing’s Sarcoma That Incorporates Bone-like ECM and Mineralization. ACS Biomater. Sci. Eng. 2020, 6, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutmacher, D.W. Biomaterials offer cancer research the third dimension. Nat. Mater. 2010, 9, 90–93. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. A Cancer Invasion Model Combined with Cancer-Associated Fibroblasts Aggregates Incorporating Gelatin Hydrogel Microspheres Containing a p53 Inhibitor. Tissue Eng. Part C Methods 2019, 25, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Fong, E.L.; Lamhamedi-Cherradi, S.E.; Burdett, E.; Ramamoorthy, V.; Lazar, A.J.; Kasper, F.K.; Farach-Carson, M.C.; Vishwamitra, D.; Demicco, E.G.; Menegaz, B.A.; et al. Modeling Ewing sarcoma tumors in vitro with 3D scaffolds. Proc. Natl. Acad. Sci. USA 2013, 110, 6500–6505. [Google Scholar] [CrossRef] [Green Version]
- Boucherit, N.; Gorvel, L.; Olive, D. 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front. Immunol. 2020, 11, 603640. [Google Scholar] [CrossRef]
- Ho, T.; Msallam, R. Tissues and Tumor Microenvironment (TME) in 3D: Models to Shed Light on Immunosuppression in Cancer. Cells 2021, 10, 831. [Google Scholar] [CrossRef]
- Ramos-Espinosa, G.; Wang, Y.; Brandner, J.M.; Schneider, S.W.; Gorzelanny, C. Melanoma Associated Chitinase 3-Like 1 Promoted Endothelial Cell Activation and Immune Cell Recruitment. Int. J. Mol. Sci. 2021, 22, 3912. [Google Scholar] [CrossRef]
- Lugo-Cintron, K.M.; Gong, M.M.; Ayuso, J.M.; Tomko, L.A.; Beebe, D.J.; Virumbrales-Munoz, M.; Ponik, S.M. Breast Fibroblasts and ECM Components Modulate Breast Cancer Cell Migration Through the Secretion of MMPs in a 3D Microfluidic Co-Culture Model. Cancers 2020, 12, 1173. [Google Scholar] [CrossRef]
- Mi, S.; Liu, Z.; Du, Z.; Yi, X.; Sun, W. Three-dimensional microfluidic tumor-macrophage system for breast cancer cell invasion. Biotechnol. Bioeng. 2019, 116, 1731–1741. [Google Scholar] [CrossRef]
- Saraiva, D.P.; Matias, A.T.; Braga, S.; Jacinto, A.; Cabral, M.G. Establishment of a 3D Co-culture With MDA-MB-231 Breast Cancer Cell Line and Patient-Derived Immune Cells for Application in the Development of Immunotherapies. Front. Oncol. 2020, 10, 1543. [Google Scholar] [CrossRef]
- Fong, E.L.; Wan, X.; Yang, J.; Morgado, M.; Mikos, A.G.; Harrington, D.A.; Navone, N.M.; Farach-Carson, M.C. A 3D in vitro model of patient-derived prostate cancer xenograft for controlled interrogation of in vivo tumor-stromal interactions. Biomaterials 2016, 77, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, D.; Xu, T.; Liu, P.; Cao, Y.; Wang, Y.; Yang, X.; Xu, X.; Wang, X.; Niu, H. Bladder cancer cells re-educate TAMs through lactate shuttling in the microfluidic cancer microenvironment. Oncotarget 2015, 6, 39196–39210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Gomez, K.E.; Wu, F.; Keysar, S.B.; Morton, J.J.; Miller, B.; Chimed, T.S.; Le, P.N.; Nieto, C.; Chowdhury, F.N.; Tyagi, A.; et al. Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells. Cancer Res. 2020, 80, 4185–4198. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2020, 20, 279–293. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Ha, A.; de Lau, W.; Yuki, K.; Santos, A.J.M.; You, C.; Geurts, M.H.; Puschhof, J.; Pleguezuelos-Manzano, C.; Peng, W.C.; et al. Next-Generation Surrogate Wnts Support Organoid Growth and Deconvolute Frizzled Pleiotropy In Vivo. Cell Stem Cell 2020, 27, 840–851.e846. [Google Scholar] [CrossRef] [PubMed]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Martinez-Silgado, A.; Akkerman, N.; Saftien, A.; Boot, C.; de Waal, A.; Beumer, J.; Dutta, D.; Heo, I.; et al. Intestinal organoid cocultures with microbes. Nat. Protoc. 2021, 16, 4633–4649. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e1519. [Google Scholar] [CrossRef] [Green Version]
- Ooft, S.N.; Weeber, F.; Schipper, L.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van de Haar, J.; Prevoo, W.; van Werkhoven, E.; Snaebjornsson, P.; et al. Prospective experimental treatment of colorectal cancer patients based on organoid drug responses. ESMO Open 2021, 6, 100103. [Google Scholar] [CrossRef] [PubMed]
- Bahcecioglu, G.; Basara, G.; Ellis, B.W.; Ren, X.; Zorlutuna, P. Breast cancer models: Engineering the tumor microenvironment. Acta Biomater. 2020, 106, 1–21. [Google Scholar] [CrossRef]
- Datta, P.; Dey, M.; Ataie, Z.; Unutmaz, D.; Ozbolat, I.T. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis. Oncol. 2020, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Lee, J.; Cuddihy, M.J.; Kotov, N.A. Three-dimensional cell culture matrices: State of the art. Tissue Eng. Part B Rev. 2008, 14, 61–86. [Google Scholar] [CrossRef] [Green Version]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [Green Version]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Barcelos, L.S.; Souza, J.B.; Marangoni, K.; Goulart, L.R.; Alonso-Goulart, V. Comparative Assay of 2D and 3D Cell Culture Models: Proliferation, Gene Expression and Anticancer Drug Response. Curr. Pharm. Des. 2018, 24, 1689–1694. [Google Scholar] [CrossRef]
- Takahashi, N.; Cho, P.; Selfors, L.M.; Kuiken, H.J.; Kaul, R.; Fujiwara, T.; Harris, I.S.; Zhang, T.; Gygi, S.P.; Brugge, J.S. 3D Culture Models with CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol. Cell 2020, 80, 828–844.e826. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.N.; Nasir, A.; Camacho, S.; Berry, D.L.; Schmidt, M.O.; Pearson, G.W.; Riegel, A.T.; Wellstein, A. Monitoring Cancer Cell Invasion and T-Cell Cytotoxicity in 3D Culture. J. Vis. Exp. 2020, e61392. [Google Scholar] [CrossRef]
- Meier-Hubberten, J.C.; Sanderson, M.P. Establishment and Analysis of a 3D Co-Culture Spheroid Model of Pancreatic Adenocarcinoma for Application in Drug Discovery. Methods Mol. Biol. 2019, 1953, 163–179. [Google Scholar] [CrossRef]
- Giannattasio, A.; Weil, S.; Kloess, S.; Ansari, N.; Stelzer, E.H.; Cerwenka, A.; Steinle, A.; Koehl, U.; Koch, J. Cytotoxicity and infiltration of human NK cells in in vivo-like tumor spheroids. BMC Cancer 2015, 15, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Assante Miranda, L.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J. Immunother. Cancer 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Di Blasio, S.; van Wigcheren, G.F.; Becker, A.; van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nat. Commun. 2020, 11, 2749. [Google Scholar] [CrossRef]
- Kross, K.W.; Heimdal, J.H.; Olsnes, C.; Olofsson, J.; Aarstad, H.J. Co-culture of head and neck squamous cell carcinoma spheroids with autologous monocytes predicts prognosis. Scand. J. Immunol. 2008, 67, 392–399. [Google Scholar] [CrossRef]
- Gopal, S.; Kwon, S.J.; Ku, B.; Lee, D.W.; Kim, J.; Dordick, J.S. 3D tumor spheroid microarray for high-throughput, high-content natural killer cell-mediated cytotoxicity. Commun. Biol. 2021, 4, 893. [Google Scholar] [CrossRef]
- Scognamiglio, G.; De Chiara, A.; Parafioriti, A.; Armiraglio, E.; Fazioli, F.; Gallo, M.; Aversa, L.; Camerlingo, R.; Cacciatore, F.; Colella, G.; et al. Patient-derived organoids as a potential model to predict response to PD-1/PD-L1 checkpoint inhibitors. Br. J. Cancer 2019, 121, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Exposito, R.; Semiannikova, M.; Griffiths, B.; Khan, K.; Barber, L.J.; Woolston, A.; Spain, G.; von Loga, K.; Challoner, B.; Patel, R.; et al. CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J. Immunother. Cancer 2019, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, E.; Hau, A.C.; Oudin, A.; Golebiewska, A.; Niclou, S.P. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front. Oncol. 2020, 10, 604121. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef]
- Wu, M.H.; Huang, S.B.; Lee, G.B. Microfluidic cell culture systems for drug research. Lab Chip 2010, 10, 939–956. [Google Scholar] [CrossRef]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef] [Green Version]
- Regehr, K.J.; Domenech, M.; Koepsel, J.T.; Carver, K.C.; Ellison-Zelski, S.J.; Murphy, W.L.; Schuler, L.A.; Alarid, E.T.; Beebe, D.J. Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 2009, 9, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Toepke, M.W.; Beebe, D.J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 2006, 6, 1484–1486. [Google Scholar] [CrossRef]
- Berthier, E.; Warrick, J.; Yu, H.; Beebe, D.J. Managing evaporation for more robust microscale assays. Part 1. Volume loss in high throughput assays. Lab Chip 2008, 8, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Araci, I.E.; Quake, S.R. Microfluidic very large scale integration (mVLSI) with integrated micromechanical valves. Lab Chip 2012, 12, 2803–2806. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, J.; Crouzier, T.; Selimovic, S.; Boudou, T.; Khademhosseini, A.; Picart, C. Gradients of physical and biochemical cues on polyelectrolyte multilayer films generated via microfluidics. Lab Chip 2013, 13, 1562–1570. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.T.; Wood, J.A.; Shah, N.M.; Hughbanks, M.L.; Russell, P.; Barakat, A.I.; Murphy, C.J. Integration of basal topographic cues and apical shear stress in vascular endothelial cells. Biomaterials 2012, 33, 4126–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, M.J.; Domansky, K.; Kaazempur-Mofrad, M.R.; Kalezi, A.; Capitano, A.; Upadhyaya, A.; Kurzawski, P.; Wack, K.E.; Stolz, D.B.; Kamm, R.; et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol. Bioeng. 2002, 78, 257–269. [Google Scholar] [CrossRef]
- Radhakrishnan, J.; Varadaraj, S.; Dash, S.K.; Sharma, A.; Verma, R.S. Organotypic cancer tissue models for drug screening: 3D constructs, bioprinting and microfluidic chips. Drug Discov. Today 2020, 25, 879–890. [Google Scholar] [CrossRef]
- Chung, S.; Yun, H.; Kamm, R.D. Nanointerstice-driven microflow. Small 2009, 5, 609–613. [Google Scholar] [CrossRef]
- Lam, S.F.; Bishop, K.W.; Mintz, R.; Fang, L.; Achilefu, S. Calcium carbonate nanoparticles stimulate cancer cell reprogramming to suppress tumor growth and invasion in an organ-on-a-chip system. Sci. Rep. 2021, 11, 9246. [Google Scholar] [CrossRef]
- Humayun, M.; Ayuso, J.M.; Brenneke, R.A.; Virumbrales-Munoz, M.; Lugo-Cintron, K.; Kerr, S.; Ponik, S.M.; Beebe, D.J. Elucidating cancer-vascular paracrine signaling using a human organotypic breast cancer cell extravasation model. Biomaterials 2021, 270, 120640. [Google Scholar] [CrossRef]
- Chen, M.B.; Hajal, C.; Benjamin, D.C.; Yu, C.; Azizgolshani, H.; Hynes, R.O.; Kamm, R.D. Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc. Natl. Acad. Sci. USA 2018, 115, 7022–7027. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.B.; Lamar, J.M.; Li, R.; Hynes, R.O.; Kamm, R.D. Elucidation of the Roles of Tumor Integrin beta1 in the Extravasation Stage of the Metastasis Cascade. Cancer Res. 2016, 76, 2513–2524. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.A.; Chen, C.S. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar] [CrossRef] [Green Version]
- Ayensa-Jimenez, J.; Perez-Aliacar, M.; Randelovic, T.; Olivan, S.; Fernandez, L.; Sanz-Herrera, J.A.; Ochoa, I.; Doweidar, M.H.; Doblare, M. Mathematical formulation and parametric analysis of in vitro cell models in microfluidic devices: Application to different stages of glioblastoma evolution. Sci. Rep. 2020, 10, 21193. [Google Scholar] [CrossRef]
- Cho, H.Y.; Choi, J.H.; Kim, K.J.; Shin, M.; Choi, J.W. Microfluidic System to Analyze the Effects of Interleukin 6 on Lymphatic Breast Cancer Metastasis. Front. Bioeng. Biotechnol. 2020, 8, 611802. [Google Scholar] [CrossRef]
- Mollica, H.; Palomba, R.; Primavera, R.; Decuzzi, P. Two-Channel Compartmentalized Microfluidic Chip for Real-Time Monitoring of the Metastatic Cascade. ACS Biomater. Sci. Eng. 2019, 5, 4834–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; Lee, J.H.; Shin, Y.; Chung, S.; Kuh, H.J. Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.D.; Kratz, A.; Park, J.G.; Barrientos, E.S.; Saini, H.; Nguyen, T.; Pockaj, B.; Mouneimne, G.; LaBaer, J.; Nikkhah, M. A Human Organotypic Microfluidic Tumor Model Permits Investigation of the Interplay between Patient-Derived Fibroblasts and Breast Cancer Cells. Cancer Res. 2019, 79, 3139–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Lin, X.; Lin, L.; Feng, Q.; Kitamori, T.; Lin, J.M.; Sun, J. Integrated Microfluidic Platform with Multiple Functions To Probe Tumor-Endothelial Cell Interaction. Anal. Chem. 2017, 89, 10037–10044. [Google Scholar] [CrossRef]
- Michna, R.; Gadde, M.; Ozkan, A.; DeWitt, M.; Rylander, M. Vascularized microfluidic platforms to mimic the tumor microenvironment. Biotechnol. Bioeng. 2018, 115, 2793–2806. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Segall, J.E.; Wu, M. Microfluidic modeling of the biophysical microenvironment in tumor cell invasion. Lab Chip 2017, 17, 3221–3233. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.B.; Leslie, M.T.; Gaskins, H.R.; Kenis, P.J.A. Methods to study the tumor microenvironment under controlled oxygen conditions. Trends Biotechnol. 2014, 32, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.F.; Shirure, V.S.; Chu, Y.E.; Soetikno, A.G.; George, S.C. Microfluidic device to attain high spatial and temporal control of oxygen. PLoS ONE 2018, 13, e0209574. [Google Scholar] [CrossRef] [Green Version]
- McMinn, P.H.; Hind, L.E.; Huttenlocher, A.; Beebe, D.J. Neutrophil trafficking on-a-chip: An in vitro, organotypic model for investigating neutrophil priming, extravasation, and migration with spatiotemporal control. Lab Chip 2019, 19, 3697–3705. [Google Scholar] [CrossRef] [PubMed]
- Aung, A.; Kumar, V.; Theprungsirikul, J.; Davey, S.K.; Varghese, S. An Engineered Tumor-on-a-Chip Device with Breast Cancer-Immune Cell Interactions for Assessing T-cell Recruitment. Cancer Res. 2020, 80, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Jaeger, M.S.; Schenkel, T.; Breinig, T.; Meyerhans, A.; Duschl, C. T cell activation on a single-cell level in dielectrophoresis-based microfluidic devices. J. Chromatogr. A 2008, 1202, 83–89. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, H.O.; Kim, K.D.; Kim, S.K.; Lee, S.K.; Jung, H. Monitoring the status of T-cell activation in a microfluidic system. Analyst 2011, 136, 2831–2836. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Truttschel, R.; Gong, M.M.; Humayun, M.; Virumbrales-Munoz, M.; Vitek, R.; Felder, M.; Gillies, S.D.; Sondel, P.; Wisinski, K.B.; et al. Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunology 2019, 8, 1553477. [Google Scholar] [CrossRef] [Green Version]
- Briones, J.; Espulgar, W.; Koyama, S.; Takamatsu, H.; Tamiya, E.; Saito, M. The future of microfluidics in immune checkpoint blockade. Cancer Gene Ther. 2020, 28, 895–910. [Google Scholar] [CrossRef]
- Kloten, V.; Lampignano, R.; Krahn, T.; Schlange, T. Circulating Tumor Cell PD-L1 Expression as Biomarker for Therapeutic Efficacy of Immune Checkpoint Inhibition in NSCLC. Cells 2019, 8, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harb, W.; Fan, A.; Tran, T.; Danila, D.C.; Keys, D.; Schwartz, M.; Ionescu-Zanetti, C. Mutational Analysis of Circulating Tumor Cells Using a Novel Microfluidic Collection Device and qPCR Assay. Transl Oncol. 2013, 6, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Po, J.W.; Ma, Y.; Balakrishna, B.; Brungs, D.; Azimi, F.; de Souza, P.; Becker, T.M. Immunomagnetic isolation of circulating melanoma cells and detection of PD-L1 status. PLoS ONE 2019, 14, e0211866. [Google Scholar] [CrossRef]
- Jan, Y.J.; Chen, J.F.; Zhu, Y.; Lu, Y.T.; Chen, S.H.; Chung, H.; Smalley, M.; Huang, Y.W.; Dong, J.; Chen, L.C.; et al. NanoVelcro rare-cell assays for detection and characterization of circulating tumor cells. Adv. Drug Deliv. Rev. 2018, 125, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, Y.; Zeng, Y.; He, M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, S.S.; Dunlay, C.J.; Simeone, D.M.; Nagrath, S. Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip 2014, 14, 1891–1900. [Google Scholar] [CrossRef]
- Iliescu, F.S.; Vrtacnik, D.; Neuzil, P.; Iliescu, C. Microfluidic Technology for Clinical Applications of Exosomes. Micromachines 2019, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Demaree, B.; Weisgerber, D.; Lan, F.; Abate, A.R. An Ultrahigh-throughput Microfluidic Platform for Single-cell Genome Sequencing. J. Vis. Exp. 2018, 57598. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merouane, A.; Rey-Villamizar, N.; Lu, Y.; Liadi, I.; Romain, G.; Lu, J.; Singh, H.; Cooper, L.J.; Varadarajan, N.; Roysam, B. Automated profiling of individual cell-cell interactions from high-throughput time-lapse imaging microscopy in nanowell grids (TIMING). Bioinformatics 2015, 31, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Yang, L.; Wei, W.; Shi, Q. Microchip-based single-cell functional proteomics for biomedical applications. Lab Chip 2017, 17, 1250–1263. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Varadarajan, N. Single-cell technologies for profiling T cells to enable monitoring of immunotherapies. Curr. Opin. Chem. Eng. 2018, 19, 142–152. [Google Scholar] [CrossRef]
- Armbrecht, L.; Muller, R.S.; Nikoloff, J.; Dittrich, P.S. Single-cell protein profiling in microchambers with barcoded beads. Microsyst. Nanoeng. 2019, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, S.; Tasoglu, S. A Bioprinted Liver-on-a-Chip for Drug Screening Applications. Trends Biotechnol. 2016, 34, 681–682. [Google Scholar] [CrossRef]
- Riley, A.; Green, V.; Cheah, R.; McKenzie, G.; Karsai, L.; England, J.; Greenman, J. A novel microfluidic device capable of maintaining functional thyroid carcinoma specimens ex vivo provides a new drug screening platform. BMC Cancer 2019, 19, 259. [Google Scholar] [CrossRef] [Green Version]
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11, 5271. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.J.; Dhingra, K.; Prabhakar, D.; Chandrasekar, V.; Natarajan, S.K.; Vasan, A.S.; Kulkarni, A.; Shafiee, H. A microfluidic platform for drug screening in a 3D cancer microenvironment. Biosens. Bioelectron. 2017, 94, 632–642. [Google Scholar] [CrossRef]
- Allen, S.G.; Chen, Y.C.; Madden, J.M.; Fournier, C.L.; Altemus, M.A.; Hiziroglu, A.B.; Cheng, Y.H.; Wu, Z.F.; Bao, L.; Yates, J.A.; et al. Macrophages Enhance Migration in Inflammatory Breast Cancer Cells via RhoC GTPase Signaling. Sci. Rep. 2016, 6, 39190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Weigelt, B.; Peterse, J.L.; van ‘t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Kim, H.; Chung, H.; Kim, J.; Choi, D.H.; Shin, Y.; Kang, Y.G.; Kim, B.M.; Seo, S.U.; Chung, S.; Seok, S.H. Macrophages-Triggered Sequential Remodeling of Endothelium-Interstitial Matrix to Form Pre-Metastatic Niche in Microfluidic Tumor Microenvironment. Adv. Sci. 2019, 6, 1900195. [Google Scholar] [CrossRef]
- Dangles, V.; Validire, P.; Wertheimer, M.; Richon, S.; Bovin, C.; Zeliszewski, D.; Vallancien, G.; Bellet, D. Impact of human bladder cancer cell architecture on autologous T-lymphocyte activation. Int. J. Cancer 2002, 98, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feder-Mengus, C.; Ghosh, S.; Weber, W.P.; Wyler, S.; Zajac, P.; Terracciano, L.; Oertli, D.; Heberer, M.; Martin, I.; Spagnoli, G.C.; et al. Multiple mechanisms underlie defective recognition of melanoma cells cultured in three-dimensional architectures by antigen-specific cytotoxic T lymphocytes. Br. J. Cancer 2007, 96, 1072–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reislander, T.; Agarkova, I.; Kelm, J.M.; et al. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunol. Immunother. 2017, 66, 129–140. [Google Scholar] [CrossRef]
- de Wit, S.; van Dalum, G.; Lenferink, A.T.; Tibbe, A.G.; Hiltermann, T.J.; Groen, H.J.; van Rijn, C.J.; Terstappen, L.W. The detection of EpCAM(+) and EpCAM(-) circulating tumor cells. Sci. Rep. 2015, 5, 12270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.C.; Reese, L.M.; Bickford, L.R.; Verbridge, S.S. Toward the Broad Adoption of 3D Tumor Models in the Cancer Drug Pipeline. ACS Biomater. Sci. Eng. 2015, 1, 877–894. [Google Scholar] [CrossRef]
- Kimlin, L.C.; Casagrande, G.; Virador, V.M. In vitro three-dimensional (3D) models in cancer research: An update. Mol. Carcinog. 2013, 52, 167–182. [Google Scholar] [CrossRef]
- Unger, C.; Kramer, N.; Walzl, A.; Scherzer, M.; Hengstschlager, M.; Dolznig, H. Modeling human carcinomas: Physiologically relevant 3D models to improve anti-cancer drug development. Adv. Drug Deliv. Rev. 2014, 79, 50–67. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Bergdorf, K.; Wolf, M.; Bharti, V.; Shattuck-Brandt, R.; Blevins, A.; Jones, C.; Phifer, C.; Lee, M.; Lowe, C.; et al. Fine-Needle Aspiration-Based Patient-Derived Cancer Organoids. iScience 2020, 23, 101408. [Google Scholar] [CrossRef]
- Joshi, P.; Lee, M.Y. High Content Imaging (HCI) on Miniaturized Three-Dimensional (3D) Cell Cultures. Biosensors 2015, 5, 768–790. [Google Scholar] [CrossRef] [Green Version]
- Smith, Q.; Gerecht, S. Going with the flow: Microfluidic platforms in vascular tissue engineering. Curr. Opin. Chem. Eng. 2014, 3, 42–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.J.; Kuen, D.S.; Chung, Y. Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
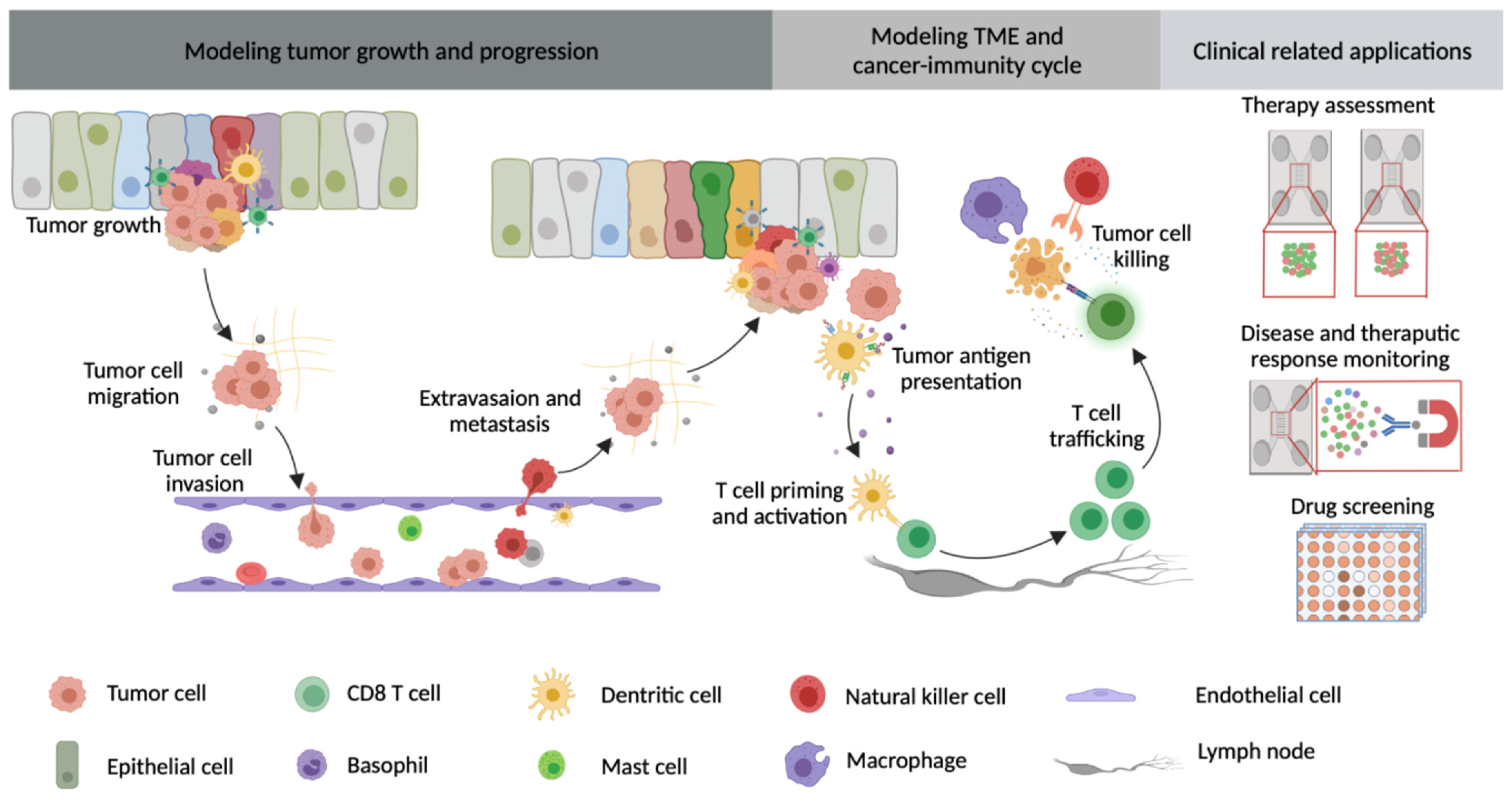

{kind=link}
| Type | Models | Material Source | Applications | Advantages | Disadvantages | Reference |
|---|---|---|---|---|---|---|
| In vivo murine models | Syngeneic tumor models | -Immune-competent mice: C57BL/6, BALB/c, FVB, etc. -Transplantable cells: B16, 4T1, CT26, etc. | -Tumor formation and progression -Evaluate antitumor immune response | -Have physiologically relevant tumor microenvironment -Easy to manipulate | -Variability of phenotype because of the site of engraftment -Lack of heterogeneity | [43,44,45,46,47] |
| Genetically engineered mouse models (GEMM) | -Immune-competent mice: C57BL/6, etc. | -Autochthonous tumor development -Evaluate antitumor immune response -Modeling immune-related adverse events (irAEs) | -Have naïve TME -Tumor initiation and progression driven by relevant genetic alterations | -Variability in tumor penetrance and latency -Low immunogenicity due to defined alterations | [48,49,50] | |
| Humanized mouse | -Immune-deficient mice: SCID, NOD, NSG, etc. | -Evaluate antitumor therapies | -Reproduce genomic heterogeneity of human disease -Have reconstituted human immune system | -Require autologous immune system reconstitution -Low rates and duration of immune reconstitution | [45,51,52,53] | |
| 2D | Coculture | -Tumor cells -TME components (macrophages, dendritic cells, fibroblast cells, etc.) | -Study the interaction between tumor and immune cells (cytokine secretion, tumor killing, etc.) | -Easy to manipulate -Can be used in high-throughput study | -Lack of native immune and stromal components -Limited reflection in tumor morphological phenotype | [54,55] |
| 3D | Spheroids | Coculture: cell lines, mouse- or patient-derived tissues, and other TME components (macrophages, T cells, etc.) | -Study the interaction between tumor and immune cells -Evaluate antitumor immune response | -Easy to manipulate -Can reflect genetic alterations and keep morphological phenotype of original tumor | -Lack of native immune and stromal components | [56,57] |
| Microfluidic devices: cell lines, mouse- or patient-derived tissues | -Study the interaction between tumor and immune cells -Evaluate the efficacy of therapeutic combinations -Profile secreted cytokines | -Require limited material (cells, media, reagents, etc.) -Can reflect genetic alterations and keep morphological phenotype of original tumor -Preserve immune cell population in TME | -Size limitation -Require microfluidic devices -Only have native tumor-infiltrating immune cells -Cannot model T-cell trafficking | [58,59,60] | ||
| Organoids | Coculture: mouse- or patient-derived tissues and other TME components (macrophages, dendritic cells, etc.) | -Evaluate antitumor immune response -Assessment of tumor organoid killing | -Easy to enrich and expand tumor organoids -Can reflect genetic alterations and keep morphological phenotype of original tumor | -Lack of native immune and stromal components | [61,62,63] | |
| ALI (Air-Liquid Interface) culture: mouse or patient-derived tissues | -Study the interaction between tumor and immune cells -Evaluate antitumor immune response -Assessment of tumor organoid killing | -Can reflect genetic alterations and keep morphological phenotype of original tumor -Preserve multiple immune cells and fibroblasts in TME | -Only have native tumor-infiltrating immune cells -Cannot model T-cell trafficking | [61,62,64] |
| Applications | Models | Experiment Design | Microfluidic Features | Reference |
|---|---|---|---|---|
| Cancer growth and progression | Tumor growth | Coculture cancer cells with fibroblasts | With fibronectin-rich matrix | [99] |
| Culture cancer cells with/without treatment of fibrin | A bifurcated microfluidic device allowing comparison between two different cell environments | [148] | ||
| Tumor migration and extravasation | Treat cancer cells with different secreted factors | Use a monolayer of endothelial cells to mimic microvasculature | [149,150] | |
| Coculture cancer cells with fibroblast | [151] | |||
| Angiogenesis | Test the effects of multiple angiogenic factors on angiogenesis | Use biomimetic model to reconstitute angiogenic sprouting in microfluidic device | [152,153] | |
| Cancer metastasis | Treat cancer cells with proinflammatory cytokine (e.g., IL-6) | Have lymph vessel–tissue–blood vessel structure | [154] | |
| Treat cancer cells with proinflammatory cytokine (e.g., TNFα) | Use a monolayer of endothelial cells to mimic microvasculature | [155] | ||
| TME and cancer–immunity cycle | TME modeling | Coculture tumor spheroids with other TME components (e.g., CAF, stroma cells, endothelial cells) | Culture spheroids | [156,157,158] |
| Vascularized system modeling | Contain interconnected microchannels to model a highly vascularized system | [159] | ||
| ECM and interstitial flow modeling | Modeling biophysical features, such as ECM and interstitial flow in TME | [160] | ||
| Oxygen concentration modeling | Include three parallel connected tissue chambers and an oxygen scavenger channel to control oxygen concentration | [161,162] | ||
| Study the interaction between tumor and immune cells | Culture murine- and patient-derived organotypic tumor spheroids (MDOTSs/PDOTSs) | [60,138] | ||
| Immune cell migration/recruitment | Identify potential factors (e.g., chemokine, cytokines) affecting immune cell migration | Integrate microscopy technology with microfluidic chips or use microfluidic devices designed for co-culture | [98,103,163,164] | |
| T lymphocyte activation | Monitor T-cell activation by analyzing CD69 expression | Use a chip containing microelectrodes to get dielectrophoretic manipulation | [165] | |
| Monitor T-cell activation by analyzing the binding of T cells to TNFα-treated human umbilical vein endothelial cells (HUVECs) | Adjustable shear stress | [166] | ||
| Clinica- related applications | Therapy assessment | Evaluate the efficacy of therapeutic combinations | Culture MDOTSs/PDOTSs | [60,138] |
| Coculture cancer spheroids with natural killer cells or antibody–cytokine regimens | Use a monolayer of endothelial cells to mimic microvasculature | [167] | ||
| Disease and therapeutic response monitoring | Analyze CTCs from patients to predict prognosis and evaluate progression-free survival and overall survival of patients | Exploit antibody-coated magnetic particles targeting EpCAM to detects and quantify CTCs | [168,169,170,171,172] | |
| Capture exosome to monitor immunotherapeutic response | Employ immunomagnetic beads/antibodies/chips to capture and measure exosomal tumor markers | [173,174,175] | ||
| Study intra-tumoral heterogeneity in microfluidic devices with scRNA-seq and understand therapeutic evasion | Incorporate different scRNA-seq techniques into microfluidic chips (e.g., droplet microfluidics, Microwell-seq microfluidics) | [176,177,178] | ||
| Monitor immune cell heterogeneity | Timelapse imaging microscopy-based microfluidic platform | [179] | ||
| Microfluidic devices integrating single- cell barcoding chip (SCBC) or antibody microarray (BOBarray) | [180,181,182] | |||
| Drug screening | Test drug toxicity with bio-printed hepatic spheroids | Use hepatic spheroids as material source to directly print liver tissue into the microfluidic device | [183] | |
| Evaluate the response of thyroid tissue to radioiodine sensitivity/adjuvant therapies in real time | Culture live-sliced human thyroid tissue | [184] | ||
| Provide dynamic and combinatorial drug screening | Culture pancreatic organoids | [185] | ||
| Chemotherapeutic drug testing and efficacy evaluation | Integration of microfluidics and electrical sensing modality | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Appelt, J.W.; Jenkins, R.W. Going with the Flow: Modeling the Tumor Microenvironment Using Microfluidic Technology. Cancers 2021, 13, 6052. https://doi.org/10.3390/cancers13236052
Xie H, Appelt JW, Jenkins RW. Going with the Flow: Modeling the Tumor Microenvironment Using Microfluidic Technology. Cancers. 2021; 13(23):6052. https://doi.org/10.3390/cancers13236052
Chicago/Turabian StyleXie, Hongyan, Jackson W. Appelt, and Russell W. Jenkins. 2021. "Going with the Flow: Modeling the Tumor Microenvironment Using Microfluidic Technology" Cancers 13, no. 23: 6052. https://doi.org/10.3390/cancers13236052
APA StyleXie, H., Appelt, J. W., & Jenkins, R. W. (2021). Going with the Flow: Modeling the Tumor Microenvironment Using Microfluidic Technology. Cancers, 13(23), 6052. https://doi.org/10.3390/cancers13236052