
Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL



Simple Summary
Abstract
1. Introduction
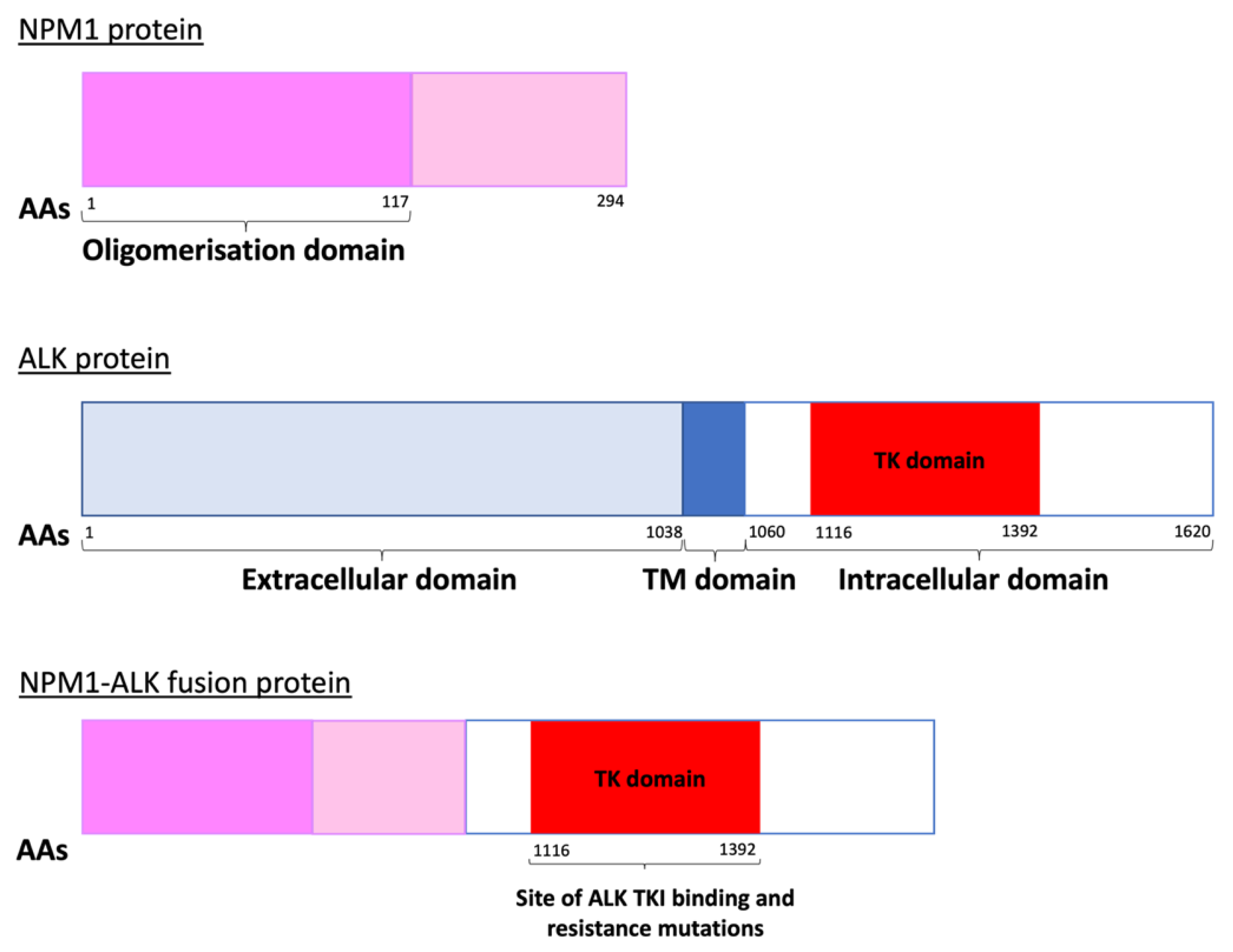
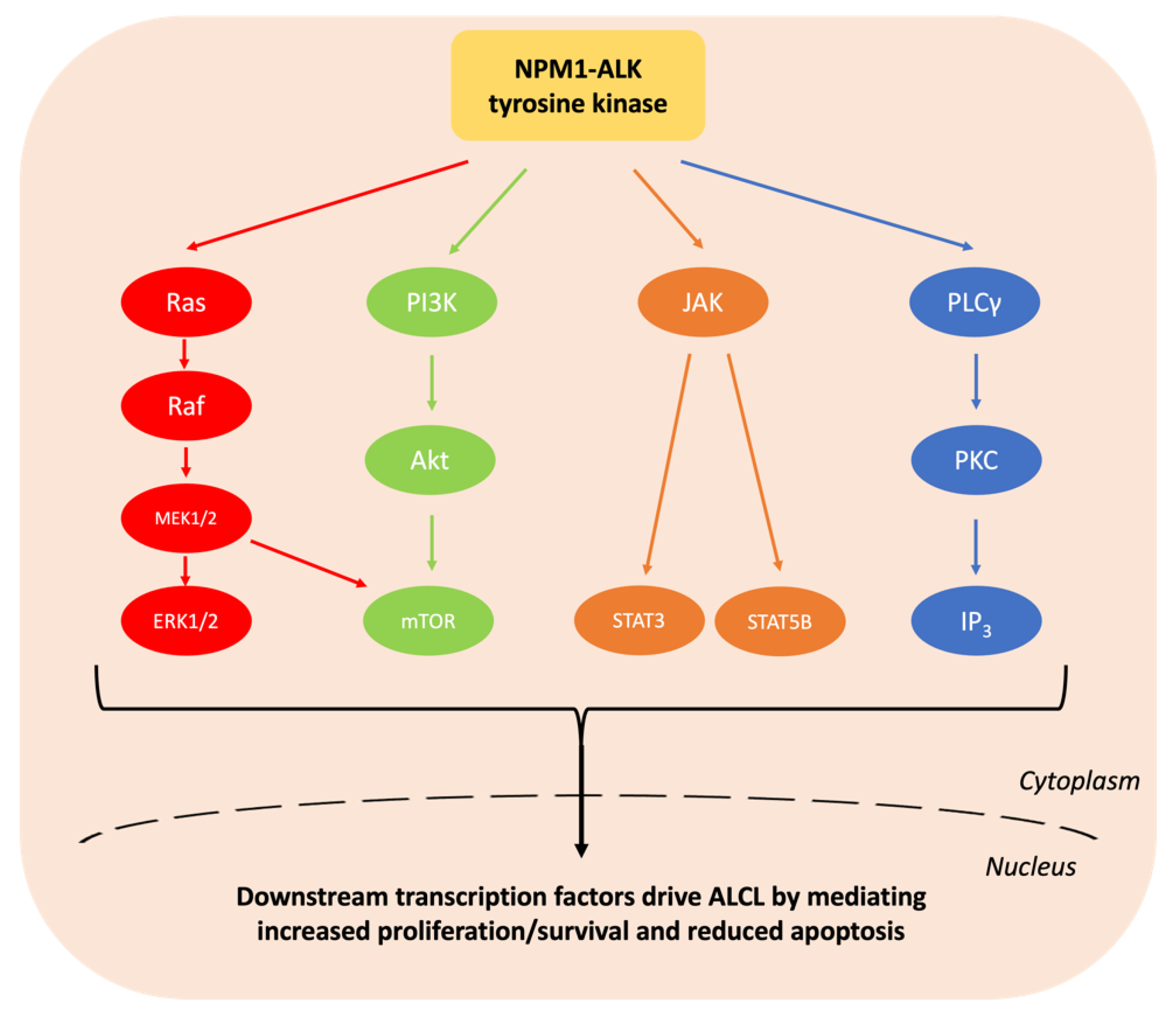
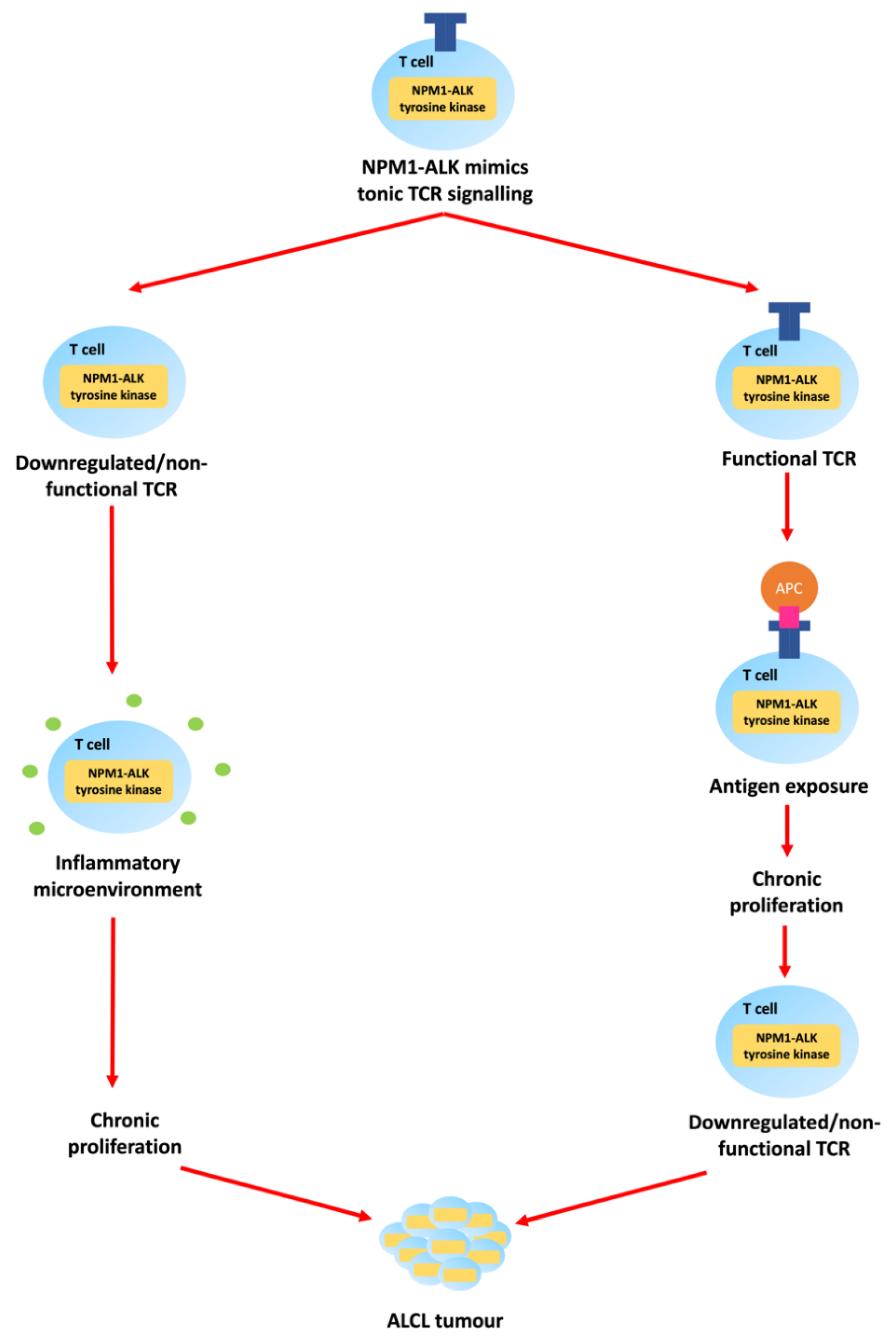
1.1. Epidemiology and Pathologenesis of Paediatric ALCL
1.2. Clinical Presentation and Management of Paediatric ALCL
2. Targeted Agents for the Treatment of ALCL
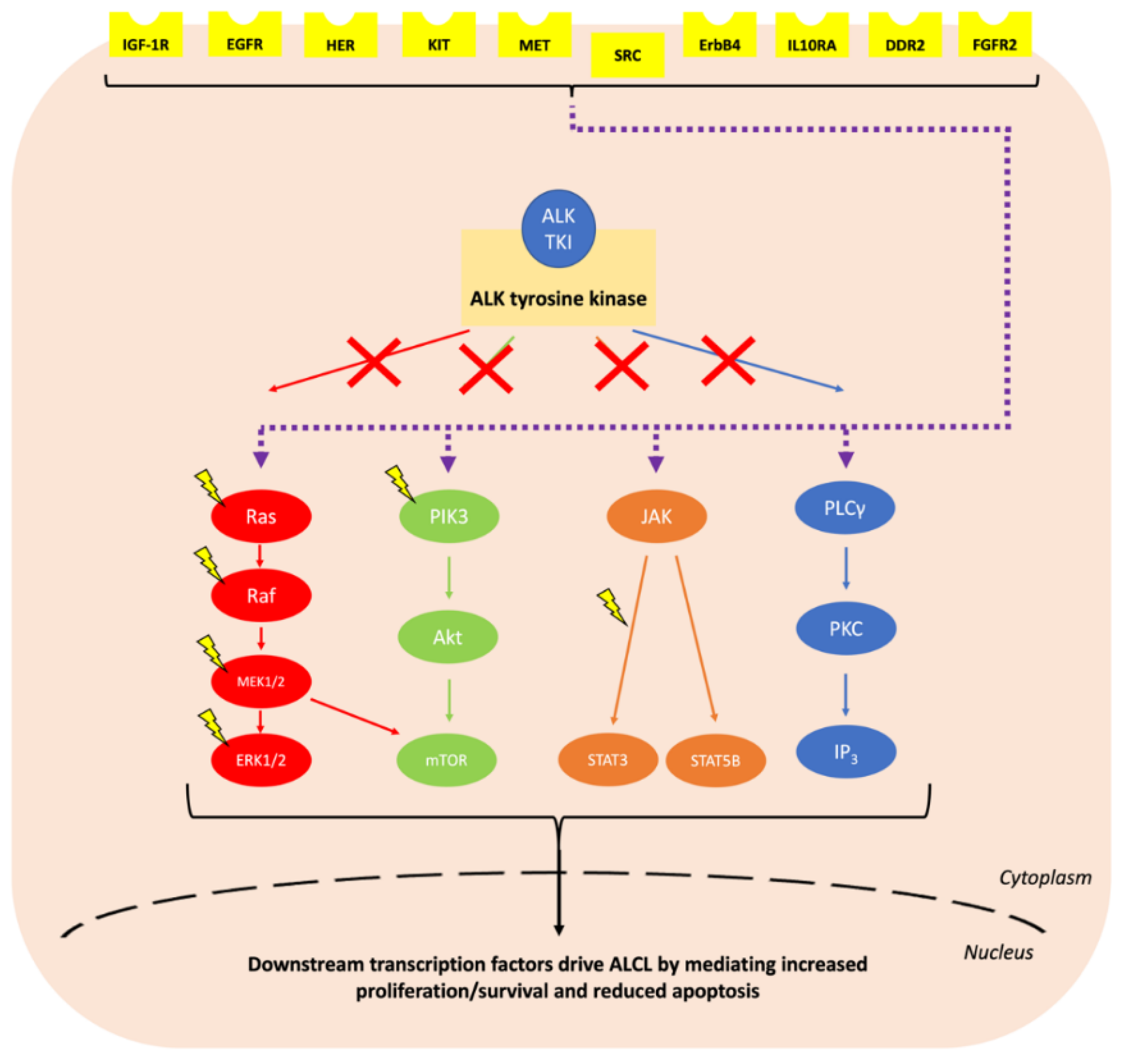
2.1. ALK Tyrosine Kinase Inhibitors
2.2. Brentuximab Vedotin
2.3. Checkpoint Inhibitors
3. Mechanisms of Resistance to ALCL Therapy
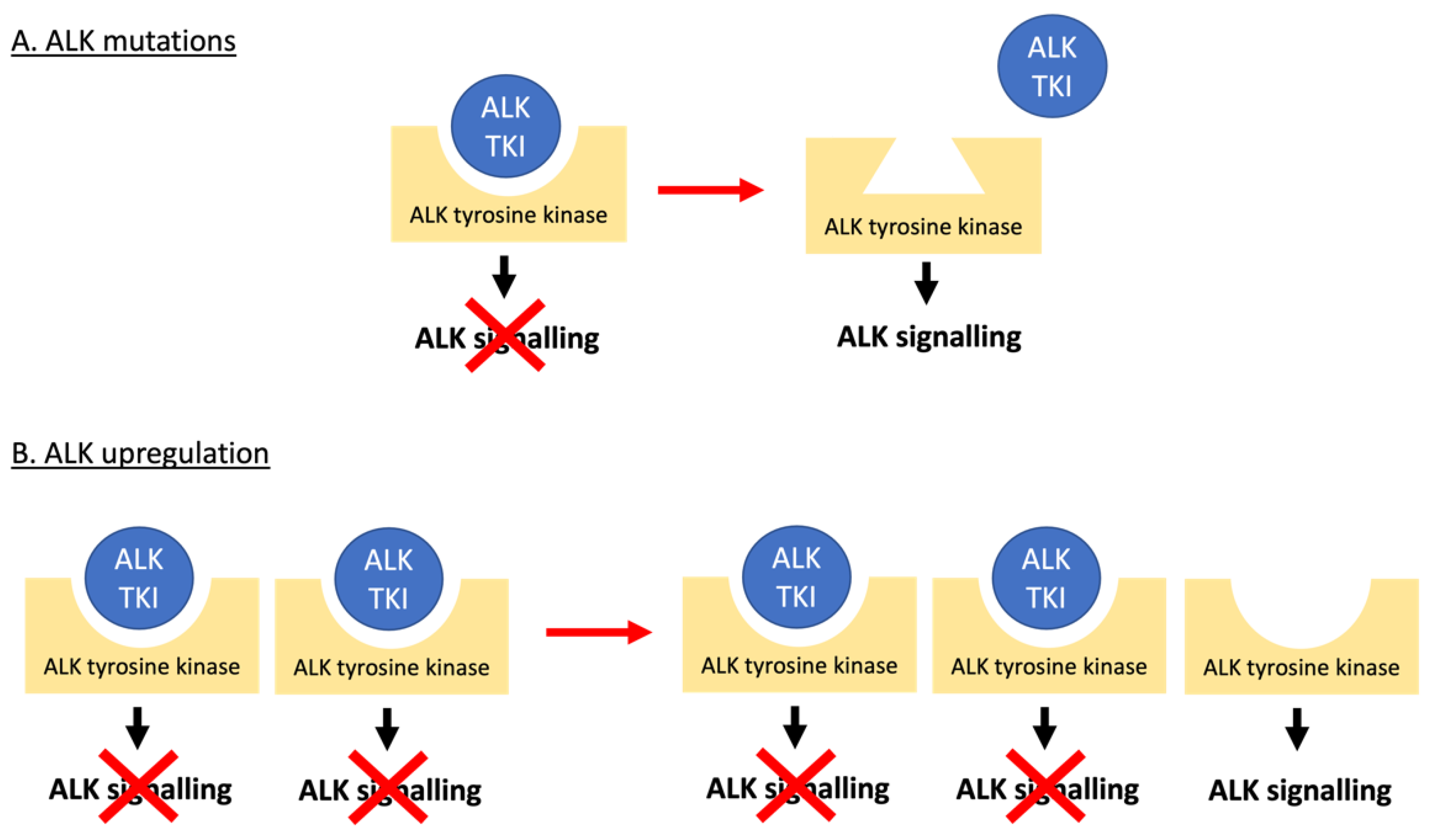
3.1. Mechanisms of Resistance to ALK Tyrosine Kinase Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALK TKI | ALK Mutation | ||
|---|---|---|---|
| Resistance | Sensitivity | Conflicting | |
| Crizotinib | C1156T [88] C1156Y [17,79,89,90,91,92,93,94] | L1198F [79,89,95,96] C1156Y/L1198F [95] | |
| D1203N [79,96,97,98] | G1202R/L1198F [91] | ||
| E1210K [79,90,99] | I1171N/L1265F [91] | ||
| F1174C [76,79,89,91,99] F1174I [91] F1174L [100,101] F1174V [91] F1245V [92] | |||
| G1128A [102] | |||
| G1202R [78,79,89,90,94] G1202del [79,89] | |||
| G1269A [79,82,89,90,94,96,99] G1269S [103] | |||
| I1171N [79,89,90,103,104,105] I1171S [79,89,90,103] I1171T [76,79,89,90,91,92,98] I1171X [106] | |||
| I1268L [91] | |||
| L1152P [103,107] | |||
| L1152R [81,94] | |||
| L1196M [17,78,79,80,82,89,90,91,92,93,94,96,98,99] L1196Q [91,96,104] | |||
| L1198P [97] | |||
| R1192P [108] | |||
| S1206C [90,103] S1206Y [78,79,90,94] | |||
| T1151K [109,110] T1151M [108] | |||
| V1180L [96] | |||
| Q1188_L1190del [111] | |||
| 1151Tins [78,90] | |||
| D1203N/E1210K [79,89] | |||
| D1203N/F1174C [79,89] | |||
| F1174L/G1269A [98] | |||
| Ceritinib | F1174L [79,90,103,107,108] | E1210K [79,89] | C1156Y [79,89,90,103,107,112] |
| F1174S [90] | F1245C [113] | D1203N [79,89,90,96] | |
| F1174V [103] | I1171T [79,89,107] | F1174C [79,89,107,112] | |
| G1123S [114] | I1268L [91] | G1202R [79,89,90,92,98,103,112] | |
| G1128A [98] | L1196Q [91] | G1269A [79,89,96,98,107,108] | |
| G1202del [79,89,103] | S1206Y [107,115] | I1171N [79,89,90,107] | |
| L1122V [96] | V1185L [91] | I1171S [79,89,90] | |
| L1152P [103] | G1269A/I1171S [116] | L1196M [79,89,90,91,96,107,115] | |
| L1152R [103,107] | G1269A/I1171N [91] | ||
| L1198F [79,89,96] | |||
| R1192P [108] | |||
| R1275Q [17] | |||
| T1151K [109] | |||
| T1151M [108] | |||
| T1151Sins [117] | |||
| Q1188_L1190del [111] | |||
| 1151Tins [103,107] | |||
| C1156Y/I1171N [79] | |||
| C1156Y/G1202del/V1180L [79] | |||
| D1203N/E1210K [79,89] | |||
| D1203N/F1174C [79,89] | |||
| E1210K/I1171T [98] | |||
| G1202R/F1174L [79] | |||
| G1202R/F1174V [92] | |||
| G1202R/L1196M [91] | |||
| Alectinib | F1174I [91] | C1156Y [79,89,118] | F1174C [76,79,89] |
| F1174L [79,90,91,103,107,108] | D1203N [79,89,96] | I1171T [76,79,89,90,91,119] | |
| F1174S [90] | E1210K [79,89] | L1196M [79,89,90,91,96,118] | |
| F1174V [91,103] | F1174L [118] | 1151Tins [118] | |
| G1202R [79,89,90,92,99,118] | G1269A [79,89,96,118] | ||
| G1202S [99] | I1268L [91] | ||
| G1202del [79,89] | L1152R [118] | ||
| G1210K [120] | L1198F [79,89] | ||
| G1269A [108] | L1256F [91] | ||
| I1171N [79,89,90,91,92] | S1206Y [118] | ||
| I1171S [79,89,90,91,108] | T1151K [110] | ||
| I1171 X [106] | V1185L [91] | ||
| L1122V [96] | I1171N/L1256F [91] | ||
| L1196Q [91] | |||
| L1198F [96] | |||
| R1192P [108] | |||
| T1151M [108] | |||
| V1180L [119] | |||
| W1295C [98] | |||
| D1203N/E1210K [79,89] | |||
| D1203N/F1174C [79,89] | |||
| F1174L/G1269A [98] | |||
| G1202R/L1196M [98] | |||
| L1196M/V1185L [91] | |||
| Brigatinib | G1202L [121] | C1156Y [79,89,107] | D1203N [79,89,90,96,107] |
| G1202del [79,89] | F1174C [79,89,107] | E1210K [79,89,90] | |
| L1122V [84,96] | F1174L [107] | G1202R [62,79,89,90,107] | |
| S1206C [90] | G1269A [96,107] | I1171N [79,89,104,107] | |
| D1203N/F1174C [89] | I1171S [79,89,106] | L1198F [79,89,96,107] | |
| D1203N/E1210K [89] | I1171T [79,89] | S1206Y [17,90,107] | |
| E1210K/S1206C [89,99,103] | L1152P [107] | ||
| F1174V/L1198F [84] | L1152R [107] | ||
| F1174L/L1198V [99] | L1196M [79,80,89,96,107] | ||
| G1202R/L1196M [122] | L1196Q [104] | ||
| V1180L [96,107] | |||
| 1151Tins [107] | |||
| G1269A/I1171S [116] | |||
| G1269A/I1171N [91] | |||
| I1171N/L1196M [91] | |||
| I1171N/L1198F [91] | |||
| I1171N/L1256F [91] | |||
| Lorlatinib | C1156F [123] | C1156Y [79,89] | E1201K [79,89,123] |
| G1128S [123] | D1203N [79,89] | G1269A [79,89,96,124] | |
| L1256F [91] | F1174C [79,89] | I1171N [79,89,123] | |
| C1156F/D1203N [123] | F1174I [123] | I1171T [79,89,123] | |
| C1156F/L1198F [124,125] | F1174L [126] | ||
| C1156Y/D1203N [125] | F1245C [126] | ||
| C1156Y/F1174C [125] | G1202del [79,89] | ||
| C1156Y/F1174I [125] | G1202K [120] | ||
| C1156Y/F1174V [125] | G1202L [121] | ||
| C1156Y/G1269A [125,127] | G1202R [79,89,128] | ||
| C1156Y/I1171T [125] | I1171S [79,89] | ||
| C1156Y/L1196M [125] | L1196M [79,89,96] | ||
| C1156Y/L1198F [125] | R1275Q [126] | ||
| C1156Y/S1256F [125] | V1180L [96] | ||
| D1203N/F1174C [79,89] | |||
| D1203N/L1196M [127] | |||
| F1174C/G1202R [91] | |||
| F1174C/G1269A [125] | |||
| F1174C/L1196M [125] | |||
| F1174L/G1202R [91,127] | |||
| G1202R/G1269A [91,98,124] | |||
| G1202R/I1171N [91] | |||
| G1202R/L1196M [92,122,125] | |||
| G1202R/L1198F [91,125] | |||
| G1269A/I1171S [116] | |||
| G1269A/I1171N [125] | |||
| G1269A/I1171T [125] | |||
| G1269A/L1196M [91,125] | |||
| G1269A/N1178H [124] | |||
| I1171N/C1156Y [91] | |||
| I1171N/L1198F [91] | |||
| I1171N/L1256F [91] | |||
| L1196M/F1174C [125] | |||
| L1196M/F1174L [125] | |||
| L1196M/F1174V [125] | |||
| L1196M/I1171S [125] | |||
| L1196M/I1179V [125] | |||
| L1196M/L1198F [125] | |||
| L1196M/L1198H [125] | |||
| L1196M/L1256F [125] | |||
3.2. Mechanisms of Resistance to Brentuximab Vedotin
3.3. Mechanisms of Resistance to Immune Checkpoint Inhibitors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prokoph, N.; Larose, H.; Lim, M.S.; Burke, G.A.A.; Turner, S.D. Treatment options for paediatric anaplastic large cell lymphoma (ALCL): Current standard and beyond. Cancers 2018, 10, 99. [Google Scholar] [CrossRef]
- Abla, O.; Alexander, S.; Attarbaschi, A.; Batchelor, T.T.; Beishuizen, A.; Bond, J.; Borkhardt, A.; Brugieres, L.; Burke, A.; Burkhardt, B.; et al. Non-Hodgkin’s Lymphoma in Childhood and Adolescence, 1st ed.; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Bischof, D.; Pulford, K.; Mason, D.Y.; Morris, S.W. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol. Cell. Biol. 1997, 17, 2312–2325. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Tooze, R.; Maclennan, K.; Alexander, D.R. Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes B-cell lymphomas with hyperactive Jun kinase. Oncogene 2003, 22, 7750–7761. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, T.I.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.A.; Brugières, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Gong, J.Z.; Guasparri, I.; Pesci, A.; Cai, J.; Liu, J.; Simmons, W.J.; Dhall, G.; Howes, J.; Piva, R.; et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003, 101, 1919–1927. [Google Scholar] [CrossRef]
- Turner, S.D.; Merz, H.; Yeung, D.; Alexander, D.R. CD2 promoter regulated nucleophosmin-anaplastic lymphoma kinase in transgenic mice causes B lymphoid malignancy. Anticancer Res. 2006, 26, 3275–3279. [Google Scholar]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef]
- Turner, S.D.; Yeung, D.; Hadfield, K.; Cook, S.J.; Alexander, D.R. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell. Signal. 2007, 19, 740–747. [Google Scholar] [CrossRef]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Anaplastic lymphoma kinase (ALK) inhibitors in the treatment of ALK-driven lung cancers. Pharmacol. Res. 2017, 117, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor resistance against ALK targeted therapy—Where it comes from and where it goes. Cancers 2018, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Trigg, R.M.; Turner, S.D. ALK in neuroblastoma: Biological and therapeutic implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef]
- Laurent, C.; Lopez, C.; Desjobert, C.; Berrebi, A.; Damm-Welk, C.; Delsol, G.; Brousset, P.; Lamant, L. Circulating t(2;5)-positive cells can be detected in cord blood of healthy newborns. Leukemia 2012, 26, 188–190. [Google Scholar] [CrossRef]
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugières, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 173, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Knörr, F.; Damm-Welk, C.; Ruf, S.; Singh, V.K.; Zimmermann, M.; Reiter, A.; Woessmann, W. Blood cytokine concentrations in pediatric patients with anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 2018, 103, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Bonzheim, I.; Geissinger, E.; Roth, S.; Zettl, A.; Marx, A.; Rosenwald, A.; Müller-Hermelink, H.K.; Rüdiger, T. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood 2004, 104, 3358–3360. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, T.I.M.; Hodson, D.J.; Macintyre, E.A.; Turner, S.D. Challenging perspectives on the cellular origins of lymphoma. Open Biol. 2016, 6, 160232. [Google Scholar] [CrossRef] [PubMed]
- Schleussner, N.; Merkel, O.; Costanza, M.; Liang, H.C.; Hummel, F.; Romagnani, C.; Durek, P.; Anagnostopoulos, I.; Hummel, M.; Jöhrens, K.; et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia 2018, 32, 1994–2007. [Google Scholar] [CrossRef] [PubMed]
- Moti, N.; Malcolm, T.; Hamoudi, R.; Mian, S.; Garland, G.; Hook, C.E.; Burke, G.A.A.; Wasik, M.A.; Merkel, O.; Kenner, L.; et al. Anaplastic large cell lymphoma-propagating cells are detectable by side population analysis and possess an expression profile reflective of a primitive origin. Oncogene 2015, 34, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Congras, A.; Hoareau-Aveilla, C.; Caillet, N.; Tosolini, M.; Villarese, P.; Cieslak, A.; Rodriguez, L.; Asnafi, V.; Macintyre, E.; Egger, G.; et al. ALK-transformed mature T lymphocytes restore early thymus progenitor features. J. Clin. Investig. 2020, 130, 6395–6408. [Google Scholar] [CrossRef]
- Kasprzycka, M.; Marzec, M.; Liu, X.; Zhang, Q.; Wasik, M.A. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc. Natl. Acad. Sci. USA 2006, 103, 9964–9969. [Google Scholar] [CrossRef]
- Zhang, Q.; Wei, F.; Wang, H.Y.; Liu, X.; Roy, D.; Xiong, Q.B.; Jiang, S.; Medvec, A.; Danet-Desnoyers, G.; Watt, C.; et al. The potent oncogene NPM-ALK mediates malignant transformation of normal human CD4(+) T lymphocytes. Am. J. Pathol. 2013, 183, 1971–1980. [Google Scholar] [CrossRef]
- Marzec, M.; Halasa, K.; Liu, X.; Wang, H.Y.; Cheng, M.; Baldwin, D.; Tobias, J.W.; Schuster, S.J.; Woetmann, A.; Zhang, Q.; et al. Malignant transformation of CD4+ T lymphocytes mediated by oncogenic kinase NPM/ALK recapitulates IL-2–induced cell signaling and gene expression reprogramming. J. Immunol. 2013, 191, 6200. [Google Scholar] [CrossRef]
- Brugières, L.; Le Deley, M.-C.; Rosolen, A.; Williams, D.; Horibe, K.; Wrobel, G.; Mann, G.; Zsiros, J.; Uyttebroeck, A.; Marky, I.; et al. Impact of the methotrexate administration dose on the need for intrathecal treatment in children and adolescents with anaplastic large-cell lymphoma: Results of a randomized trial of the EICNHL group. J. Clin. Oncol. 2009, 27, 897–903. [Google Scholar] [CrossRef]
- Mussolin, L.; Pillon, M.; d’Amore, E.S.; Santoro, N.; Lombardi, A.; Fagioli, F.; Zanesco, L.; Rosolen, A. Prevalence and clinical implications of bone marrow involvement in pediatric anaplastic large cell lymphoma. Leukemia 2005, 19, 1643–1647. [Google Scholar] [CrossRef]
- Williams, D.; Mori, T.; Reiter, A.; Woessman, W.; Rosolen, A.; Wrobel, G.; Zsiros, J.; Uyttebroeck, A.; Marky, I.; Le Deley, M.C.; et al. Central nervous system involvement in anaplastic large cell lymphoma in childhood: Results from a multicentre European and Japanese study. Pediatr. Blood Cancer 2013, 60, E118–E121. [Google Scholar] [CrossRef]
- Mussolin, L.; Le Deley, M.C.; Carraro, E.; Damm-Welk, C.; Attarbaschi, A.; Williams, D.; Burke, A.; Horibe, K.; Nakazawa, A.; Wrobel, G.; et al. Prognostic factors in childhood anaplastic large cell lymphoma: Long term results of the international ALCL99 trial. Cancers 2020, 12, 2747. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, G.; Mauguen, A.; Rosolen, A.; Reiter, A.; Williams, D.; Horibe, K.; Brugières, L.; Le Deley, M.C. Safety assessment of intensive induction therapy in childhood anaplastic large cell lymphoma: Report of the ALCL99 randomised trial. Pediatr. Blood Cancer 2011, 56, 1071–1077. [Google Scholar] [CrossRef]
- Ruf, R.; Brugieres, L.; Pillon, M.; Zimmermann, M.; Attarbaschi, A.; Melgrenn, K.; Williams, D.; Uyttebroeck, A.; Wrobel, G.; Reiter, A.; et al. Risk-adapted therapy for patients with relapsed or refractory ALCL—Final report of the prospective ALCL-relapse trial of the EICNHL. Br. J. Haematol. 2015, 171, 35. [Google Scholar]
- EMC. Vinblastine Sulphate 1 mg/mL Injection. Published December 2020. Available online: https://www.medicines.org.uk/emc/product/1422/smpc#gref (accessed on 15 May 2021).
- Brugieres, L.; Pacquement, H.; Le Deley, M.C.; Leverger, G.; Lutz, P.; Paillard, C.; Baruchel, A.; Frappaz, D.; Nelken, B.; Lamant, L.; et al. Single-drug vinblastine as salvage treatment for refractory or relapsed anaplastic large-cell lymphoma: A report from the French Society of Pediatric Oncology. J. Clin. Oncol. 2009, 27, 5056–5061. [Google Scholar] [CrossRef]
- Le Deley, M.C.; Rosolen, A.; Williams, D.M.; Horibe, K.; Wrobel, G.; Attarbaschi, A.; Zsiros, J.; Uyttebroeck, A.; Marky, I.M.; Lamant, L.; et al. Vinblastine in children and adolescents with high-risk anaplastic large-cell lymphoma: Results of the randomized ALCL99-vinblastine trial. J. Clin. Oncol. 2010, 28, 3987–3993. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Kraveka, J.M.; Weitzman, S.; Lowe, E.; Smith, L.; Lynch, J.C.; Chang, M.; Kinney, M.C.; Perkins, S.L.; Laver, J.; et al. Advanced stage anaplastic large cell lymphoma in children and adolescents: Results of ANHL0131, a randomized phase III trial of APO versus a modified regimen with vinblastine: A report from the children’s oncology group. Pediatr. Blood Cancer 2014, 61, 2236–2242. [Google Scholar] [CrossRef]
- Li, S.; Yang, J.; Wang, J.; Gao, W.; Ding, Y.; Jia, Z. Down-regulation of miR-210-3p encourages chemotherapy resistance of renal cell carcinoma via modulating ABCC1. Cell Biosci. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Mickisch, G.H.; Roehrich, K.; Koessig, J.; Forster, S.; Tschada, R.K.; Alken, P.M. Mechanisms and modulation of multidrug resistance in primary human renal cell carcinoma. J. Urol. 1990, 144, 755–759. [Google Scholar] [CrossRef]
- Struski, S.; Cornillet-Lefebvre, P.; Doco-Fenzy, M.; Dufer, J.; Ulrich, E.; Masson, L.; Michel, N.; Gruson, N.; Potron, G. Cytogenetic characterization of chromosomal rearrangement in a human vinblastine-resistant CEM cell line: Use of comparative genomic hybridization and fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2002, 132, 51–54. [Google Scholar] [CrossRef]
- Holmes, J.; Jacobs, A.; Carter, G.; Janowska-Wieczorek, A.; Padua, R.A. Multidrug resistance in haemopoietic cell lines, myelodysplastic syndromes and acute myeloblastic leukaemia. Br. J. Haematol. 1989, 72, 40–44. [Google Scholar] [CrossRef]
- Zamora, J.M.; Beck, W.T. Chloroquine enhancement of anticancer drug cytotoxicity in multiple drug resistant human leukemic cells. Biochem. Pharmacol. 1986, 35, 4303–4310. [Google Scholar] [CrossRef]
- Syed, S.K.; Christopherson, R.I.; Roufogalis, B.D. Reversal of vinblastine transport by chlorpromazine in membrane vesicles from multidrug-resistant human CCRF-CEM leukaemia cells. Br. J. Cancer 1998, 78, 321–327. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R.; Chen, Z.S., Jr. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Obey, T.B.; Lyle, C.S.; Chambers, T.C. Role of c-Jun in cellular sensitivity to the microtubule inhibitor vinblastine. Biochem. Biophys. Res. Commun. 2005, 335, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jaffrezou, J.P.; Tsuchiya, E.; Duran, G.E.; Chen, K.G.; Derry, W.B.; Wilson, L.; Jordan, M.A.; Sikic, B.I. Resistance to microtubule-targeted cytotoxins in a K562 leukemia cell variant associated with altered tubulin expression and polymerization. Bull. Cancer 2004, 91, E81–E112. [Google Scholar] [PubMed]
- Balis, F.M.; Thompson, P.A.; Mosse, Y.P.; Blaney, S.M.; Minard, C.G.; Weigel, B.J.; Fox, E. First-dose and steady-state pharmacokinetics of orally administered crizotinib in children with solid tumors: A report on ADVL0912 from the Children’s Oncology Group Phase 1/Pilot Consortium. Cancer Chemother. Pharmacol. 2017, 79, 181–187. [Google Scholar] [CrossRef]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef]
- Orthofer, M.; Valsesia, A.; Mägi, R.; Wang, Q.P.; Kaczanowska, J.; Kozieradzki, I.; Leopoldi, A.; Cikes, D.; Zopf, L.M.; Tretiakov, E.O.; et al. Identification of ALK in Thinness. Cell 2020, 181, 1246–1262.e22. [Google Scholar] [CrossRef]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- FDA. FDA Approves Crizotinib for Children and Young Adults with Relapsed or Refractory, Systemic Anaplastic Large Cell Lymphoma. Published January 2021. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-crizotinib-children-and-young-adults-relapsed-or-refractory-systemic-anaplastic-large (accessed on 15 May 2021).
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children’s Oncology Group study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Brugières, L.; Houot, R.; Cozic, N.; De La Fouchardière, C.; Morschhauser, F.; Brice, P.; Laboure, N.A.; Auvrignon, A.; Aladjidi, N.; Kolb, B.; et al. Crizotinib in advanced ALK+ anaplastic large cell lymphoma in children and adults: Results of the Acs© phase II trial. Blood 2017, 130 (Suppl. 1), 2831. [Google Scholar]
- Fukano, R.; Mori, T.; Sekimizu, M.; Choi, I.; Kada, A.; Saito, A.M.; Asada, R.; Takeuchi, K.; Terauchi, T.; Tateishi, U.; et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci. 2020, 111, 4540–4547. [Google Scholar] [CrossRef] [PubMed]
- Greengard, E.; Mosse, Y.P.; Liu, X.; Minard, C.G.; Reid, J.M.; Voss, S.; Wilner, K.; Fox, E.; Balis, F.; Blaney, S.M.; et al. Safety, tolerability and pharmacokinetics of crizotinib in combination with cytotoxic chemotherapy for pediatric patients with refractory solid tumors or anaplastic large cell lymphoma (ALCL): A Children’s Oncology Group phase 1 consortium study (ADVL1212). Cancer Chemother. Pharmacol. 2020, 86, 829–840. [Google Scholar] [CrossRef] [PubMed]
- EU Clinical Trials Register. A phase 1B of Crizotinib Either in Combination or as Single Agent in Pediatric Patients with ALK, ROS1 or MET Positive Malignancies. Published May 2016. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2015-005437-53/NL (accessed on 31 May 2021).
- ClinicalTrials.gov. Brentuximab Vedotin or Crizotinib and Combination Chemotherapy in Treating Patients with Newly Diagnosed Stage II-IV Anaplastic Large Cell Lymphoma. Published May 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT01979536 (accessed on 31 May 2021).
- Huber, R.M.; Hansen, K.H.; Paz-Ares Rodríguez, L.; West, H.L.; Reckamp, K.L.; Leighl, N.B.; Tiseo, M.; Smit, E.F.; Kim, D.W.; Gettinger, S.N.; et al. Brigatinib in crizotinib-refractory ALK+ NSCLC: 2-year follow-up on systemic and intracranial outcomes in the phase 2 ALTA trial. J. Thorac. Oncol. 2020, 15, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.J.; Reilly, A.F.; Lim, M.S.; Gross, T.G.; Saguilig, L.; Barkauskas, D.A.; Wu, R.; Alexander, S.; Bollard, C.M. Brentuximab vedotin in combination with chemotherapy for pediatric patients with ALK+ ALCL: Results of COG trial ANHL12P1. Blood 2021, 137, 3595–3603. [Google Scholar] [CrossRef]
- Chihara, D.; Miljkovic, M.; Iyer, S.P.; Vega, F. Targeted based therapy in nodal T-cell lymphomas. Leukemia 2021, 35, 956–967. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Leonard, J.P.; Younes, A.; Rosenblatt, J.D.; Brice, P.; Bartlett, N.L.; Bosly, A.; Pinter-Brown, L.; Kennedy, D.; Sievers, E.L.; et al. A Phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br. J. Haematol. 2009, 146, 171–179. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017, 130, 2709–2717. [Google Scholar] [CrossRef]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef]
- Locatelli, F.; Mauz-Koerholz, C.; Neville, K.; Llort, A.; Beishuizen, A.; Daw, S.; Pillon, M.; Aladjidi, N.; Klingebiel, T.; Landman-Parker, J.; et al. Brentuximab vedotin for paediatric relapsed or refractory Hodgkin’s lymphoma and anaplastic large-cell lymphoma: A multicentre, open-label, phase 1/2 study. Lancet Haematol. 2018, 5, e450–e461. [Google Scholar] [CrossRef]
- Burke, G.A.A. Brentuximab vedotin: Frontline help in ALCL. Blood 2021, 137, 3581–3582. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef] [PubMed]
- Hebart, H.; Lang, P.; Woessmann, W. Nivolumab for refractory anaplastic large cell lymphoma: A case report. Ann. Intern. Med. 2016, 165, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Abbou, S.; Minard-Colin, V.; Geoerger, B.; Scoazec, J.Y.; Vassal, G.; Jaff, N.; Heuberger, L.; Valteau-Couanet, D.; Brugieres, L. Efficacy of nivolumab in a patient with systemic refractory ALK+ anaplastic large cell lymphoma. Pediatr. Blood Cancer 2018, 65, e26902. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Mussolin, L.; Brugieres, L. Abrupt relapse of ALK-positive lymphoma after discontinuation of crizotinib. N. Engl. J. Med. 2016, 374, 95–96. [Google Scholar] [CrossRef]
- Zdzalik, D.; Dymek, B.; Grygielewicz, P.; Gunerka, P.; Bujak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K. Activating mutations in ALK kinase domain confer resistance to structurally unrelated ALK inhibitors in NPM-ALK-positive anaplastic large-cell lymphoma. J. Cancer Res. Clin. Oncol. 2014, 140, 589–598. [Google Scholar] [CrossRef]
- Andraos, E.; Dignac, J.; Meggetto, F. NPM-ALK: A driver of lymphoma pathogenesis and a therapeutic target. Cancers 2021, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–4782. [Google Scholar] [CrossRef]
- Jamme, P.; Descarpentries, C.; Gervais, R.; Dansin, E.; Wislez, M.; Grégoire, V.; Richard, N.; Baldacci, S.; Rabbe, N.; Kyheng, M.; et al. Relevance of detection of mechanisms of resistance to ALK inhibitors in ALK-rearranged NSCLC in routine practice. Clin. Lung Cancer 2019, 20, 297–304.e1. [Google Scholar] [CrossRef]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor AP26113 in human NPM-ALK-positive anaplastic large cell lymphoma. Mol. Cancer Res. 2015, 13, 775–783. [Google Scholar] [CrossRef]
- Amin, A.D.; Rajan, S.S.; Liang, W.S.; Pongtornpipat, P.; Groysman, M.J.; Tapia, E.O.; Peters, T.L.; Cuyugan, L.; Adkins, J.; Rimsza, L.M.; et al. Evidence suggesting that discontinuous dosing of ALK kinase inhibitors may prolong control of ALK+ tumors. Cancer Res. 2015, 75, 2916–2927. [Google Scholar] [CrossRef]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–6865. [Google Scholar] [CrossRef]
- Rajan, S.S.; Amin, A.D.; Li, L.; Rolland, D.C.; Li, H.; Kwon, D.; Kweh, M.F.; Arumov, A.; Roberts, E.R.; Yan, A.; et al. The mechanism of cancer drug addiction in ALK-positive T-cell lymphoma. Oncogene 2020, 39, 2103–2117. [Google Scholar] [CrossRef]
- Singh, A.; Chen, H. Optimal care for patients with anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer: A review on the role and utility of ALK inhibitors. Cancer Manag. Res. 2020, 12, 6615–6628. [Google Scholar] [CrossRef]
- Bui, K.T.; Cooper, W.A.; Kao, S.; Boyer, M. Targeted molecular treatments in non-small cell lung cancer: A clinical guide for oncologists. J. Clin. Med. 2018, 7, 192. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Araki, M.; Sakashita, T.; Ma, B.; Kanada, R.; Yanagitani, N.; Horiike, A.; Koike, S.; Oh-Hara, T.; Watanabe, K.; et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 2019, 41, 105–119. [Google Scholar] [CrossRef]
- Yanagitani, N.; Uchibori, K.; Koike, S.; Tsukahara, M.; Kitazono, S.; Yoshizawa, T.; Horiike, A.; Ohyanagi, F.; Tambo, Y.; Nishikawa, S.; et al. Drug resistance mechanisms in Japanese anaplastic lymphoma kinase-positive non-small cell lung cancer and the clinical responses based on the resistant mechanisms. Cancer Sci. 2020, 111, 932–939. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1749. [Google Scholar] [CrossRef]
- Fontana, D.; Ceccon, M.; Gambacorti-Passerini, C.; Mologni, L. Activity of second-generation ALK inhibitors against crizotinib-resistant mutants in an NPM-ALK model compared to EML4-ALK. Cancer Med. 2015, 4, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, W.; Sun, H.; Pang, L.; Yin, B. Mutation-mediated influences on binding of anaplastic lymphoma kinase to crizotinib decoded by multiple replica Gaussian accelerated molecular dynamics. J. Comput. Aided. Mol. Des. 2020, 34, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.D.; Madhavan, S. A Computational approach for prioritizing selection of therapies targeting drug resistant variation in anaplastic lymphoma kinase. AMIA Summits Transl. Sci. Proc. 2018, 2017, 160–167. [Google Scholar] [PubMed]
- Heuckmann, J.M.; Hölzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grütter, C.; et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Chiang, C.L.; Hung, J.Y.; Lee, M.H.; Su, W.C.; Wu, S.Y.; Wei, Y.F.; Lee, K.Y.; Tseng, Y.H.; Su, J.; et al. Resistance profiles of anaplastic lymphoma kinase tyrosine kinase inhibitors in advanced non-small-cell lung cancer: A multicenter study using targeted next-generation sequencing. Eur. J. Cancer 2021, 156, 1–11. [Google Scholar] [CrossRef]
- McCoach, C.E.; Le, A.T.; Gowan, K.; Jones, K.; Schubert, L.; Doak, A.; Estrada-Bernal, A.; Davies, K.D.; Merrick, D.T.; Bunn, P.A., Jr.; et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin. Cancer Res. 2018, 24, 3334–3347. [Google Scholar] [CrossRef]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Merguerian, M.D.; Rowe, S.P.; Pratilas, C.A.; Chen, A.R.; Ladle, B.H. Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Mol. Case Stud. 2021, 7, a006064. [Google Scholar] [CrossRef]
- Ai, X.; Niu, X.; Chang, L.; Chen, R.; Ou, S.I.; Lu, S. Next generation sequencing reveals a novel ALK G1128A mutation resistant to crizotinib in an ALK-Rearranged NSCLC patient. Lung Cancer 2018, 123, 83–86. [Google Scholar] [CrossRef]
- Gristina, V.; La Mantia, M.; Iacono, F.; Galvano, A.; Russo, A.; Bazan, V. The emerging therapeutic landscape of ALK inhibitors in non-small cell lung cancer. Pharmaceuticals 2020, 13, 474. [Google Scholar] [CrossRef]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef]
- Sehgal, K.; Peters, M.L.B.; VanderLaan, P.A.; Rangachari, D.; Kobayashi, S.S.; Costa, D.B. Activity of brigatinib in the setting of alectinib resistance mediated by ALK I1171S in ALK-rearranged lung cancer. J. Thorac. Oncol. 2019, 14, e1–e3. [Google Scholar] [CrossRef]
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.D.; Li, L.; Rajan, S.S.; Gokhale, V.; Groysman, M.J.; Pongtornpipat, P.; Tapia, E.O.; Wang, M.; Schatz, J.H. TKI sensitivity patterns of novel kinase-domain mutations suggest therapeutic opportunities for patients with resistant ALK+ tumors. Oncotarget 2016, 7, 23715–23729. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Cui, J.J.; Fernandez-Rocha, M.; Schrock, A.B.; Ali, S.M.; Ou, S.I. Identification of a novel T1151K ALK mutation in a patient with ALK-rearranged NSCLC with prior exposure to crizotinib and ceritinib. Lung Cancer 2017, 110, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Schrock, A.B.; Bosemani, T.; Benn, B.S.; Ali, S.M.; Ou, S.I. Dramatic response to alectinib in a lung cancer patient with a novel VKORC1L1-ALK fusion and an acquired ALK T1151K mutation. Lung Cancer 2018, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, M.; Chaudhari, K.; Nathany, S.; Talwar, V. Identification of a novel resistance ALK p.(Q1188_L1190del) deletion in a patient with ALK-rearranged non–small-cell lung cancer. Cancer Genet. 2021, 256, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-addicted non-small-cell lung cancer: Treatment opportunities and future perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- Kodityal, S.; Elvin, J.A.; Squillace, R.; Agarwal, N.; Miller, V.A.; Ali, S.M.; Klempner, S.J.; Ou, S.H.I. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer 2016, 92, 19–21. [Google Scholar] [CrossRef]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a novel ALK G1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J. Thorac. Oncol. 2015, 10, e55–e57. [Google Scholar] [CrossRef]
- Ceccon, M. Ceritinib as a promising therapy for ALK related diseases. Transl. Lung Cancer Res. 2014, 3, 376–378. [Google Scholar]
- Takahashi, K.; Seto, Y.; Okada, K.; Uematsu, S.; Uchibori, K.; Tsukahara, M.; Oh-Hara, T.; Fujita, N.; Yanagitani, N.; Nishio, M.; et al. Overcoming resistance by ALK compound mutation (I1171S + G1269A) after sequential treatment of multiple ALK inhibitors in non-small cell lung cancer. Thorac. Cancer 2020, 11, 581–587. [Google Scholar] [CrossRef]
- Guo, J.; Guo, L.; Sun, L.; Wu, Z.; Ye, J.; Liu, J.; Zuo, Q. Capture-based ultra-deep sequencing in plasma ctDNA reveals the resistance mechanism of ALK inhibitors in a patient with advanced ALK-positive NSCLC. Cancer Biol. Ther. 2018, 19, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef]
- Yang, P.; Cao, R.; Bao, H.; Wu, X.; Yang, L.; Zhu, D.; Zhang, L.; Peng, L.; Cai, Y.; Zhang, W.; et al. Identification of novel Alectinib-resistant ALK mutation G1202K with sensitization to lorlatinib: A case report and in silico structural modelling. Onco Targets Ther. 2021, 14, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Li, T.; Wang, P.; Lizaso, A.; Huang, D. The efficacy of lorlatinib in a lung adenocarcinoma patient with a novel ALK G1202L mutation: A case report. Cancer Biol. Ther. 2021, 22, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; Cortinovis, D.; Agustoni, F.; Arosio, G.; Villa, M.; Cordani, N.; Bidoli, P.; Bisson, W.H.; Pagni, F.; Piazza, R.; et al. A compound L1196M/G1202R ALK mutation in a patient with ALK-positive lung cancer with acquired resistance to brigatinib also confers primary resistance to lorlatinib. J. Thorac. Oncol. 2019, 14, e257–e259. [Google Scholar] [CrossRef]
- Mologni, L.; Ceccon, M.; Pirola, A.; Chiriano, G.; Piazza, R.; Scapozza, L.; Gambacorti-Passerini, C. NPM/ALK mutants resistant to ASP3026 display variable sensitivity to alternative ALK inhibitors but succumb to the novel compound PF-06463922. Oncotarget 2015, 6, 5720–5734. [Google Scholar] [CrossRef]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef]
- Pastor, E.R.; Mousa, S.A. Current management of neuroblastoma and future direction. Crit. Rev. Oncol. Hematol. 2019, 138, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.Y.; et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin. Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretić, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef]
- Shi, P.; Lai, R.; Lin, Q.; Iqbal, A.S.; Young, L.C.; Kwak, L.W.; Ford, R.J.; Amin, H.M. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood 2009, 114, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Rossing, H.H.; Grauslund, M.; Urbanska, E.M.; Melchior, L.C.; Rask, C.K.; Costa, J.C.; Skov, B.G.; Sørensen, J.B.; Santoni-Rugiu, E. Concomitant occurrence of EGFR (epidermal growth factor receptor) and KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutations in an ALK (anaplastic lymphoma kinase)-positive lung adenocarcinoma patient with acquired resistance to crizotinib: A case report. BMC Res. Notes 2013, 6, 489. [Google Scholar]
- Sánchez-Herrero, E.; Serna-Blasco, R.; Ivanchuk, V.; García-Campelo, R.; Dómine Gómez, M.; Sánchez, J.M.; Massutí, B.; Reguart, N.; Camps, C.; Sanz-Moreno, S.; et al. NGS-based liquid biopsy profiling identifies mechanisms of resistance to ALK inhibitors: A step toward personalized NSCLC treatment. Mol. Oncol. 2021, 15, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, M.; Yasuda, H.; Tani, T.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Nukaga, S.; Hirano, T.; Kawada, I.; et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol. Cancer Res. 2017, 15, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, A.; Yamada, T.; Nanjo, S.; Takeuchi, S.; Ebi, H.; Kita, K.; Matsumoto, K.; Seiji, Y. Receptor ligand-triggered resistance to alectinib and its circumvention by Hsp90 inhibition in EML4-ALK lung cancer cells. Oncotarget 2014, 5, 4920–4928. [Google Scholar] [CrossRef] [PubMed]
- Minari, R.; Gnetti, L.; Lagrasta, C.A.; Squadrilli, A.; Bordi, P.; Azzoni, C.; Bottarelli, L.; Cosenza, A.; Ferri, L.; Caruso, G.; et al. Emergence of a HER2-amplified clone during disease progression in an ALK-rearranged NSCLC patient treated with ALK-inhibitors: A case report. Transl. Lung Cancer Res. 2020, 9, 787–792. [Google Scholar] [CrossRef]
- Dong, X.; Fernandez-Salas, E.; Li, E.; Wang, S. Elucidation of resistance mechanisms to second-generation ALK inhibitors alectinib and ceritinib in non-small cell lung cancer cells. Neoplasia 2016, 18, 162–171. [Google Scholar] [CrossRef]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Banno, E.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Activated MET acts as a salvage signal after treatment with alectinib, a selective ALK inhibitor, in ALK-positive non-small cell lung cancer. Int. J. Oncol. 2015, 46, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Ozasa, H.; Aoki, W.; Aburaya, S.; Funazo, T.; Furugaki, K.; Yoshimura, Y.; Ajimizu, H.; Okutani, R.; Yasuda, Y.; et al. Alectinib resistance in ALK-rearranged lung cancer by dual salvage signaling in a clinically paired resistance model. Mol. Cancer Res. 2019, 17, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, C.; Peng, T.; Hu, C.; Lu, C.; Li, L.; Wang, Y.; Han, R.; Feng, M.; Sun, F.; et al. Metformin reduces HGF-induced resistance to alectinib via the inhibition of Gab1. Cell Death Dis. 2020, 11, 111. [Google Scholar] [CrossRef]
- Shi, R.; Filho, S.N.M.; Li, M.; Fares, A.; Weiss, J.; Pham, N.A.; Ludkovski, O.; Raghavan, V.; Li, Q.; Ravi, D.; et al. BRAF V600E mutation and MET amplification as resistance pathways of the second-generation anaplastic lymphoma kinase (ALK) inhibitor alectinib in lung cancer. Lung Cancer 2020, 146, 78–85. [Google Scholar] [CrossRef]
- Fan, P.D.; Narzisi, G.; Jayaprakash, A.D.; Venturini, E.; Robine, N.; Smibert, P.; Germer, S.; Yu, H.A.; Jordan, E.J.; Paik, P.K.; et al. YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics. Proc. Natl. Acad. Sci. USA 2018, 115, E6030–E6038. [Google Scholar] [CrossRef] [PubMed]
- Prokoph, N.; Probst, N.A.; Lee, L.C.; Monahan, J.M.; Matthews, J.D.; Liang, H.C.; Bahnsen, K.; Montes-Mojarro, I.A.; Karaca-Atabay, E.; Sharma, G.G.; et al. IL10RA modulates crizotinib sensitivity in NPM1-ALK+ anaplastic large cell lymphoma. Blood 2020, 136, 1657–1669. [Google Scholar] [CrossRef]
- Karaca-Atabay, E.; Mecca, C.; Wang, Q.; Ambrogio, C.; Mota, I.; Prokoph, N.; Mura, G.; Martinengo, C.; Patrucco, E.; Leonardi, G.; et al. Tyrosine phosphatases regulate resistance to ALK inhibitors in ALK+ anaplastic large cell lymphoma. Blood 2021, in press. [Google Scholar] [CrossRef]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef]
- Mengoli, M.C.; Barbieri, F.; Bertolini, F.; Tiseo, M.; Rossi, G. K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung: Report of two cases and review of the literature. Lung Cancer 2016, 93, 55–58. [Google Scholar] [CrossRef]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef]
- Larose, H.; Prokoph, N.; Matthews, J.D.; Schlederer, M.; Högler, S.; Alsulami, A.F.; Ducray, S.P.; Nuglozeh, E.; Fazaludeen, F.M.S.; Elmouna, A.; et al. Whole exome sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target. Haematologica 2021, 106, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Trigg, R.M.; Lee, L.C.; Prokoph, N.; Jahangiri, L.; Reynolds, C.P.; Amos Burke, G.A.; Probst, N.A.; Han, M.; Matthews, J.D.; Lim, H.K.; et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat. Commun. 2019, 10, 5428. [Google Scholar] [CrossRef] [PubMed]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Sogabe, S.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 1430–1436. [Google Scholar] [CrossRef]
- Urbanska, E.M.; Sørensen, J.B.; Melchior, L.C.; Costa, J.C.; Santoni-Rugiu, E. Changing ALK-TKI-resistance mechanisms in rebiopsies of ALK-rearranged NSCLC: ALK- and BRAF-mutations followed by epithelial-mesenchymal transition. Int. J. Mol. Sci. 2020, 21, 2847. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Cho, B.C.; Kim, H.R.; Lee, H.J.; Shim, H.S. A case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J. Thorac. Oncol. 2016, 11, e55–e58. [Google Scholar] [CrossRef]
- Fujita, S.; Masago, K.; Katakami, N.; Yatabe, Y. Transformation to SCLC after treatment with the ALK inhibitor alectinib. J. Thorac. Oncol. 2016, 11, e67–e72. [Google Scholar] [CrossRef]
- Levacq, D.; D’Haene, N.; de Wind, R.; Remmelink, M.; Berghmans, T. Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: A new mechanism of resistance to ALK inhibitors. Lung Cancer 2016, 102, 38–41. [Google Scholar] [CrossRef]
- Coleman, N.; Wotherspoon, A.; Yousaf, N.; Popat, S. Transformation to neuroendocrine carcinoma as a resistance mechanism to lorlatinib. Lung Cancer 2019, 134, 117–120. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Dries, R.; Sengupta, S.; Seruggia, D.; Gao, Y.; Sharma, B.; Huang, H.; Moreau, L.; McLane, M.; Day, D.S.; et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 2019, 572, 676–680. [Google Scholar] [CrossRef]
- Berko, E.R.; Mossé, Y.P. Thrown for a loop: Awakening BORIS to evade ALK inhibition therapy. Cancer Cell 2019, 36, 345–347. [Google Scholar] [CrossRef]
- Cai, C.; Long, Y.; Li, Y.; Huang, M. Coexisting of COX7A2L-ALK, LINC01210-ALK, ATP13A4-ALK and acquired SLCO2A1-ALK in a lung adenocarcinoma with rearrangements loss during the treatment of crizotinib and ceritinib: A case report. Onco Targets Ther. 2020, 13, 8313–8316. [Google Scholar] [CrossRef] [PubMed]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef]
- Ji, C.; Zhang, L.; Cheng, Y.; Patel, R.; Wu, H.; Zhang, Y.; Wang, M.; Ji, S.; Belani, C.P.; Yang, J.M.; et al. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol. Ther. 2014, 15, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Moia, R.; Boggione, P.; Mahmoud, A.M.; Kodipad, A.A.; Adhinaveni, R.; Sagiraju, S.; Patriarca, A.; Gaidano, G. Targeting p53 in chronic lymphocytic leukemia. Expert Opin. Ther. Targets 2020, 24, 1239–1250. [Google Scholar] [CrossRef]
- Moia, R.; Patriarca, A.; Schipani, M.; Ferri, V.; Favini, C.; Sagiraju, S.; Al Essa, W.; Gaidano, G. Precision medicine management of chronic lymphocytic leukemia. Cancers 2020, 12, 642. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Otomo, R.; Matsushima-Hibiya, Y.; Suzuki, H.; Nakajima, A.; Abe, N.; Tomiyama, A.; Ichimura, K.; Matsuda, K.; Watanabe, T.; et al. The p53 activator overcomes resistance to ALK inhibitors by regulating p53-target selectivity in ALK-driven neuroblastomas. Cell Death Discov. 2018, 4, 56. [Google Scholar] [CrossRef]
- Rassidakis, G.Z.; Thomaides, A.; Wang, S.; Jiang, Y.; Fourtouna, A.; Lai, R.; Medeiros, L.J. p53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia 2005, 19, 1663–1669. [Google Scholar] [CrossRef]
- Cui, Y.-X.; Kerby, A.; McDuff, F.K.E.; Ye, H.; Turner, S.D. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood 2009, 113, 5217–5227. [Google Scholar] [CrossRef]
- Drakos, E.; Atsaves, V.; Schlette, E.; Li, J.; Papanastasi, I.; Rassidakis, G.Z.; Medeiros, L.J. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009, 23, 2290–2299. [Google Scholar] [CrossRef]
- Redaelli, S.; Ceccon, M.; Antolini, L.; Rigolio, R.; Pirola, A.; Peronaci, M.; Gambacorti-Passerini, C.; Mologni, L. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 2016, 7, 72886–72897. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Yoda, S.; Lennerz, J.K.; Langenbucher, A.; Lin, J.J.; Rooney, M.M.; Prutisto-Chang, K.; Oh, A.; Adams, N.A.; Yeap, B.Y.; et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin. Cancer Res. 2020, 26, 2535–2545. [Google Scholar] [CrossRef]
- Chihara, D.; Wong, S.; Feldman, T.; Fanale, M.A.; Sanchez, L.; Connors, J.M.; Savage, K.J.; Oki, Y. Outcome of patients with relapsed or refractory anaplastic large cell lymphoma who have failed brentuximab vedotin. Hematol. Oncol. 2019, 37, 35–38. [Google Scholar] [CrossRef]
- Arai, H.; Furuichi, S.; Nakamura, Y.; Ichikawa, M.; Mitani, K. ALK-negative anaplastic large cell lymphoma with loss of CD30 expression during treatment with brentuximab vedotin. Rinsho Ketsueki 2016, 57, 634–637. [Google Scholar]
- Fordham, A.M.; Xie, J.; Gifford, A.J.; Wadham, C.; Morgan, L.T.; Mould, E.V.A.; Fadia, M.; Zhai, L.; Massudi, H.; Ali, Z.S.; et al. CD30 and ALK combination therapy has high therapeutic potency in RANBP2-ALK-rearranged epithelioid inflammatory myofibroblastic sarcoma. Br. J. Cancer 2020, 123, 1101–1113. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef]
- Wei, W.; Lin, Y.; Song, Z.; Xiao, W.; Chen, L.; Yin, J.; Zhou, Y.; Barta, S.K.; Petrus, M.; Waldmann, T.A.; et al. A20 and RBX1 regulate brentuximab vedotin sensitivity in hodgkin lymphoma models. Clin. Cancer Res. 2020, 26, 4093–4106. [Google Scholar] [CrossRef]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.N.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 overcomes resistance to brentuximab vedotin in hodgkin lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Oh, M.S.; Giles, F.J. Molecular biomarkers of primary and acquired resistance to T-cell-mediated immunotherapy in cancer: Landscape, clinical implications, and future directions. Oncologist 2018, 23, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wu, Z.X.; Assaraf, Y.G.; Chen, Z.S.; Wang, L. Overcoming anti-cancer drug resistance via restoration of tumor suppressor gene function. Drug Resist. Updat. 2021, 57, 100770. [Google Scholar] [CrossRef]
- Cretella, D.; Digiacomo, G.; Giovannetti, E.; Cavazzoni, A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers 2019, 11, 1318. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, Y.; Zhang, J.; Jin, X.; Wu, H.; Huang, H. Fructose-1,6-bisphosphatase loss modulates STAT3-dependent expression of PD-L1 and cancer immunity. Theranostics 2020, 10, 1033–1045. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- De Souza, A. Finding the hot spot: Identifying immune sensitive gastrointestinal tumors. Transl. Gastroenterol. Hepatol. 2020, 5, 48. [Google Scholar] [CrossRef]
- Ross-Macdonald, P.; Walsh, A.M.; Chasalow, S.D.; Ammar, R.; Papillon-Cavanagh, S.; Szabo, P.M.; Choueiri, T.K.; Sznol, M.; Wind-Rotolo, M. Molecular correlates of response to nivolumab at baseline and on treatment in patients with RCC. J. Immunother. Cancer 2021, 9, e001506. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Vagia, E.; Viveiros, P.; Kang, C.Y.; Lee, J.Y.; Gim, G.; Cho, S.; Choi, H.; Kim, L.; Park, I.; et al. Overcoming acquired resistance to PD-1 inhibitor with the addition of metformin in small cell lung cancer (SCLC). Cancer Immunol. Immunother. 2021, 70, 961–965. [Google Scholar] [CrossRef] [PubMed]
- De Wispelaere, W.; Annibali, D.; Tuyaerts, S.; Lambrechts, D.; Amant, F. Resistance to immune checkpoint blockade in uterine leiomyosarcoma: What can we learn from other cancer types? Cancers 2021, 13, 2040. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Tsuchihashi, K.; Tsuji, K.; Kito, Y.; Tanoue, K.; Ohmura, H.; Ito, M.; Isobe, T.; Ariyama, H.; Kusaba, H.; et al. Prominent PD-L1-positive M2 macrophage infiltration in gastric cancer with hyper-progression after anti-PD-1 therapy: A case report. Medicine 2021, 100, e25773. [Google Scholar] [CrossRef]
- Viveiros, P.; Burns, M.; Davis, A.; Oh, M.; Park, K.; Jain, S.; Chae, Y.K. EP1.04-12 Response to combination of metformin and nivolumab in a NSCLC patient whose disease previously progressed on nivolumab. J. Thorac. Oncol. 2019, 14, S976. [Google Scholar] [CrossRef]
- Cabrera, C.M.; Jiménez, P.; Cabrera, T.; Esparza, C.; Ruiz-Cabello, F.; Garrido, F. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: Beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens 2003, 61, 211–219. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]





| Protein Alteration (Upregulation Unless Otherwise Specified) | ALK TKI | Disease |
|---|---|---|
| IGF-1R | Crizotinib [129,130] | NSCLC and ALCL |
| Epidermal growth factor receptor (EGFR) | Crizotinib [78,81,98,131,132] Ceritinib [98,133] Alectinib [98,132,134] Lorlatinib [124,132] | NSCLC |
| Lorlatinib [124] | Neuroblastoma | |
| Human epidermal growth factor receptor (HER), including via increased neuregulin 1 ligand | Ceritinib and alectinib [135,136] | NSCLC |
| KIT proto-oncogene receptor tyrosine kinase (KIT), including via increased stem cell factor (SCF) ligand | Crizotinib [78] Ceritinib [98] | NSCLC |
| MET proto-oncogene receptor tyrosine kinase (MET), including via increased hepatocyte growth factor (HGF) ligand | Alectinib [134,137,138,139,140] Ceritinib and lorlatinib [79,98,117] | NSCLC |
| SRC proto-oncogene, non-receptor tyrosine kinase (SRC) | Crizotinib [141] Alectinib [138] | NSCLC |
| Discoidin domain receptor tyrosine kinase 2 (DDR2) | Alectinib [79] | NSCLC |
| Fibroblast growth factor receptor 2 (FGFR2) | Ceritinib [79] | NSCLC |
| ERb-B4 receptor tyrosine kinase 4 (ErbB4) | Lorlatinib [124] | Neuroblastoma |
| Interleukin 10 receptor subunit alpha (IL10RA) | Crizotinib [142] Alectinib [142] Brigatinib [142] Lorlatinib [142] | ALCL |
| Protein tyrosine phosphatase non-receptor tyrosine kinase 1/2 (PTPN1/2) loss | Crizotinib [143] | ALCL |
| Event | Impact on the Tumour Microenvironment |
|---|---|
| Increased AXL receptor tyrosine kinase (AXL) expression | Increases regulatory T cells, MDSCs and M2 macrophages [185] |
| Increased Wnt signalling | Decreases tumour infiltrating lymphocytes [185] |
| Loss of Phosphatase and tensin homolog (PTEN) | Induces vascular endothelial growth factor (VEGF) production and reduces T cell infiltration [176,185] |
| Loss of functional beta 2 microglobulin | Dysfunctional CD8+ T cells [175,188] |
| Hypoxia | Dysfunctional CD8+ T cells [184,187] |
| Upregulation of T cell immunoglobulin and mucin-domain containing-3 (Tim-3) | Dysfunctional T helper 1 (Th1) cells and reduced cytokine expression [189,190] |
| Reduced expression of absent in melanoma 2 (AIM2) | Decreases inflammation [181] |
| Reduced expression of poliovirus receptor-related immunoglobulin domain containing protein (PVRIG) | Dysfunctional CD8+ T cells [181] |
| Increased expression of mannosidase alpha class 2A member 1 (MAN2A1) | Altered Th1/T-helper 2 (Th2) axis towards Th2 expression [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hare, L.; Burke, G.A.A.; Turner, S.D. Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL. Cancers 2021, 13, 6003. https://doi.org/10.3390/cancers13236003
Hare L, Burke GAA, Turner SD. Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL. Cancers. 2021; 13(23):6003. https://doi.org/10.3390/cancers13236003
Chicago/Turabian StyleHare, Lucy, G. A. Amos Burke, and Suzanne D. Turner. 2021. "Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL" Cancers 13, no. 23: 6003. https://doi.org/10.3390/cancers13236003
APA StyleHare, L., Burke, G. A. A., & Turner, S. D. (2021). Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL. Cancers, 13(23), 6003. https://doi.org/10.3390/cancers13236003