
Chromosome Imbalances in Neuroblastoma—Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future



Abstract
Simple Summary
Abstract
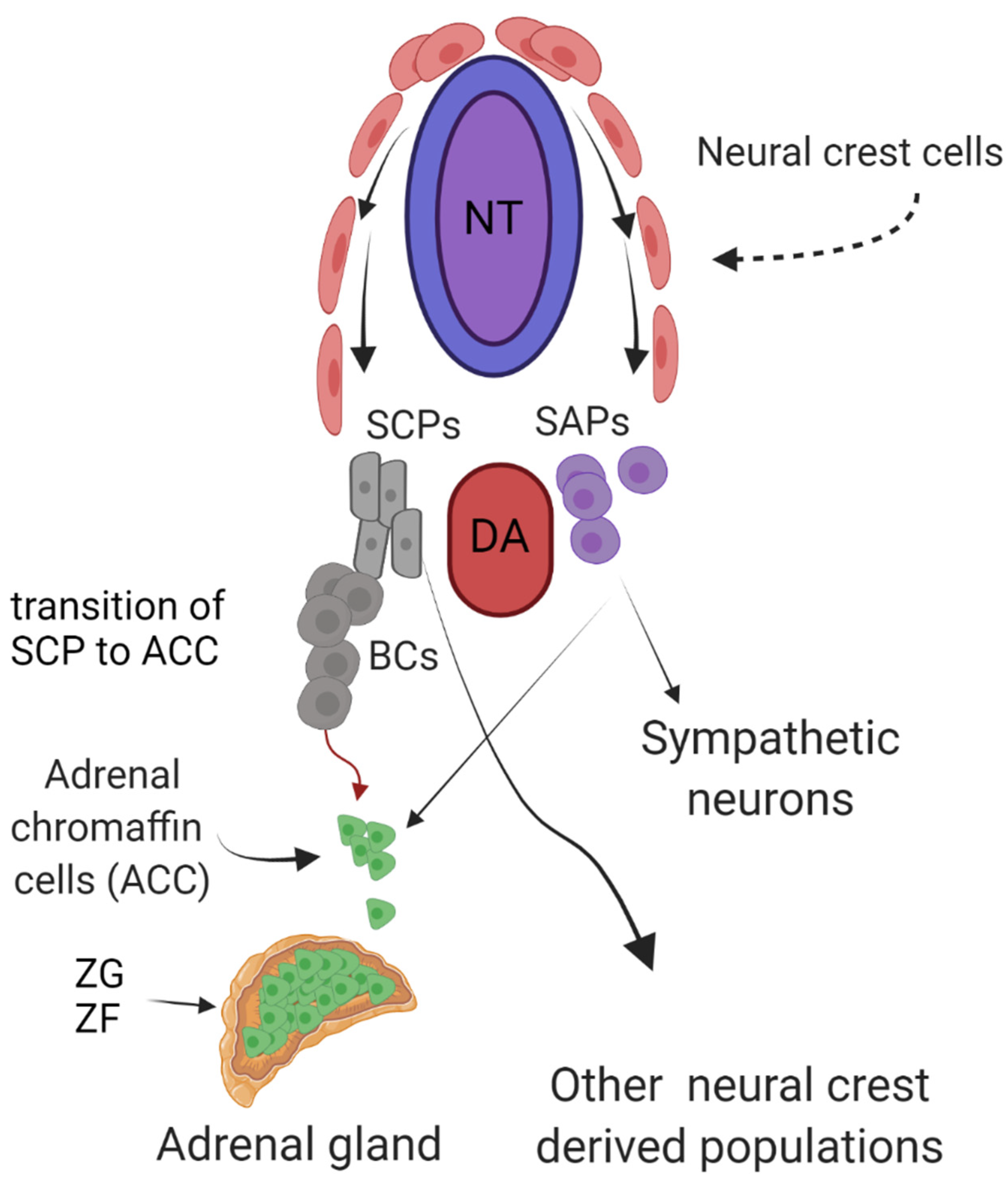
1. Introduction
2. Novel Mechanisms Underlying the Contribution of 1p36 Deletion to MYCN Neuroblastoma
3. Working Alone or in Cooperation? 2p-gain and the ALKAL2, MYCN, and ALK Troika
4. New Candidate Tumor Suppressor Genes and Molecular Mechanisms Shed Light on 11q Deletion Neuroblastoma
5. Friends and Foes—Implications for the Future
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Huber, K.; Kalcheim, C.; Unsicker, K. The development of the chromaffin cell lineage from the neural crest. Auton. Neurosci. 2009, 151, 10–16. [Google Scholar] [CrossRef]
- Saito, D.; Takase, Y.; Murai, H.; Takahashi, Y. The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science 2012, 336, 1578–1581. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Bedoya-Reina, O.C.; Li, W.; Arceo, M.; Plescher, M.; Bullova, P.; Pui, H.; Kaucka, M.; Kharchenko, P.; Martinsson, T.; Holmberg, J.; et al. Single-nuclei transcriptomes from human adrenal gland reveal distinct cellular identities of low and high-risk neuroblastoma tumors. Nat. Commun. 2021, 12, 5309. [Google Scholar] [CrossRef]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.D.; Ye, C.J.; Yao, X.Y.; Luo, W.Q.; Cheng, X.M.; Miao, J.J.; Wang, J.F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733.e6. [Google Scholar] [CrossRef]
- Hanemaaijer, E.S.; Margaritis, T.; Sanders, K.; Bos, F.L.; Candelli, T.; Al-Saati, H.; van Noesel, M.M.; Meyer-Wentrup, F.A.G.; van de Wetering, M.; Holstege, F.C.P.; et al. Single-cell atlas of developing murine adrenal gland reveals relation of Schwann cell precursor signature to neuroblastoma phenotype. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Ali, F.R.; Marcos, D.; Chernukhin, I.; Woods, L.M.; Parkinson, L.M.; Wylie, L.A.; Papkovskaia, T.D.; Davies, J.D.; Carroll, J.S.; Philpott, A. Dephosphorylation of the Proneural Transcription Factor ASCL1 Re-Engages a Latent Post-Mitotic Differentiation Program in Neuroblastoma. Mol. Cancer Res. 2020, 18, 1759–1766. [Google Scholar] [CrossRef]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar] [CrossRef]
- Ritenour, L.E.; Randall, M.P.; Bosse, K.R.; Diskin, S.J. Genetic susceptibility to neuroblastoma: Current knowledge and future directions. Cell Tissue Res. 2018, 372, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Kastriti, M.E.; Kameneva, P.; Adameyko, I. Stem cells, evolutionary aspects and pathology of the adrenal medulla: A new developmental paradigm. Mol. Cell. Endocrinol. 2020, 518, 110998. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef]
- Stainczyk, S.A.; Westermann, F. Neuroblastoma-Telomere maintenance, deregulated signaling transduction and beyond. Int. J. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. MYCN Amplification in Neuroblastoma: A Paradigm for the Clinical Use of an Oncogene. Pathol. Oncol. Res. 1997, 3, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Siaw, J.T.; Javanmardi, N.; Van den Eynden, J.; Lind, D.E.; Fransson, S.; Martinez-Monleon, A.; Djos, A.; Sjoberg, R.M.; Ostensson, M.; Caren, H.; et al. 11q Deletion or ALK Activity Curbs DLG2 Expression to Maintain an Undifferentiated State in Neuroblastoma. Cell Rep. 2020, 32, 108171. [Google Scholar] [CrossRef]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Mlakar, V.; Jurkovic Mlakar, S.; Lopez, G.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. 11q deletion in neuroblastoma: A review of biological and clinical implications. Mol. Cancer 2017, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Corvetta, D.; Chayka, O.; Gherardi, S.; D’Acunto, C.W.; Cantilena, S.; Valli, E.; Piotrowska, I.; Perini, G.; Sala, A. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. J. Biol. Chem. 2013, 288, 8332–8341. [Google Scholar] [CrossRef]
- Higashi, M.; Sakai, K.; Fumino, S.; Aoi, S.; Furukawa, T.; Tajiri, T. The roles played by the MYCN, Trk, and ALK genes in neuroblastoma and neural development. Surg. Today 2019, 49, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Sekhon, G.; Goldstein, M.N. Chromosomal aberrations in human neuroblastomas. Cancer 1977, 40, 2256–2263. [Google Scholar] [CrossRef]
- Mathew, P.; Valentine, M.B.; Bowman, L.C.; Rowe, S.T.; Nash, M.B.; Valentine, V.A.; Cohn, S.L.; Castleberry, R.P.; Brodeur, G.M.; Look, A.T. Detection of MYCN gene amplification in neuroblastoma by fluorescence in situ hybridization: A pediatric oncology group study. Neoplasia 2001, 3, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Vendrell, E.; Aiza, G.; Nistal, M.; Pestana, A.; Peinado, M.A.; Castresana, J.S. Determination of genomic damage in neuroblastic tumors by arbitrarily primed PCR: MYCN amplification as a marker for genomic instability in neuroblastomas. Neuropathology 2006, 26, 165–169. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Caren, H.; Kryh, H.; Nethander, M.; Sjoberg, R.M.; Trager, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Gastier-Foster, J.M.; Mann, M.; Naranjo, A.H.; Van Ryn, C.; Bagatell, R.; Matthay, K.K.; London, W.B.; Irwin, M.S.; Shimada, H.; et al. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer 2017, 123, 4224–4235. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Umehara, S.; Gerbing, R.B.; Stram, D.O.; Brodeur, G.M.; Seeger, R.C.; Lukens, J.N.; Matthay, K.K.; Shimada, H. Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: A report from the Children’s Cancer Group. Cancer 2001, 92, 2699–2708. [Google Scholar] [CrossRef]
- Schwab, M.; Westermann, F.; Hero, B.; Berthold, F. Neuroblastoma: Biology and molecular and chromosomal pathology. Lancet Oncol. 2003, 4, 472–480. [Google Scholar] [CrossRef]
- Maris, J.M.; Guo, C.; Blake, D.; White, P.S.; Hogarty, M.D.; Thompson, P.M.; Rajalingam, V.; Gerbing, R.; Stram, D.O.; Matthay, K.K.; et al. Comprehensive analysis of chromosome 1p deletions in neuroblastoma. Med. Pediatric Oncol. 2001, 36, 32–36. [Google Scholar] [CrossRef]
- Schlisio, S.; Kenchappa, R.S.; Vredeveld, L.C.; George, R.E.; Stewart, R.; Greulich, H.; Shahriari, K.; Nguyen, N.V.; Pigny, P.; Dahia, P.L.; et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008, 22, 884–893. [Google Scholar] [CrossRef]
- Munirajan, A.K.; Ando, K.; Mukai, A.; Takahashi, M.; Suenaga, Y.; Ohira, M.; Koda, T.; Hirota, T.; Ozaki, T.; Nakagawara, A. KIF1Bbeta functions as a haploinsufficient tumor suppressor gene mapped to chromosome 1p36.2 by inducing apoptotic cell death. J. Biol. Chem. 2008, 283, 24426–24434. [Google Scholar] [CrossRef]
- Hogarty, M.D.; Maris, J.M.; White, P.S.; Guo, C.; Brodeur, G.M. Analysis of genomic imprinting at 1p35-36 in neuroblastoma. Med. Pediatric Oncol. 2001, 36, 52–55. [Google Scholar] [CrossRef]
- Tolbert, V.P.; Coggins, G.E.; Maris, J.M. Genetic susceptibility to neuroblastoma. Curr. Opin. Genet. Dev. 2017, 42, 81–90. [Google Scholar] [CrossRef]
- Garcia-Lopez, J.; Wallace, K.; Otero, J.H.; Olsen, R.; Wang, Y.D.; Finkelstein, D.; Gudenas, B.L.; Rehg, J.E.; Northcott, P.; Davidoff, A.M.; et al. Large 1p36 Deletions Affecting Arid1a Locus Facilitate Mycn-Driven Oncogenesis in Neuroblastoma. Cell Rep. 2020, 30, 454–464.e5. [Google Scholar] [CrossRef]
- Shi, H.; Tao, T.; Abraham, B.J.; Durbin, A.D.; Zimmerman, M.W.; Kadoch, C.; Look, A.T. ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci. Adv. 2020, 6, eaaz3440. [Google Scholar] [CrossRef]
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A., Jr.; Papadopoulos, N.; et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013, 45, 12–17. [Google Scholar] [CrossRef]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. elife 2015, 4, e09811. [Google Scholar] [CrossRef]
- Fadeev, A.; Mendoza-Garcia, P.; Irion, U.; Guan, J.; Pfeifer, K.; Wiessner, S.; Serluca, F.; Singh, A.P.; Nusslein-Volhard, C.; Palmer, R.H. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. USA 2018, 115, E630–E638. [Google Scholar] [CrossRef]
- Borenas, M.; Umapathy, G.; Lai, W.Y.; Lind, D.E.; Witek, B.; Guan, J.; Mendoza-Garcia, P.; Masudi, T.; Claeys, A.; Chuang, T.P.; et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. 2021, 40, e105784. [Google Scholar] [CrossRef]
- Mo, E.S.; Cheng, Q.N.; Reshetnyak, A.V.; Schlessinger, J.; Nicoli, S. Alk and Ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase Ltk. Proc. Natl. Acad. Sci. USA 2017, 114, 12027–12032. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Caren, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef]
- Martinsson, T.; Eriksson, T.; Abrahamsson, J.; Caren, H.; Hansson, M.; Kogner, P.; Kamaraj, S.; Schonherr, C.; Weinmar, J.; Ruuth, K.; et al. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res. 2011, 71, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Fadeev, A.; Krauss, J.; Singh, A.P.; Nusslein-Volhard, C. Zebrafish Leucocyte tyrosine kinase controls iridophore establishment, proliferation and survival. Pigment Cell Melanoma Res. 2016, 29, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Cazes, A.; Lopez-Delisle, L.; Tsarovina, K.; Pierre-Eugene, C.; De Preter, K.; Peuchmaur, M.; Nicolas, A.; Provost, C.; Louis-Brennetot, C.; Daveau, R.; et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 2688–2702. [Google Scholar] [CrossRef]
- Ono, S.; Saito, T.; Terui, K.; Yoshida, H.; Enomoto, H. Generation of conditional ALK F1174L mutant mouse models for the study of neuroblastoma pathogenesis. Genesis 2019, 57, e23323. [Google Scholar] [CrossRef]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci. Transl. Med. 2012, 4, 141ra91. [Google Scholar] [CrossRef]
- Schonherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Nafady, A.; Takatori, A.; Kishida, S.; Ohira, M.; Suenaga, Y.; Hossain, S.; Akter, J.; Ogura, A.; Nakamura, Y.; et al. ALK is a MYCN target gene and regulates cell migration and invasion in neuroblastoma. Sci. Rep. 2013, 3, 3450. [Google Scholar] [CrossRef]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALK(F1174L) Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK Collaborates with MYCN in Neuroblastoma Pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Lennartsson, J.; Westermark, B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef]
- Zhang, H.; Pao, L.I.; Zhou, A.; Brace, A.D.; Halenbeck, R.; Hsu, A.W.; Bray, T.L.; Hestir, K.; Bosch, E.; Lee, E.; et al. Deorphanization of the human leukocyte tyrosine kinase (LTK) receptor by a signaling screen of the extracellular proteome. Proc. Natl. Acad. Sci. USA 2014, 111, 15741–15745. [Google Scholar] [CrossRef] [PubMed]
- Javanmardi, N.; Fransson, S.; Djos, A.; Umapathy, G.; Ostensson, M.; Milosevic, J.; Borenas, M.; Hallberg, B.; Kogner, P.; Martinsson, T.; et al. Analysis of ALK, MYCN and the ALK ligand ALKAL2 (FAM150B/AUGalpha) in neuroblastoma patient samples with chromosome arm 2p rearrangements. Genes Chromosomes Cancer 2019, 59, 50–57. [Google Scholar] [CrossRef]
- Jeison, M.; Ash, S.; Halevy-Berko, G.; Mardoukh, J.; Luria, D.; Avigad, S.; Feinberg-Gorenshtein, G.; Goshen, Y.; Hertzel, G.; Kapelushnik, J.; et al. 2p24 Gain region harboring MYCN gene compared with MYCN amplified and nonamplified neuroblastoma: Biological and clinical characteristics. Am. J. Pathol. 2010, 176, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Say, B.; Carpenter, N.J.; Giacoia, G.; Jegathesan, S. Agenesis of the lung associated with a chromosome abnormality (46,XX,2p+). J. Med. Genet. 1980, 17, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.S.; Pearson, J.; Willatt, L.; Andrews, T.; Beach, R.; Green, A. Germline duplication of chromosome 2p and neuroblastoma. J. Med. Genet. 1997, 34, 949–951. [Google Scholar] [CrossRef][Green Version]
- Dowa, Y.; Yamamoto, T.; Abe, Y.; Kobayashi, M.; Hoshino, R.; Tanaka, K.; Aida, N.; Take, H.; Kato, K.; Tanaka, Y.; et al. Congenital neuroblastoma in a patient with partial trisomy of 2p. J. Pediatric Hematol. Oncol. 2006, 28, 379–382. [Google Scholar] [CrossRef]
- Nagano, H.; Kano, Y.; Kobuchi, S.; Kajitani, T. A case of partial 2p trisomy with neuroblastoma. Jpn. J. Hum. Genet. 1980, 25, 39–45. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morgenstern, D.A.; Soh, S.Y.; Stavropoulos, D.J.; Bowdin, S.; Baruchel, S.; Malkin, D.; Meyn, M.S.; Irwin, M.S. Metachronous neuroblastoma in an infant with germline translocation resulting in partial trisomy 2p: A role for ALK? J. Pediatric Hematol. Oncol. 2014, 36, e193–e196. [Google Scholar] [CrossRef]
- Bader, S.A.; Fasching, C.; Brodeur, G.M.; Stanbridge, E.J. Dissociation of suppression of tumorigenicity and differentiation in vitro effected by transfer of single human chromosomes into human neuroblastoma cells. Cell Growth Differ. 1991, 2, 245–255. [Google Scholar] [PubMed]
- Srivatsan, E.S.; Ying, K.L.; Seeger, R.C. Deletion of chromosome 11 and of 14q sequences in neuroblastoma. Genes Chromosomes Cancer 1993, 7, 32–37. [Google Scholar] [CrossRef]
- Caren, H.; Erichsen, J.; Olsson, L.; Enerback, C.; Sjoberg, R.M.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: Four cases of homozygous deletions of the CDKN2A gene. BMC Genom. 2008, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Attiyeh, E.F.; London, W.B.; Mosse, Y.P.; Wang, Q.; Winter, C.; Khazi, D.; McGrady, P.W.; Seeger, R.C.; Look, A.T.; Shimada, H.; et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N. Engl. J. Med. 2005, 353, 2243–2253. [Google Scholar] [CrossRef]
- Spitz, R.; Hero, B.; Simon, T.; Berthold, F. Loss in chromosome 11q identifies tumors with increased risk for metastatic relapses in localized and 4S neuroblastoma. Clin. Cancer Res. 2006, 12, 3368–3373. [Google Scholar] [CrossRef]
- Johnson, A.F.; Nguyen, H.T.; Veitia, R.A. Causes and effects of haploinsufficiency. Biol. Rev. 2019, 94, 1774–1785. [Google Scholar] [CrossRef]
- Fero, M.L.; Randel, E.; Gurley, K.E.; Roberts, J.M.; Kemp, C.J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998, 396, 177–180. [Google Scholar] [CrossRef]
- Inoue, K.; Zindy, F.; Randle, D.H.; Rehg, J.E.; Sherr, C.J. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001, 15, 2934–2939. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Su, T.; Santella, R.M.; Weinstein, I.B. Hint1 is a haplo-insufficient tumor suppressor in mice. Oncogene 2006, 25, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Ebus, M.E.; Koster, J.; van Sluis, P.; van Noesel, C.J.; Versteeg, R.; Caron, H.N. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008, 68, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Nicoletti, F.; Vecchio, G.M.; Parenti, R.; Magro, G. Cyclin D1 in pediatric neuroblastic tumors: A microarray analysis. Acta Histochem. 2015, 117, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef]
- Mandriota, S.J.; Valentijn, L.J.; Lesne, L.; Betts, D.R.; Marino, D.; Boudal-Khoshbeen, M.; London, W.B.; Rougemont, A.L.; Attiyeh, E.F.; Maris, J.M.; et al. Ataxia-telangiectasia mutated (ATM) silencing promotes neuroblastoma progression through a MYCN independent mechanism. Oncotarget 2015, 6, 18558–18576. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; White, P.S.; Weiss, M.J.; Hogarty, M.D.; Thompson, P.M.; Stram, D.O.; Gerbing, R.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M.; et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999, 18, 4948–4957. [Google Scholar] [CrossRef]
- Plantaz, D.; Vandesompele, J.; Van Roy, N.; Lastowska, M.; Bown, N.; Combaret, V.; Favrot, M.C.; Delattre, O.; Michon, J.; Benard, J.; et al. Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int. J. Cancer 2001, 91, 680–686. [Google Scholar] [CrossRef]
- Maris, J.M.; Guo, C.; White, P.S.; Hogarty, M.D.; Thompson, P.M.; Stram, D.O.; Gerbing, R.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M. Allelic deletion at chromosome bands 11q14-23 is common in neuroblastoma. Med. Pediatric Oncol. 2001, 36, 24–27. [Google Scholar] [CrossRef]
- Lopez, G.; Conkrite, K.L.; Doepner, M.; Rathi, K.S.; Modi, A.; Vaksman, Z.; Farra, L.M.; Hyson, E.; Noureddine, M.; Wei, J.S.; et al. Somatic structural variation targets neurodevelopmental genes and identifies SHANK2 as a tumor suppressor in neuroblastoma. Genome Res. 2020, 30, 1228–1242. [Google Scholar] [CrossRef]
- Keane, S.; Ameen, S.; Lindlof, A.; Ejeskar, K. Low DLG2 gene expression, a link between 11q-deleted and MYCN-amplified neuroblastoma, causes forced cell cycle progression, and predicts poor patient survival. Cell Commun. Signal. 2020, 18, 65. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkins, E.S.; Haas-Kogan, D.; Gerbing, R.B.; London, W.B.; Villablanca, J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Torres, E.M.; Williams, B.R.; Amon, A. Aneuploidy: Cells losing their balance. Genetics 2008, 179, 737–746. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef]
- Fransson, S.; Hansson, M.; Ruuth, K.; Djos, A.; Berbegall, A.; Javanmardi, N.; Abrahamsson, J.; Palmer, R.H.; Noguera, R.; Hallberg, B.; et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: Translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosomes Cancer 2015, 54, 99–109. [Google Scholar] [CrossRef]
- Cazes, A.; Louis-Brennetot, C.; Mazot, P.; Dingli, F.; Lombard, B.; Boeva, V.; Daveau, R.; Cappo, J.; Combaret, V.; Schleiermacher, G.; et al. Characterization of Rearrangements Involving the ALK Gene Reveals a Novel Truncated Form Associated with Tumor Aggressiveness in Neuroblastoma. Cancer Res. 2013, 73, 195–204. [Google Scholar] [CrossRef]
- Okubo, J.; Takita, J.; Chen, Y.; Oki, K.; Nishimura, R.; Kato, M.; Sanada, M.; Hiwatari, M.; Hayashi, Y.; Igarashi, T.; et al. Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene 2012, 31, 4667–4676. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- De Munck, S.; Provost, M.; Kurikawa, M.; Omori, I.; Mukohyama, J.; Felix, J.; Bloch, Y.; Abdel-Wahab, O.; Bazan, J.F.; Yoshimi, A.; et al. Structural basis of cytokine-mediated activation of ALK family receptors. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Liptay, M.; Barbosa, J.S.; Rottenberg, S. Replication Fork Remodeling and Therapy Escape in DNA Damage Response-Deficient Cancers. Front. Oncol. 2020, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Taschner-Mandl, S. Neuroblastoma and the epigenome. Cancer Metastasis Rev. 2021, 40, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Federico, S.; Chen, X.; Fan, Y.; Xu, B.; Stewart, E.; Zhou, X.; Jeon, J.; Griffiths, L.; Nguyen, R.; et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun 2020, 11, 913. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Profile | Survival 5 Years after Diagnosis |
|---|---|
| MNA | 46% |
| 11q- | 48% |
| MNA and 11q- | 0% |
| 17q+ (without MNA or 11q-) | 66% |
| Other segmental | 92% |
| Numerical only | 95% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, J.; Hallberg, B.; Palmer, R.H. Chromosome Imbalances in Neuroblastoma—Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers 2021, 13, 5897. https://doi.org/10.3390/cancers13235897
Guan J, Hallberg B, Palmer RH. Chromosome Imbalances in Neuroblastoma—Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers. 2021; 13(23):5897. https://doi.org/10.3390/cancers13235897
Chicago/Turabian StyleGuan, Jikui, Bengt Hallberg, and Ruth H. Palmer. 2021. "Chromosome Imbalances in Neuroblastoma—Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future" Cancers 13, no. 23: 5897. https://doi.org/10.3390/cancers13235897
APA StyleGuan, J., Hallberg, B., & Palmer, R. H. (2021). Chromosome Imbalances in Neuroblastoma—Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers, 13(23), 5897. https://doi.org/10.3390/cancers13235897