
Multiparametric Circulating Tumor Cell Analysis to Select Targeted Therapies for Breast Cancer Patients

 , , , , ,
, , , , ,  , ,
, ,  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Intervention
2.3. Enrichment and Enumeration of CTCs
2.4. Immunofluorescence Analysis
2.5. CTC Isolation
2.6. Whole-Genome Amplification
2.7. Whole-Exome Sequencing
2.8. Clonal Reconstruction
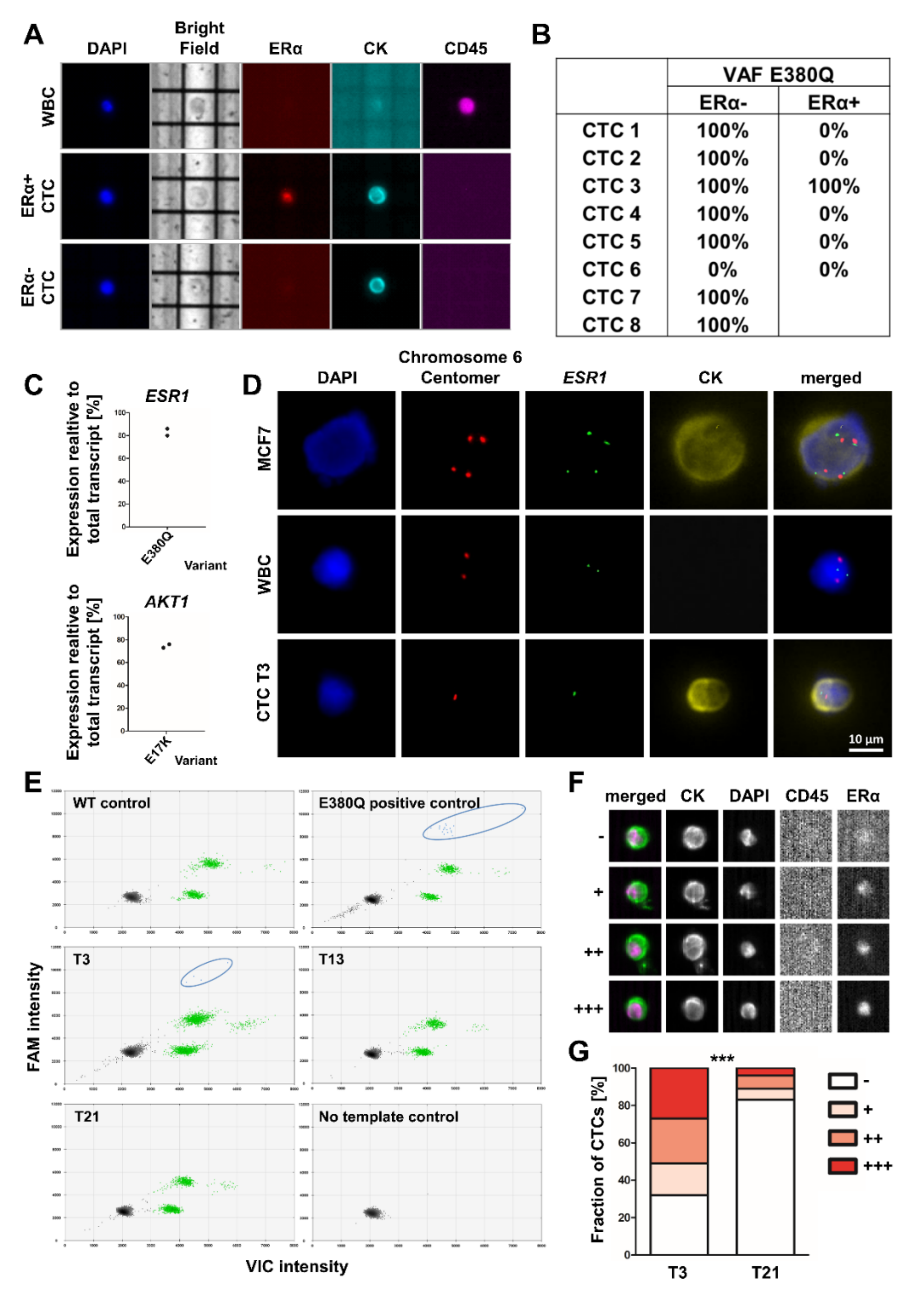
2.9. Sequencing of ESR1 and AKT1 from Single CTCs
2.10. Sequencing of PIK3CA Hotspots by ddPCR
2.11. Array Comparative Genome Hybridization
2.12. FISH Analysis
2.13. Multiplex ESR1-ddPCR from cfDNA and Data Analysis
2.14. RNA Sequencing
2.15. CTC Culture
2.16. Statistical Analysis
3. Results
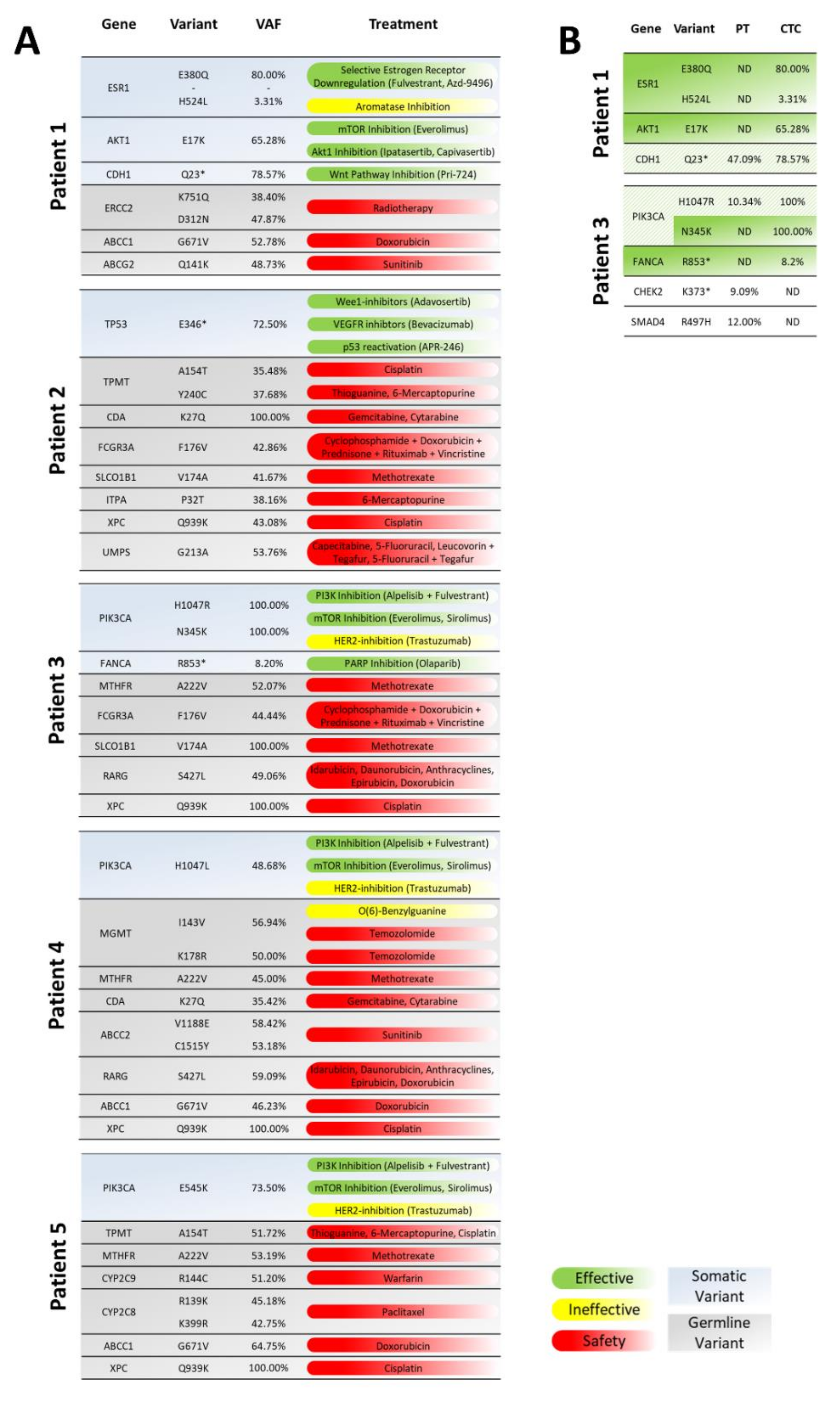
3.1. Whole-Exome Sequencing of CTCs to Provide CTC-Based Treatment Recommendations
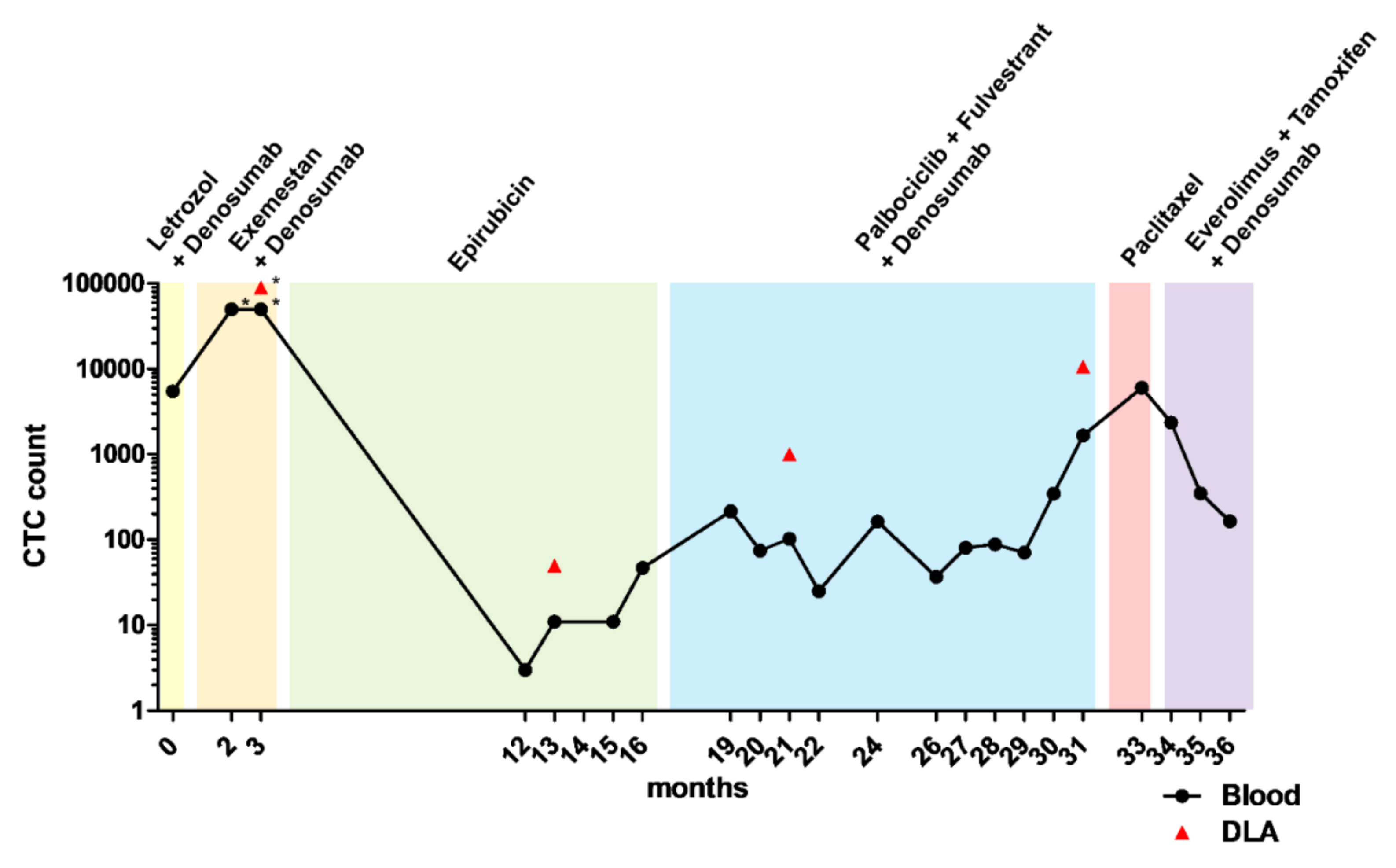
3.2. Analysis of Mutations and Chromosomal Aberrations during the Course of the Disease
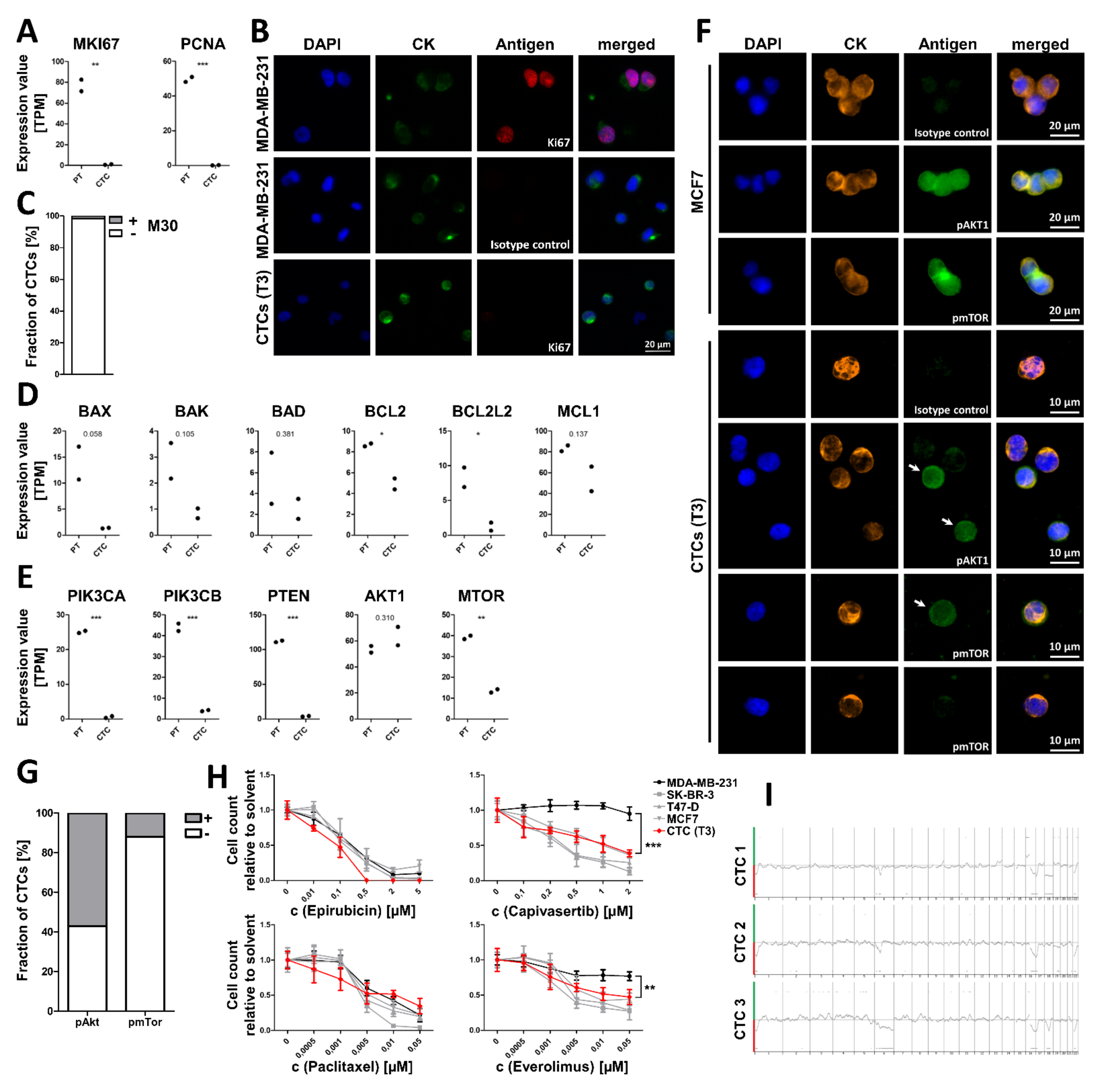
3.3. CTCs Exhibit No Indications for Proliferation but Show Reduced Apoptosis
3.4. In Vitro treatment of Cultured CTCs
3.5. CTC-Based Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, F.; Franken, A.; Fehm, T.; Neubauer, H. Navigation through inter- and intratumoral heterogeneity of endocrine resistance mechanisms in breast cancer: A potential role for Liquid Biopsies? Tumor Biol. 2017, 39, 1010428317731511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pellè, E.; Quaresmini, D.; Tucci, M. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Weber, B.; Meldgaard, P.; Hager, H.; Wu, L.; Wei, W.; Tsai, J.; Khalil, A.; Nexo, E. Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer 2014, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Grützmann, R.; Molnar, B.; Pilarsky, C.; Habermann, J.K.; Schlag, P.M.; Saeger, H.D.; Miehlke, S.; Stolz, T.; Model, F.; Roblick, U.J.; et al. Sensitive Detection of Colorectal Cancer in Peripheral Blood by Septin 9 DNA Methylation Assay. PLoS ONE 2008, 3, e3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazdur, R. FDA Oncology Update. Am. Health Drugs Benefits 2019, 12, 198–200. [Google Scholar]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nolé, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Janni, W.J.; Rack, B.; Terstappen, L.W.M.M.; Pierga, J.-Y.; Taran, F.-A.; Fehm, T.; Hall, C.; de Groot, M.R.; Bidard, F.-C.; Friedl, T.W.P.; et al. Pooled Analysis of the Prognostic Relevance of Circulating Tumor Cells in Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 2583–2593. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Ph, R.; Miller, M.C.; Reuben, J.M.; Ph, D.; Doyle, G.V.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Smerage, J.B.; Barlow, W.E.; Hortobagyi, G.N.; Winer, E.P.; Leyland-Jones, B.; Srkalovic, G.; Tejwani, S.; Schott, A.F.; Rourke, M.A.O.; Lew, D.L.; et al. Circulating Tumor Cells and Response to Chemotherapy in Metastatic Breast Cancer: SWOG S0500. J. Clin. Oncol. 2014, 32, 3483–3490. [Google Scholar] [CrossRef]
- Pestrin, M.; Bessi, S.; Puglisi, F.; Minisini, A.M.; Masci, G.; Battelli, N.; Ravaioli, A.; Gianni, L.; Di, R.; Carlo, M.; et al. Final results of a multicenter phase II clinical trial evaluating the activity of single-agent lapatinib in patients with HER2- negative metastatic breast cancer and HER2-positive circulating tumor cells. A proof-of-concept study. Breast Cancer Res. Treat. 2012, 134, 283–289. [Google Scholar] [CrossRef]
- Stebbing, J.; Payne, R.; Reise, J.; Frampton, A.E.; Avery, M.; Woodley, L.; Leo, A.D.; Pestrin, M.; Krell, J.; Coombes, R.C. The Efficacy of Lapatinib in Metastatic Breast Cancer with HER2 Non-Amplified Primary Tumors and EGFR Positive Circulating Tumor Cells: A Proof-Of-Concept Study. PLoS ONE 2013, 8, e62543. [Google Scholar] [CrossRef] [Green Version]
- Bidard, F.C. Clinical utility of circulating tumor cell count as a tool to chose between first line hormone therapy and chemotherapy for ER+ HER2- metastatic breast cancer: Results of the phase III STIC CTC trial. In Proceedings of the SABCS, San Antonio, TX, USA, 4–8 December 2018. [Google Scholar]
- Thill, M.; Liedtke, C.; Müller, V.; Janni, W.; Schmidt, M. AGO Recommendations for the Diagnosis and Treatment of Patients with Advanced and Metastatic Breast Cancer: Update 2018. Breast Care 2018, 13, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Fehm, T.N.; Meier-Stiegen, F.; Driemel, C.; Jäger, B.; Reinhardt, F.; Naskou, J.; Franken, A.; Neubauer, H.; Neves, R.P.L.; van Dalum, G.; et al. Diagnostic Leukapheresis for CTC Analysis in Breast Cancer Patients: CTC Frequency, Clinical Experiences and Recommendations for Standardized Reporting. Cytom. Part A 2018, 93, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.C.; Niederacher, D.; Topp, S.A.; Honisch, E.; Schumacher, S.; Schmitz, N.; Zacarias Fohrding, L.; Vay, C.; Hoffmann, I.; Kasprowicz, N.S.; et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16580–16585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.H.D.; Schneck, H.; Decker, Y.; Schömer, S.; Franken, A.; Endris, V.; Pfarr, N.; Weichert, W.; Niederacher, D.; Fehm, T.; et al. Isolation and characterization of circulating tumor cells using a novel workflow combining the CellSearch® system and the CellCelectorTM. Biotechnol. Prog. 2017, 33, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Velds, A.; Kemper, K.; Ranzani, M.; Bombardelli, L.; Hoogstraat, M.; Nevedomskaya, E.; Xu, G.; de Ruiter, J.; Lolkema, M.P.; et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marass, F.; Mouliere, F.; Yuan, K.; Rosenfeld, N.; Markowetz, F. A phylogenetic latent feature model for clonal deconvolution. Ann. Appl. Stat. 2016, 10, 2377–2404. [Google Scholar] [CrossRef]
- Franken, A.; Honisch, E.; Reinhardt, F.; Meier-Stiegen, F.; Yang, L.; Jaschinski, S.; Esposito, I.; Alberter, B.; Polzer, B.; Huebner, H.; et al. Detection of ESR1 Mutations in Single Circulating Tumor Cells on Estrogen Deprivation Therapy but Not in Primary Tumors from Metastatic Luminal Breast Cancer Patients. J. Mol. Diagn. 2019, 22, 111–121. [Google Scholar] [CrossRef]
- Möhlendick, B.; Bartenhagen, C.; Behrens, B.; Honisch, E.; Raba, K.; Knoefel, W.T.; Stoecklein, N.H. A Robust Method to Analyze Copy Number Alterations of Less than 100 kb in Single Cells Using Oligonucleotide Array CGH. PLoS ONE 2013, 8, e67031. [Google Scholar]
- Jeannot, E.; Darrigues, L.; Michel, M.; Pierga, M.S.J.; Rampanou, A.; Melaabi, S.; Benoist, C.; Bièche, I.; El, R.; Aurélien, A.; et al. A single droplet digital PCR for ESR1 activating mutations detection in plasma. Oncogene 2020, 39, 2987–2995. [Google Scholar] [CrossRef] [PubMed]
- Franken, A.; Driemel, C.; Behrens, B.; Meier-stiegen, F.; Endris, V.; Stenzinger, A.; Niederacher, D.; Fischer, J.C.; Stoecklein, N.H.; Ruckhaeberle, E.; et al. Label-Free Enrichment and Molecular Characterization of Viable Circulating Tumor Cells from Diagnostic Leukapheresis Products. Clin. Chem. 2019, 65, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Gala, K.; Fanning, S.; King, T.A.; Hudis, C.; et al. ESR1 ligand binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Emma, J.; Bando, H.; El-khoueiry, A.B.; Mita, A.; Schellens, J.H.M.; et al. AKT Inhibition in Solid Tumors With AKT1 Mutations. J. Clin. Oncol. 2019, 35, 2251–2262. [Google Scholar] [CrossRef]
- Zardavas, D.; Fumagalli, D.; Loi, S. Phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway inhibition: A breakthrough in the management of luminal (ER+/HER2-) breast cancers? Curr. Opin. Oncol. 2012, 24, 623–634. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Serafino, A.; Sferrazza, G.; Colini Baldeschi, A.; Nicotera, G.; Andreola, F.; Pittaluga, E.; Pierimarchi, P. Developing drugs that target the Wnt pathway: Recent approaches in cancer and neurodegenerative diseases. Expert Opin. Drug Discov. 2017, 12, 169–186. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; De Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; Van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53 -Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. TP53 Alterations Correlate with Response to VEGF/VEGFR Inhibitors: Implications for Targeted Therapeutics. Mol. Cancer Ther. 2016, 15, 2475–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, K.; Liebner, D.; Chen, J.L. TP53 mutational status is predictive of pazopanib response in advanced sarcomas. Ann. Oncol. 2016, 27, 539–543. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in Vivo: A First-in-Human Study With p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sakr, R.A.; Giri, D.; Patil, S.; Heguy, A.; Morrow, M.; Modi, S.; Norton, L.; Rosen, N.; Hudis, C.; et al. Frequent Mutational Activation of the PI3K-AKT Pathway in Trastuzumab-Resistant Breast Cancer. Clin. Cancer Res. 2012, 18, 6784–6791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Warne, P.H.; Downward, J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 2011, 30, 3222–3233. [Google Scholar] [CrossRef]
- Nakanishi, K.; Yang, Y.; Pierce, A.J.; Taniguchi, T.; Digweed, M.; D’Andrea, A.D.; Wang, Z.; Jasin, M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. USA 2005, 102, 1110–1115. [Google Scholar] [CrossRef] [Green Version]
- Wilkes, D.C.; Sailer, V.; Xue, H.; Cheng, H.; Collins, C.C.; Gleave, M.; Wang, Y.; Demichelis, F.; Beltran, H.; Rubin, M.A.; et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyde, A.; Reiter, J.G.; Naxerova, K.; Nowak, M.A. Consecutive seeding and transfer of genetic diversity in metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 14129–14137. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; De Placido, S.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res. 2018, 78, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Troxell, M.L. PIK3CA/AKT1 Mutations in Breast Carcinoma: A Comprehensive Review of Experimental and Clinical Studies Clinical & Experimental Pathology. J. Clin. Exp. Pathol. 2012, 43, 2207–2212. [Google Scholar]
- Turner, N.C.; Alarcón, E.; Armstrong, A.C.; Philco, M.; López Chuken, Y.A.; Sablin, M.-P.; Tamura, K.; Gómez Villanueva, A.; Pérez-Fidalgo, J.A.; Cheung, S.Y.A.; et al. BEECH: A dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer. Ann. Oncol. 2019, 30, 774–780. [Google Scholar] [CrossRef]
- Jones, R.H.; Carucci, M.; Casbard, A.C.; Butler, R.; Alchami, F.; Bale, C.J.; Bezecny, P.; Joffe, J.; Moon, S.; Twelves, C.; et al. Capivasertib (AZD5363) plus fulvestrant versus placebo plus fulvestrant after relapse or progression on an aromatase inhibitor in metastatic ER-positive breast cancer (FAKTION): A randomized, double-blind, placebo-controlled, phase II trial. J. Clin. Oncol. 2019, 37, 1005. [Google Scholar] [CrossRef]
- Bidard, F.C.; Sabatier, R.; Berger, F.; Pistilli, B.; Dalenc, F.; De La Motte Rouge, T.; Frenel, J.-S.; Dubot, C.; Ladoire, S.; Ferrero, J.-M.; et al. PADA-1: A randomized, open label, multicentric phase III trial to evaluate the safety and efficacy of palbociclib in combination with hormone therapy driven by circulating DNA ESR1 mutation monitoring in ER-positive, HER2-negative metastatic breast cancer. J. Clin. Oncol. 2018, 36, TPS1105. [Google Scholar] [CrossRef]
- Turner, N.; Bye, H.; Kernaghan, S.; Proszek, P.; Fribbens, C.; Moretti, L.; Morden, J.; Snowdon, C.; Macpherson, I.; Wardley, A.; et al. Abstract OT1-06-03: The plasmaMATCH trial: A multiple parallel cohort, open-label, multi-centre phase II clinical trial of ctDNA screening to direct targeted therapies in patients with advanced breast cancer (CRUK/15/010). Cancer Res. 2018, 78, OT1-06. [Google Scholar]
- Lianidou, E.; Pantel, K. Liquid biopsies. Genes Chromosom. Cancer 2019, 58, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Alberter, B.; Klein, C.A.; Polzer, B. Single-cell analysis of CTCs with diagnostic precision: Opportunities and challenges for personalized medicine. Expert Rev. Mol. Diagn. 2016, 16, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Faugeroux, V.; Pailler, E.; Oulhen, M.; Deas, O.; Brulle-Soumare, L.; Hervieu, C.; Marty, V.; Alexandrova, K.; Andree, K.C.; Stoecklein, N.H.; et al. Genetic characterization of a unique neuroendocrine transdifferentiation prostate circulating tumor cell-derived eXplant model. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkala, E.; Sollier-Christen, E.; Renier, C.; Rosàs-Canyelles, E.; Che, J.; Heirich, K.; Duncombe, T.A.; Vlassakis, J.; Yamauchi, K.A.; Huang, H.; et al. Profiling protein expression in circulating tumor cells using microfluidic western blotting. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gorges, T.M.; Kuske, A.; Röck, K.; Mauermann, O.; Müller, V.; Peine, S.; Verpoort, K.; Novosadova, V.; Kubista, M.; Riethdorf, S.; et al. Accession of Tumor Heterogeneity by Multiplex Transcriptome Profiling of Single Circulating Tumor Cells. Clin. Chem. 2016, 62, 1504–1515. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age at CTC Sequencing | Type | M | G | ER | PR | HER2/neu | Chemo-Therapy | Radiation | Endocrine Therapy | CTC Count per 7.5 mL Blood | Cells Used for WES | Material CTCs Were Obtained from for WES |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | 74 | Invasive-lob | Bone, bladder * | 2 | + | + | − | −* | + | + | Approx. 50,000 | 20,000 CTCs | DLA product |
| Patient 2 | 70 | Invasive-lob | Bone, liver | 2 | + | − | − | + | + | + | 2687 | 8 CTCs (WGA) | Blood |
| Patient 3 | 64 | Invasive-lob | Bone, LN, ovary, pleura | 2 | + | + | − | + | + | + | 583 | 6 CTCs (WGA) | Blood |
| Patient 4 | 72 | Invasive-lob | Bone, BM | 2 | + | + | − | + | + | + | 8000 | 15 CTCs (WGA) | Blood |
| Patient 5 | 51 | NST | Bone, liver | 2 | + | + | − | + | + | + | 94 | 9 CTCs (WGA) | Blood |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franken, A.; Behrens, B.; Reinhardt, F.; Yang, L.; Rivandi, M.; Marass, F.; Jaeger, B.; Krawczyk, N.; Cieslik, J.-P.; Honisch, E.; et al. Multiparametric Circulating Tumor Cell Analysis to Select Targeted Therapies for Breast Cancer Patients. Cancers 2021, 13, 6004. https://doi.org/10.3390/cancers13236004
Franken A, Behrens B, Reinhardt F, Yang L, Rivandi M, Marass F, Jaeger B, Krawczyk N, Cieslik J-P, Honisch E, et al. Multiparametric Circulating Tumor Cell Analysis to Select Targeted Therapies for Breast Cancer Patients. Cancers. 2021; 13(23):6004. https://doi.org/10.3390/cancers13236004
Chicago/Turabian StyleFranken, André, Bianca Behrens, Florian Reinhardt, Liwen Yang, Mahdi Rivandi, Francesco Marass, Bernadette Jaeger, Natalia Krawczyk, Jan-Philipp Cieslik, Ellen Honisch, and et al. 2021. "Multiparametric Circulating Tumor Cell Analysis to Select Targeted Therapies for Breast Cancer Patients" Cancers 13, no. 23: 6004. https://doi.org/10.3390/cancers13236004
APA StyleFranken, A., Behrens, B., Reinhardt, F., Yang, L., Rivandi, M., Marass, F., Jaeger, B., Krawczyk, N., Cieslik, J.-P., Honisch, E., Asperger, H., Jeannot, E., Proudhon, C., Beerenwinkel, N., Schölermann, N., Esposito, I., Dietzel, F., Stoecklein, N. H., Niederacher, D., ... Neubauer, H. (2021). Multiparametric Circulating Tumor Cell Analysis to Select Targeted Therapies for Breast Cancer Patients. Cancers, 13(23), 6004. https://doi.org/10.3390/cancers13236004