
Identification of Gene Co-Expression Networks Associated with Consensus Molecular Subtype-1 of Colorectal Cancer

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Gene Network Analysis
2.2. Pathway Analysis
2.3. Gene Expression Analysis
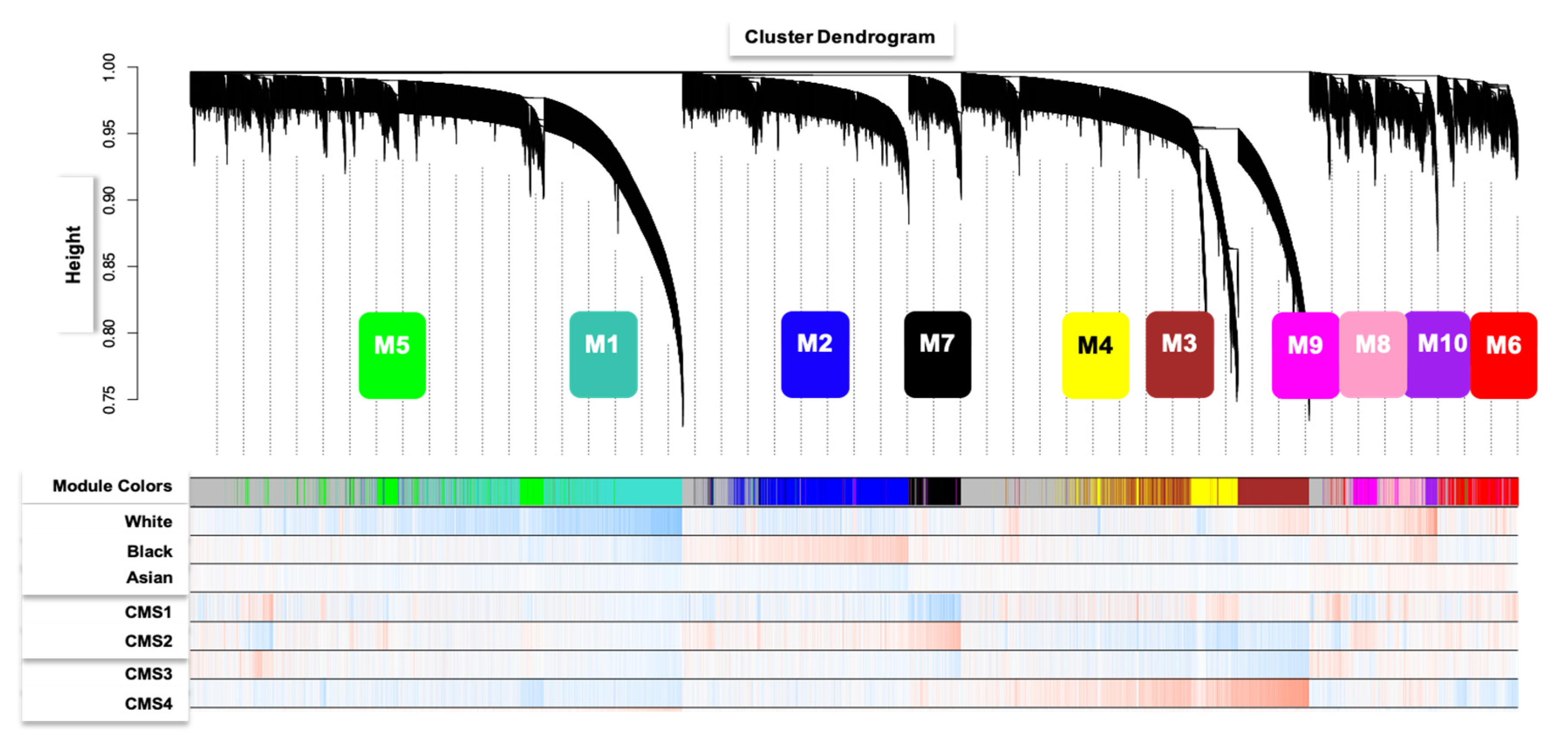
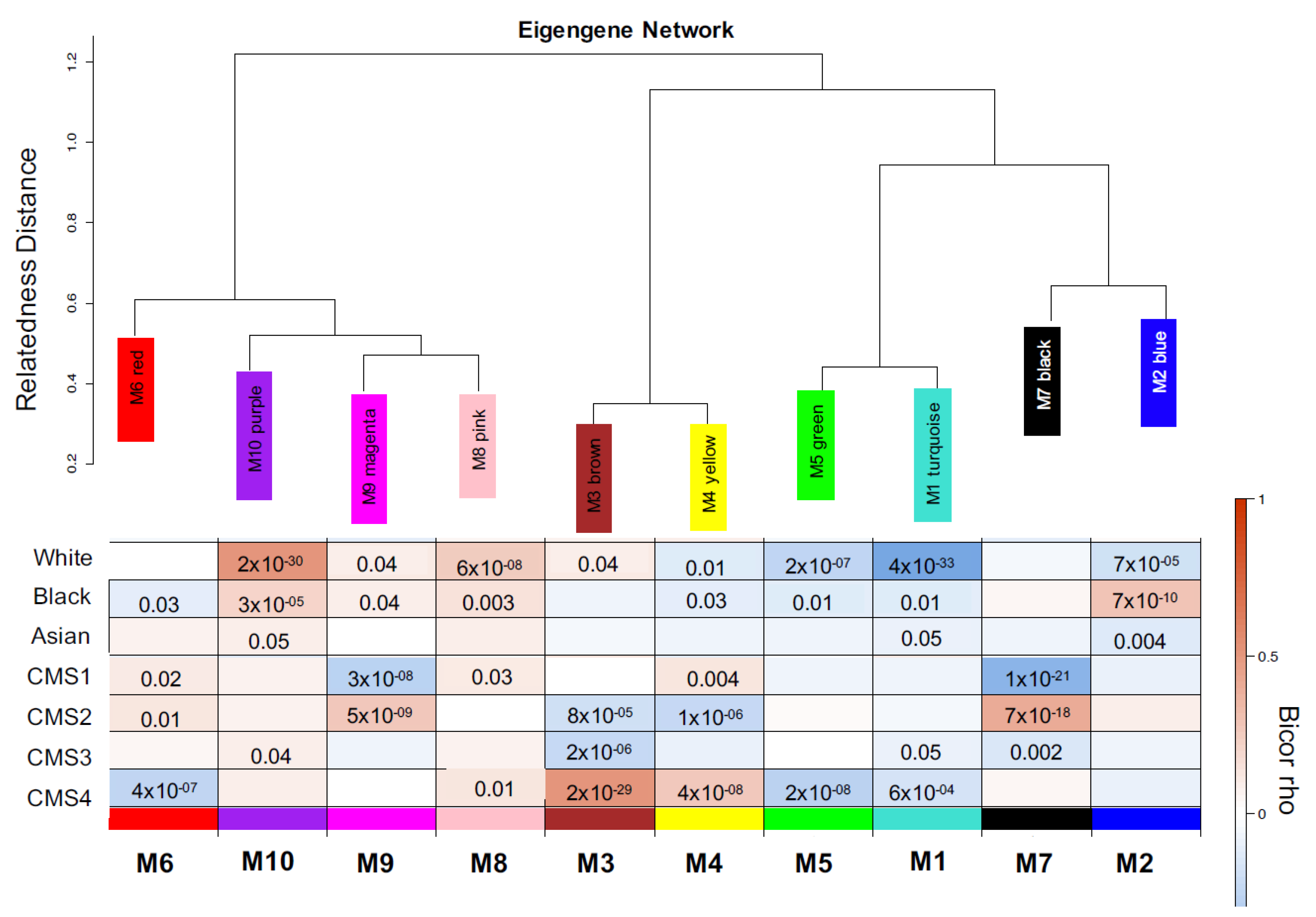
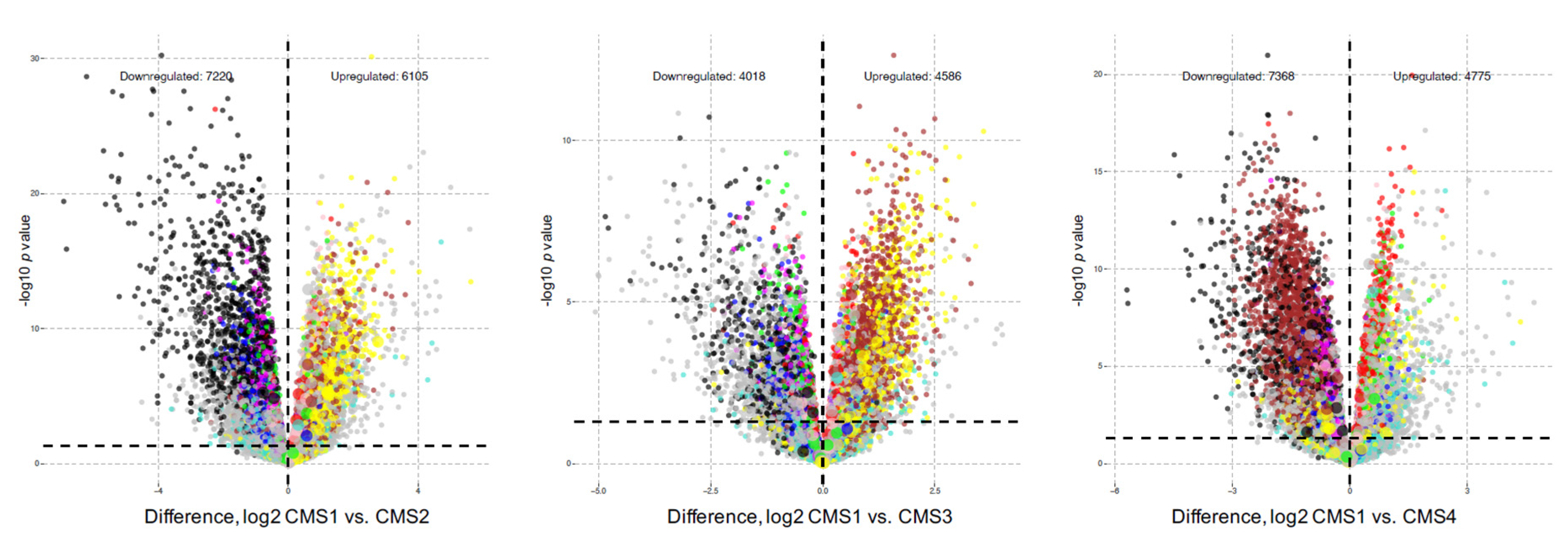
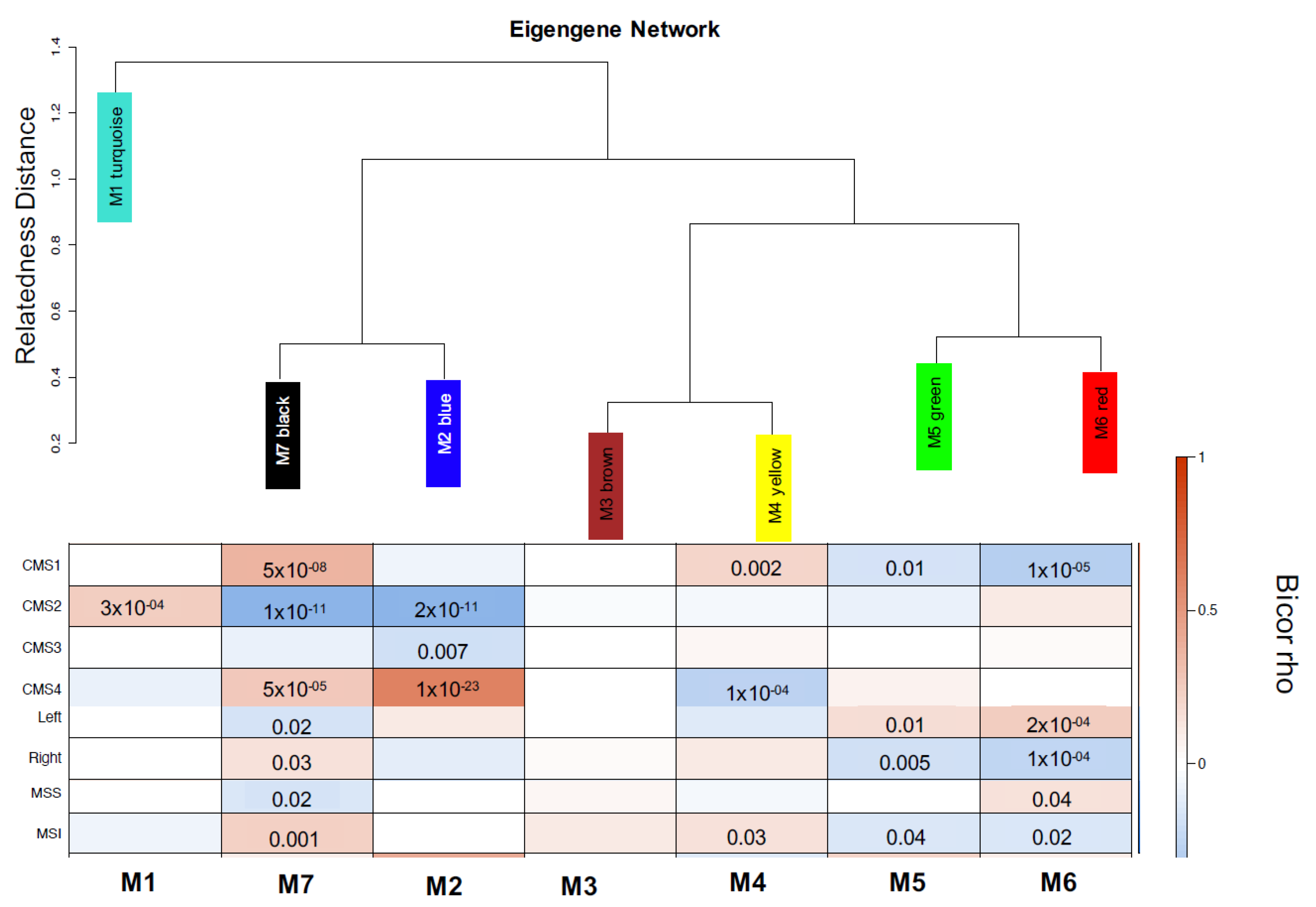
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfer, L. Cytokines, inflammation and colon cancer. Curr. Cancer Drug Targets 2011, 11, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shay, J.W. Multiple roles of APC and its therapeutic implications in colorectal cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.C.; Wolff, R.K.; Samowitz, W.S.; Herrick, J.S. The MAPK-signaling pathway in colorectal cancer: Dysregulated genes and their association with micrornas. Cancer Inform. 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Wang, W.; Kandimalla, R.; Huang, H.; Zhu, L.; Li, Y.; Gao, F.; Goel, A.; Wang, X. Molecular subtyping of colorectal cancer: Recent progress, new challenges and emerging opportunities. In Seminars in Cancer Biology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 55, pp. 37–52. [Google Scholar]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus molecular subtypes of colorectal cancer and their clinical implications. Int. Biol. Biomed. J. 2017, 3, 105. [Google Scholar]
- Tougeron, D.; Sueur, B.; Sefrioui, D.; Gentilhomme, L.; Lecomte, T.; Aparicio, T.; DES Guetz, G.; Artru, P.; De La Fouchardiere, C.; Moulin, V. A large multicenter study evaluating prognosis and chemosensitivity of metastatic colorectal cancers with microsatellite instability. J. Clin. Oncol. 2017, 35, 3536. [Google Scholar] [CrossRef]
- Evrard, C.; Tachon, G.; Randrian, V.; Karayan-Tapon, L.; Tougeron, D. Microsatellite instability: Diagnosis, heterogeneity, discordance, and clinical impact in colorectal cancer. Cancers 2019, 11, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.; Roszik, J.; Kanikarla-Marie, P.; Davis, J.S.; Morris, J.; Kopetz, S.; Menter, D.G. The potential role of platelets in the consensus molecular subtypes of colorectal cancer. Cancer Metastasis Rev. 2017, 36, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Ohandjo, A.Q.; Liu, Z.; Dammer, E.B.; Dill, C.D.; Griffen, T.L.; Carey, K.M.; Hinton, D.E.; Meller, R.; Lillard, J.W. Transcriptome network analysis identifies CXCL13-CXCR5 signaling modules in the prostate tumor immune microenvironment. Sci. Rep. 2019, 9, 14963. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G., Jr.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Nosho, K.; Irahara, N.; Meyerhardt, J.A.; Baba, Y.; Shima, K.; Glickman, J.N.; Ferrone, C.R.; Mino-Kenudson, M.; Tanaka, N. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin. Cancer Res. 2009, 15, 6412–6420. [Google Scholar] [CrossRef] [Green Version]
- Hamann, A.; Syrbe, U. T-cell trafficking into sites of inflammation. Rheumatology 2000, 39, 696–699. [Google Scholar] [CrossRef] [Green Version]
- Janakiram, N.B.; Rao, C.V. The role of inflammation in colon cancer. Inflamm. Cancer 2014, 816, 25–52. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Richmond, A. Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Mevizou, R.; Sirvent, A.; Roche, S. Control of Tyrosine Kinase Signalling by Small Adaptors in Colorectal Cancer. Cancers 2019, 11, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Aranda, M.; Redondo, M. Targeting receptor kinases in colorectal cancer. Cancers 2019, 11, 433. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Loupakis, F.; Yang, D.; Yau, L.; Feng, S.; Cremolini, C.; Zhang, W.; Maus, M.K.; Antoniotti, C.; Langer, C.; Scherer, S.J. Primary tumor location as a prognostic factor in metastatic colorectal cancer. JNCI J. Natl. Cancer Inst. 2015, 107, dju427. [Google Scholar] [CrossRef]
- Loree, J.M.; Pereira, A.A.; Lam, M.; Willauer, A.N.; Raghav, K.; Dasari, A.; Morris, V.K.; Advani, S.; Menter, D.G.; Eng, C. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin. Cancer Res. 2018, 24, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Tran, B.; Kopetz, S.; Tie, J.; Gibbs, P.; Jiang, Z.Q.; Lieu, C.H.; Agarwal, A.; Maru, D.M.; Sieber, O.; Desai, J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011, 117, 4623–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xicola, R.M.; Manojlovic, Z.; Augustus, G.J.; Kupfer, S.S.; Emmadi, R.; Alagiozian-Angelova, V.; Triche, T., Jr.; Salhia, B.; Carpten, J.; Llor, X. Lack of APC somatic mutation is associated with early-onset colorectal cancer in African Americans. Carcinogenesis 2018, 39, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Bellio, H.; Fumet, J.D.; Ghiringhelli, F. Targeting BRAF and RAS in Colorectal Cancer. Cancers 2021, 13, 2201. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk: Implications for tuberous sclerosisand cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfo, V.; Rodia, M.T.; Pucci, M.; Dall’Ora, M.; Santi, S.; Solmi, R.; Roth, L.; Lindzen, M.; Bonafè, M.; Bertotti, A. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget 2016, 7, 72167. [Google Scholar] [CrossRef]
- Johnson, S.M.; Gulhati, P.; Rampy, B.A.; Han, Y.; Rychahou, P.G.; Doan, H.Q.; Weiss, H.L.; Evers, B.M. Novel expression patterns of PI3K/Akt/mTOR signaling pathway components in colorectal cancer. J. Am. Coll. Surg. 2010, 210, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun. Signal. 2018, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Curnock, A.P.; Logan, M.K.; Ward, S.G. Chemokine signalling: Pivoting around multiple phosphoinositide 3-kinases. Immunology 2002, 105, 125–136. [Google Scholar] [CrossRef] [PubMed]
- El-Haibi, C.P.; Sharma, P.; Singh, R.; Gupta, P.; Taub, D.D.; Singh, S.; Lillard, J.W., Jr. Differential G protein subunit expression by prostate cancer cells and their interaction with CXCR5. Mol. Cancer 2013, 12, 64. [Google Scholar] [CrossRef] [Green Version]
- El Haibi, C.P.; Sharma, P.K.; Singh, R.; Johnson, P.R.; Suttles, J.; Singh, S.; Lillard, J.W. PI3Kp110-, Src-, FAK-dependent and DOCK2-independent migration and invasion of CXCL13-stimulated prostate cancer cells. Mol. Cancer 2010, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, X.; Guo, H.; Fu, L.; Pan, G.; Sun, Y. CXCL13-CXCR5 axis promotes the growth and invasion of colon cancer cells via PI3K/AKT pathway. Mol. Cell. Biochem. 2015, 400, 287–295. [Google Scholar] [CrossRef] [PubMed]
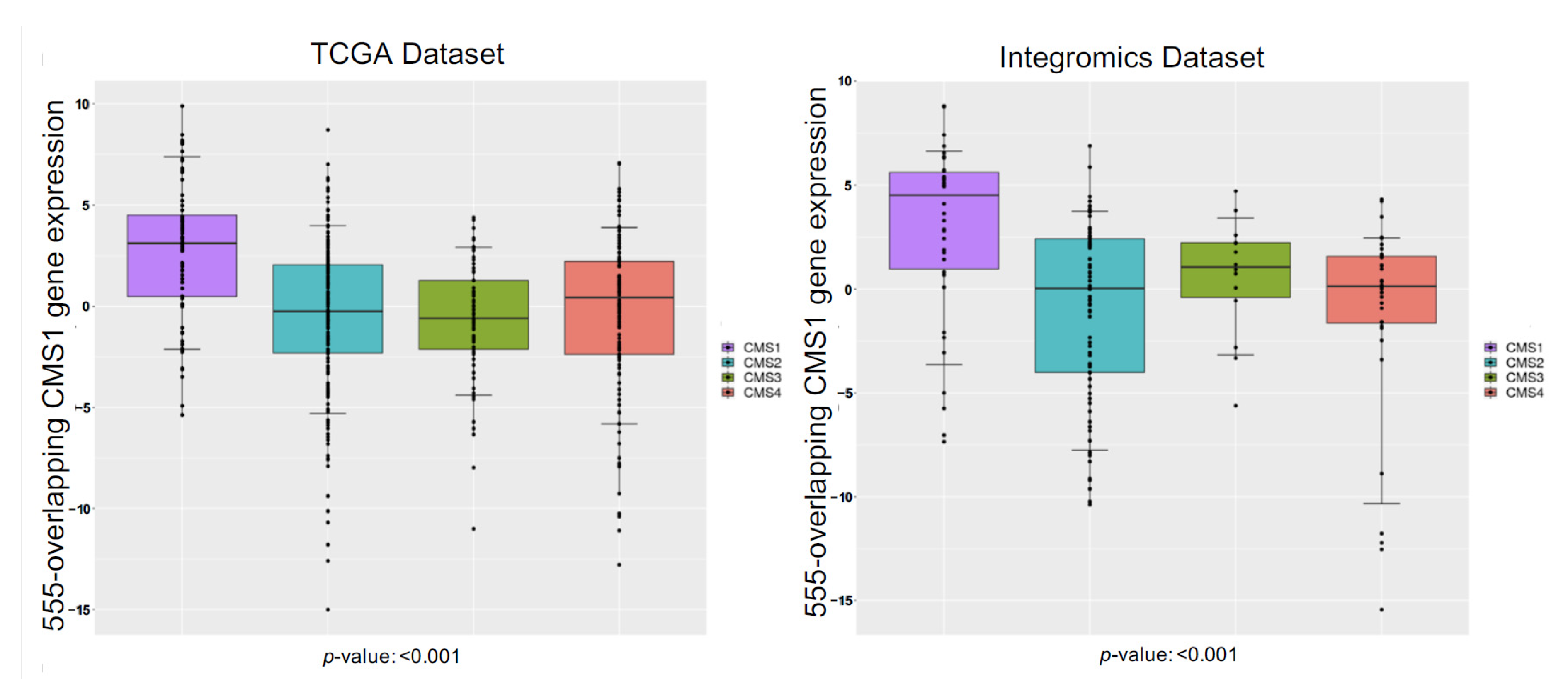
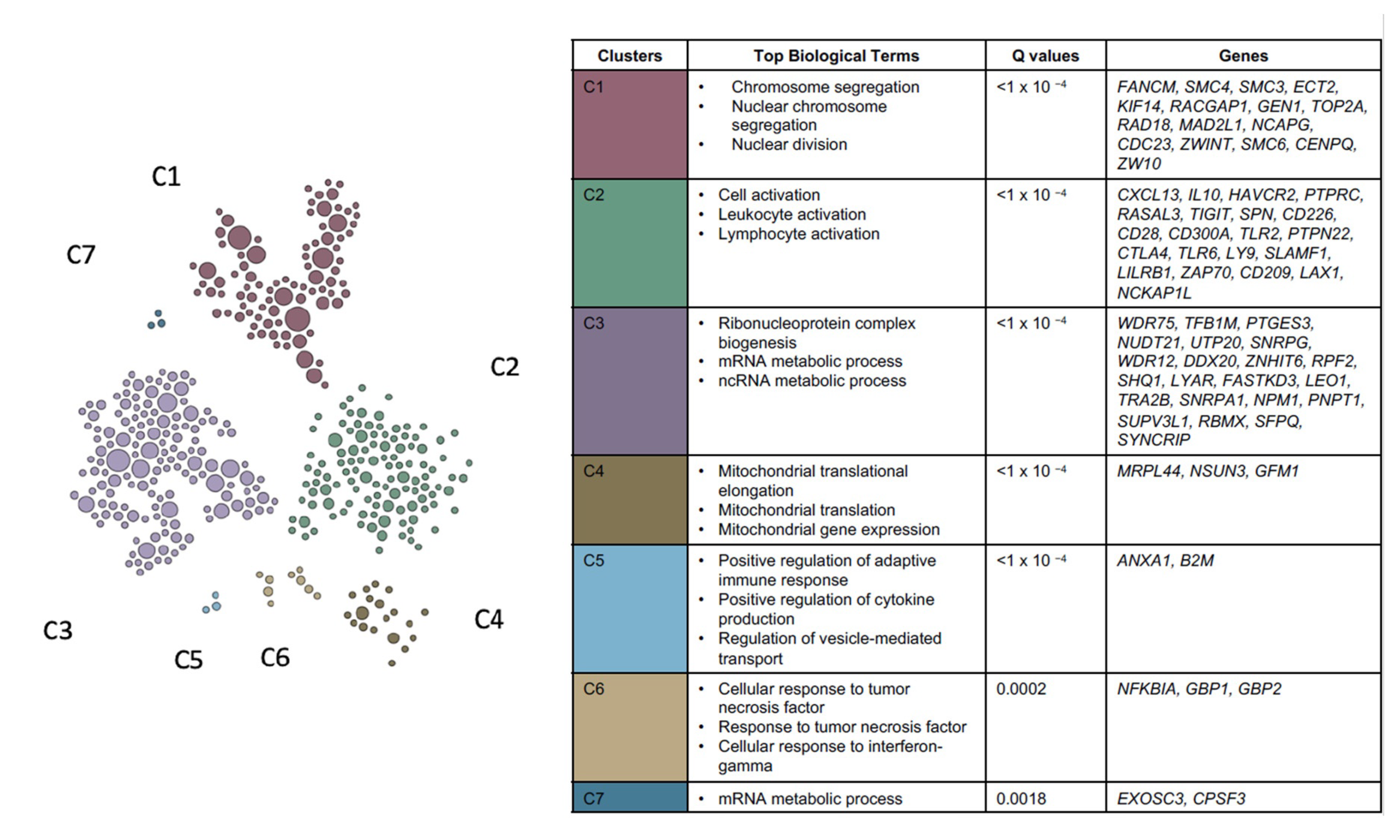

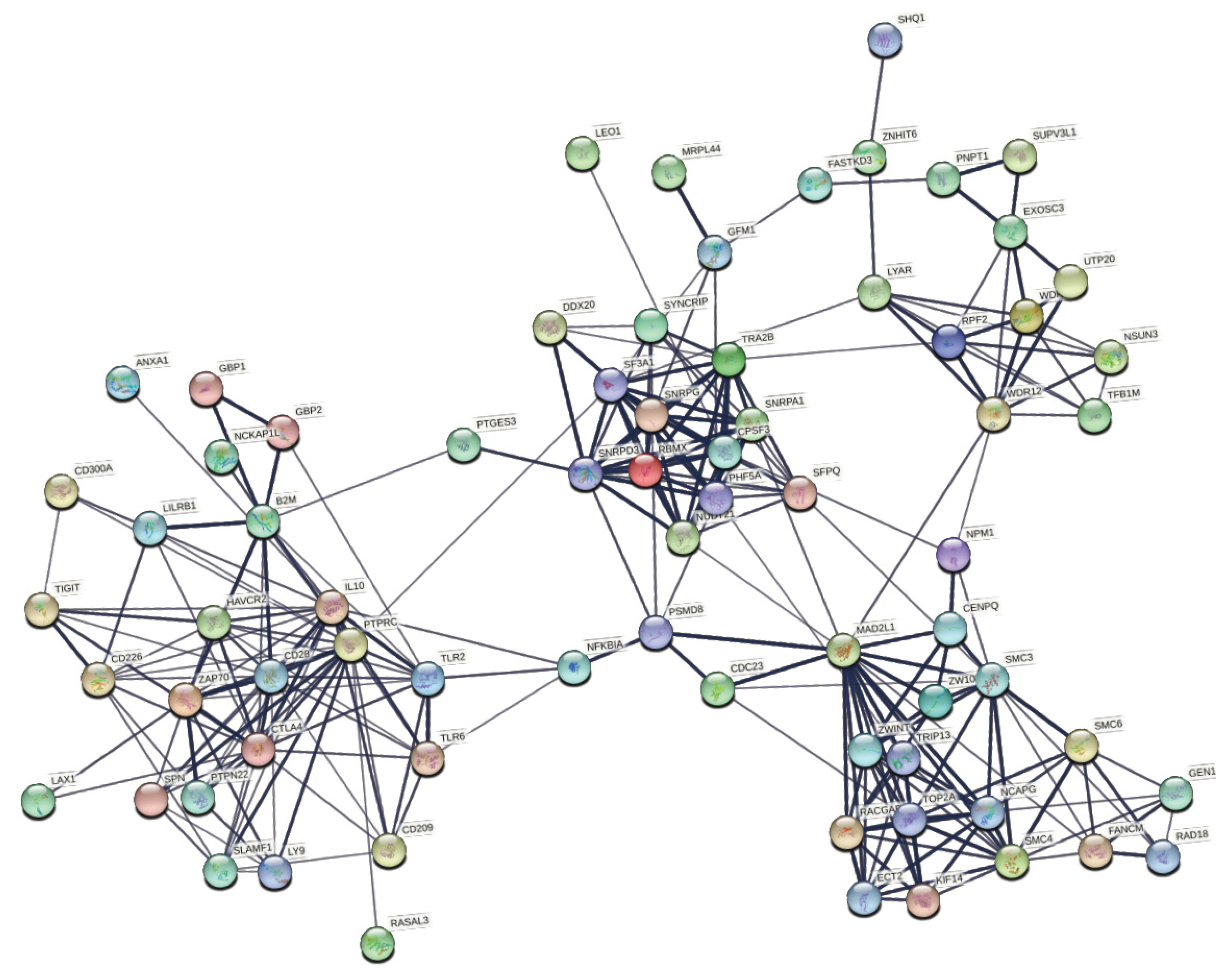

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Top 5 Biological Processes | False Discovery Rate | |
|---|---|---|---|
| Positively correlated with CMS1 | Yellow p-value = 0.004 |
|
|
| Red p-value = 0.02 |
|
| |
| Pink p-value = 0.03 |
|
| |
| Negatively correlated with CMS1 | Black p-value = 1 × 10−21 |
|
|
| Magenta p-value = 3 × 10−8 |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunez, S.K.; Young, C.D.; Griffen, T.L.; Ohandjo, A.Q.; McKinney, L.P.; Kopetz, S.; Lillard, J.W., Jr. Identification of Gene Co-Expression Networks Associated with Consensus Molecular Subtype-1 of Colorectal Cancer. Cancers 2021, 13, 5824. https://doi.org/10.3390/cancers13225824
Nunez SK, Young CD, Griffen TL, Ohandjo AQ, McKinney LP, Kopetz S, Lillard JW Jr. Identification of Gene Co-Expression Networks Associated with Consensus Molecular Subtype-1 of Colorectal Cancer. Cancers. 2021; 13(22):5824. https://doi.org/10.3390/cancers13225824
Chicago/Turabian StyleNunez, Sha’Kayla K., Corey D. Young, Ti’ara L. Griffen, Adaugo Q. Ohandjo, Lawrence P. McKinney, Scott Kopetz, and James W. Lillard, Jr. 2021. "Identification of Gene Co-Expression Networks Associated with Consensus Molecular Subtype-1 of Colorectal Cancer" Cancers 13, no. 22: 5824. https://doi.org/10.3390/cancers13225824
APA StyleNunez, S. K., Young, C. D., Griffen, T. L., Ohandjo, A. Q., McKinney, L. P., Kopetz, S., & Lillard, J. W., Jr. (2021). Identification of Gene Co-Expression Networks Associated with Consensus Molecular Subtype-1 of Colorectal Cancer. Cancers, 13(22), 5824. https://doi.org/10.3390/cancers13225824