
Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Multicellular Spheroid (MCS) Growth Assay
2.3. MCS Cell Evasion Assay
2.4. MCS Paracellular Permeability Assay
2.5. Immunostaining of Frozen MCS Sections and Confocal Microscopy Analysis
2.6. MCS Viability Assay
2.7. Western Blot Analysis
2.8. Intracellular Pathscan Signalling Array
2.9. In Vivo Experiments
2.10. TCGA Dataset Analysis
2.11. Statistical Analysis
3. Results
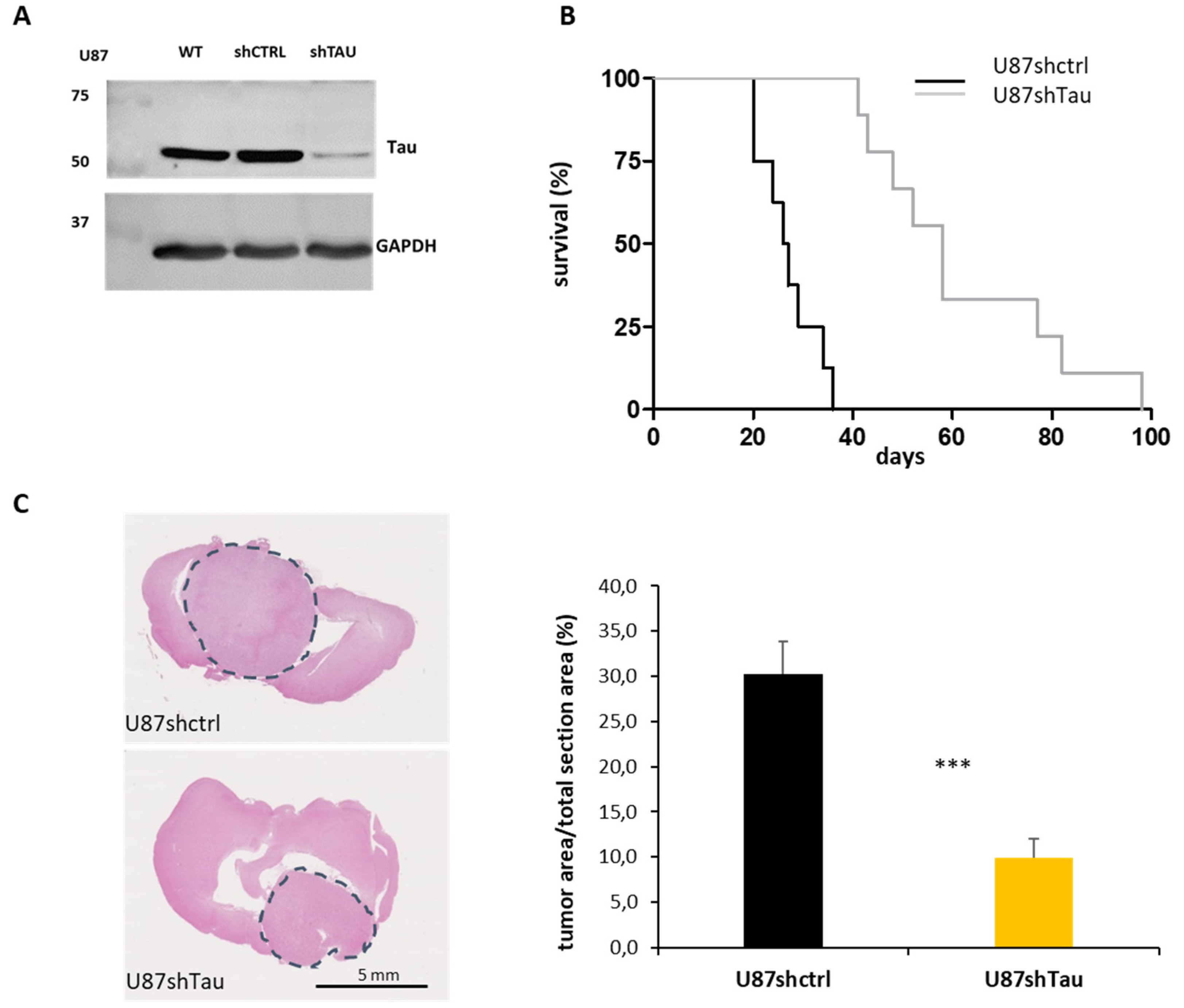
3.1. Tau Down-Regulation Reduces In Vivo Glioblastoma Tumorigenicity
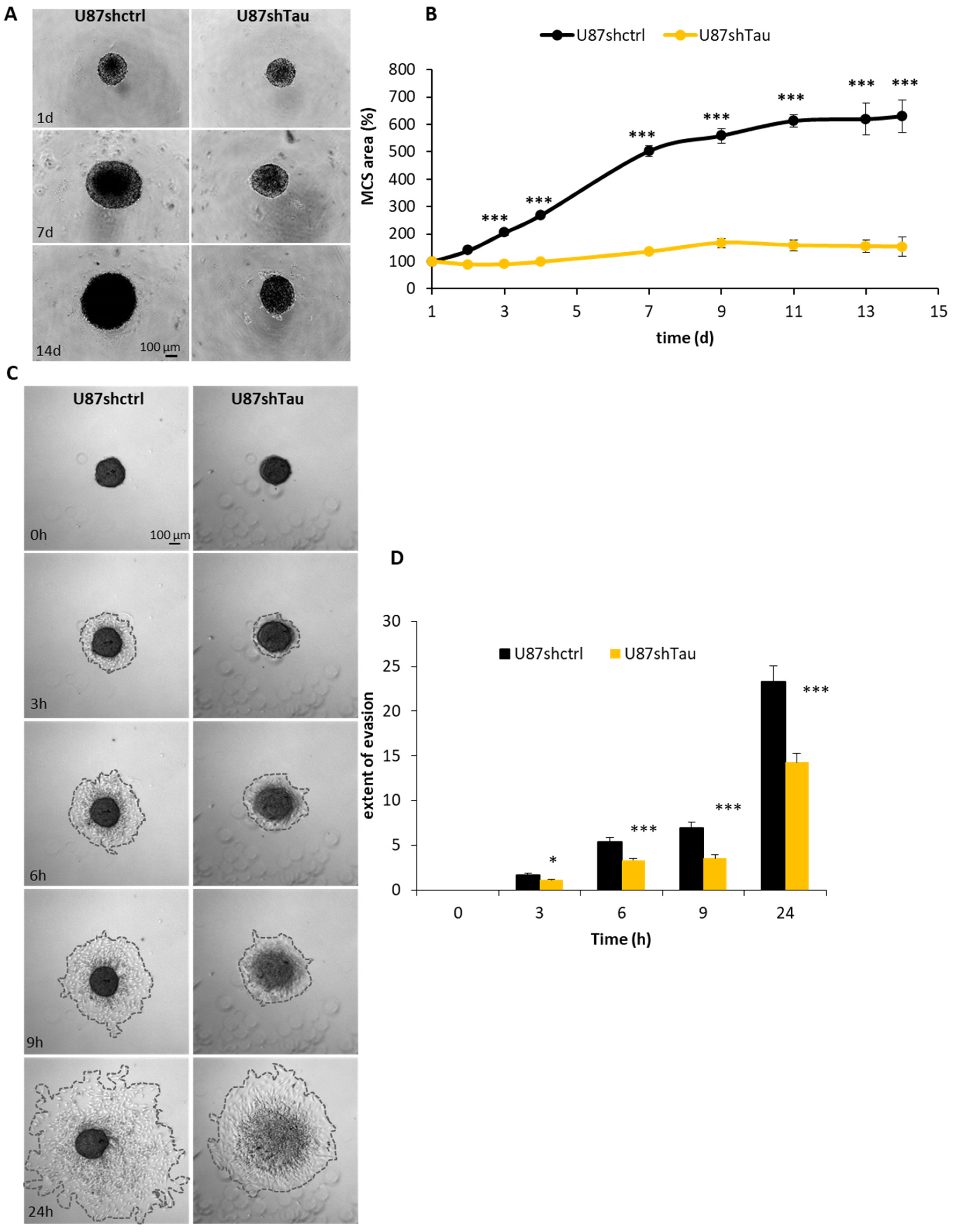
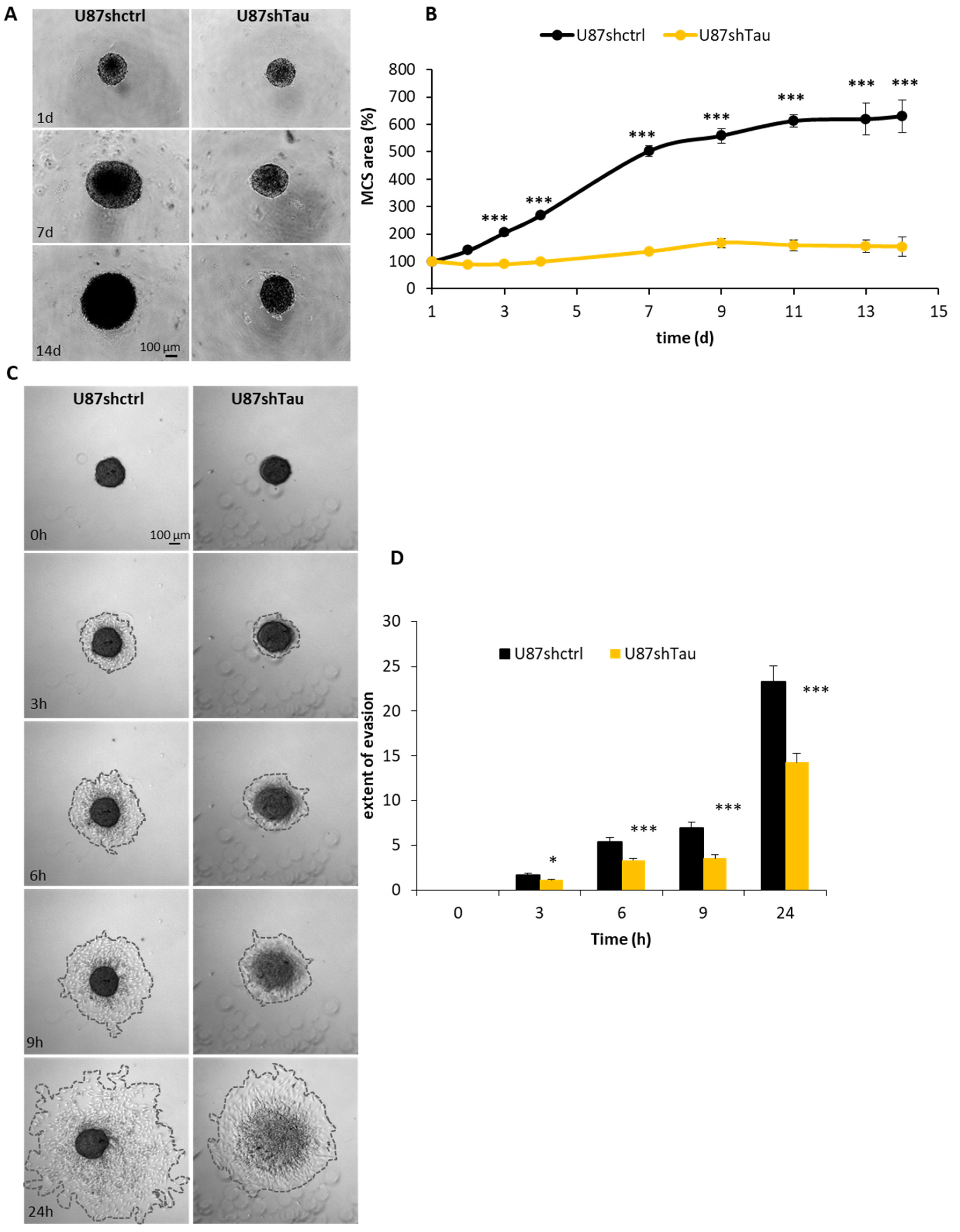
3.2. Tau Down-Regulation Hinders Multicellular Spheroid Growth and Cell Evasion
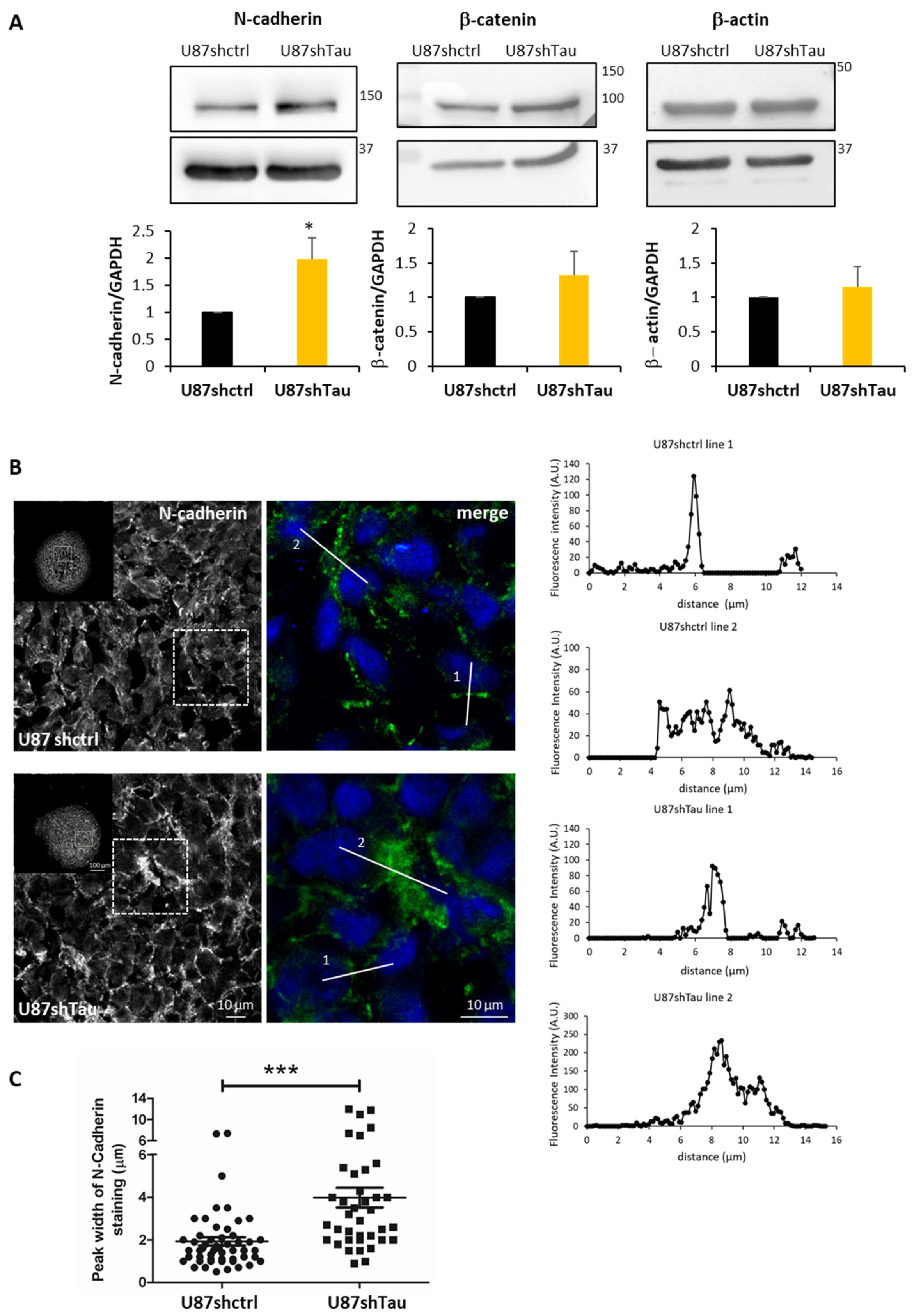
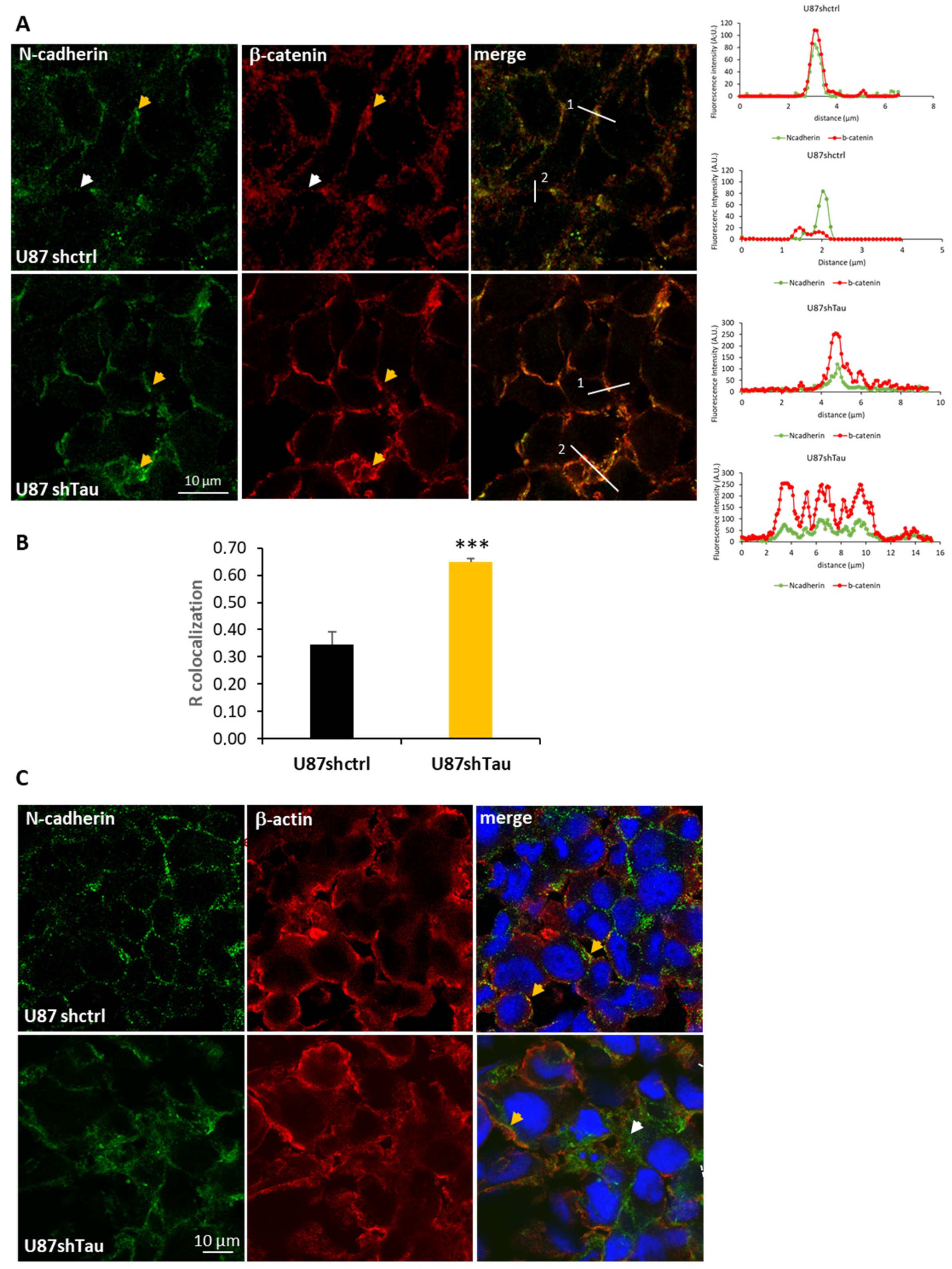
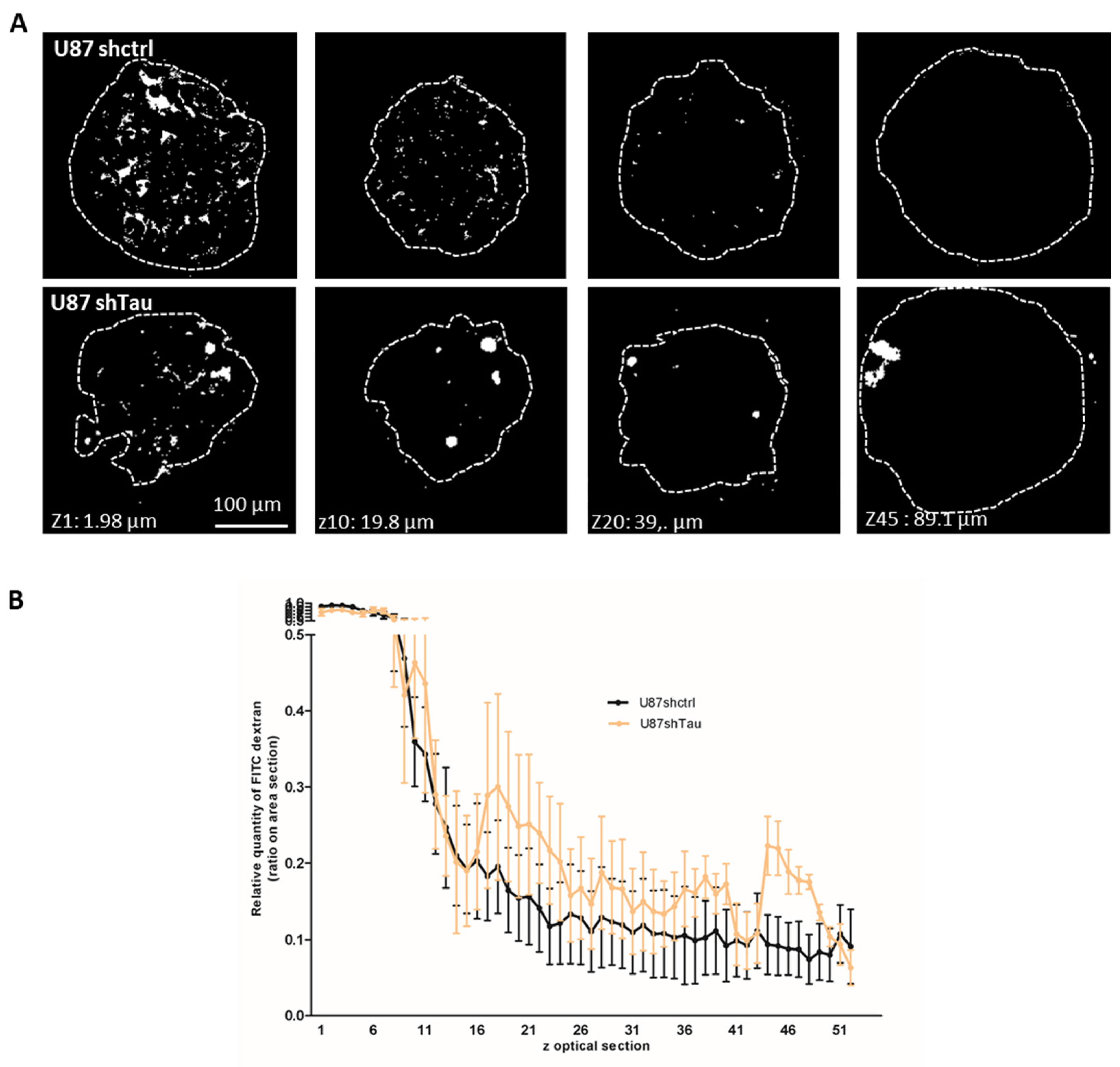
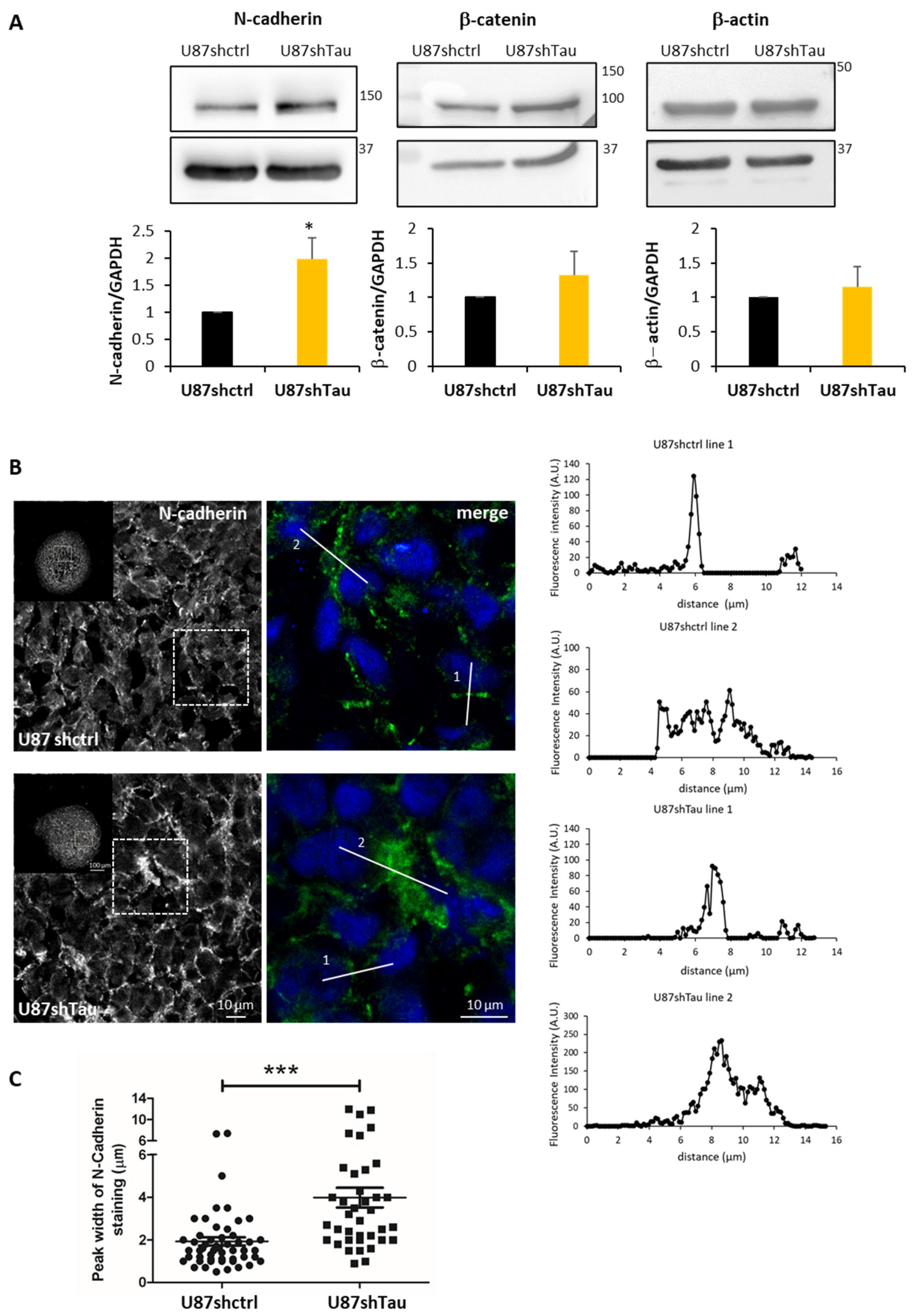
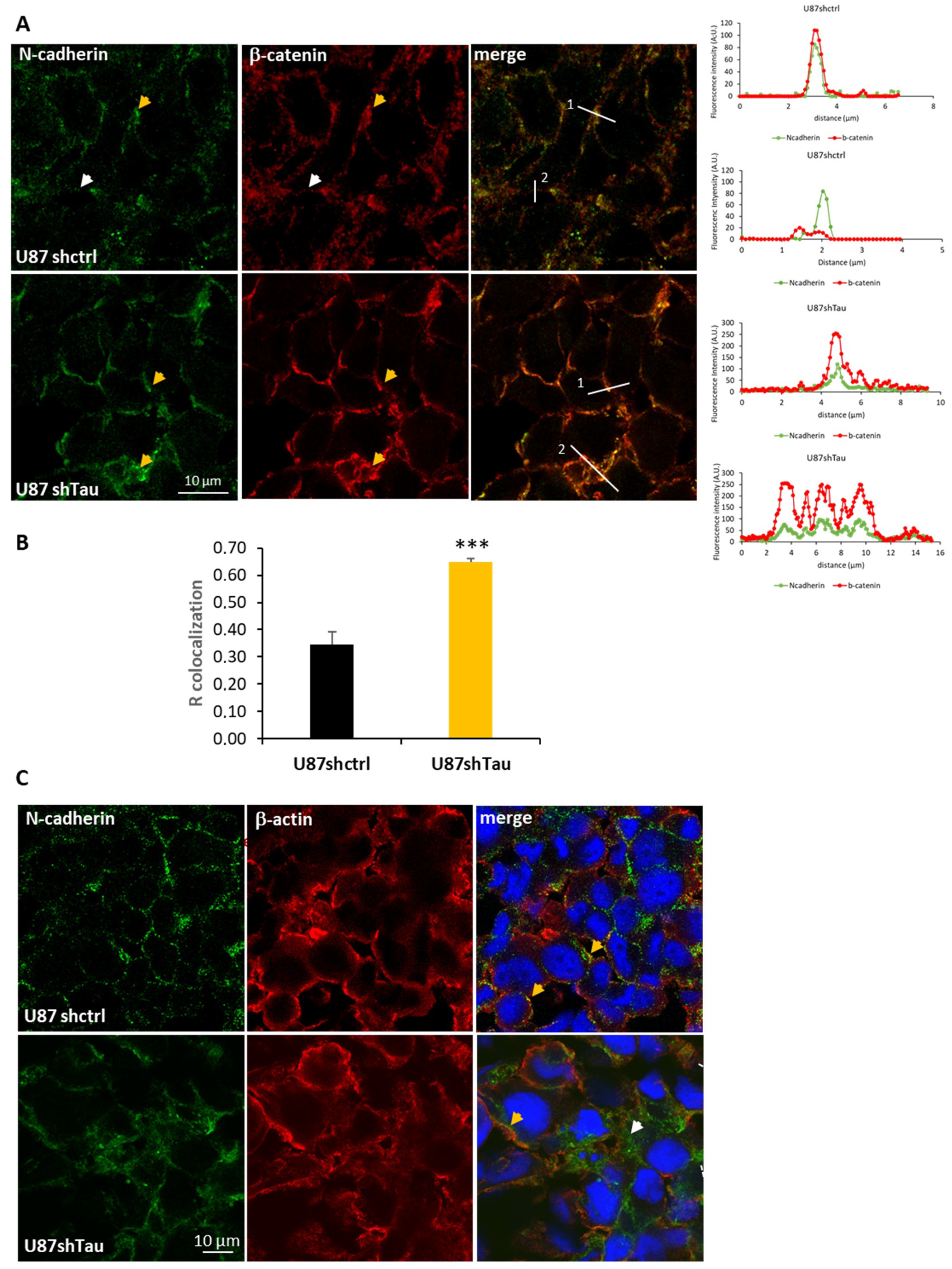
3.3. Tau Down-Regulation Decreases Multicellular Spheroid Paracellular Permeability and Induces N-Cadherin Mislocalization
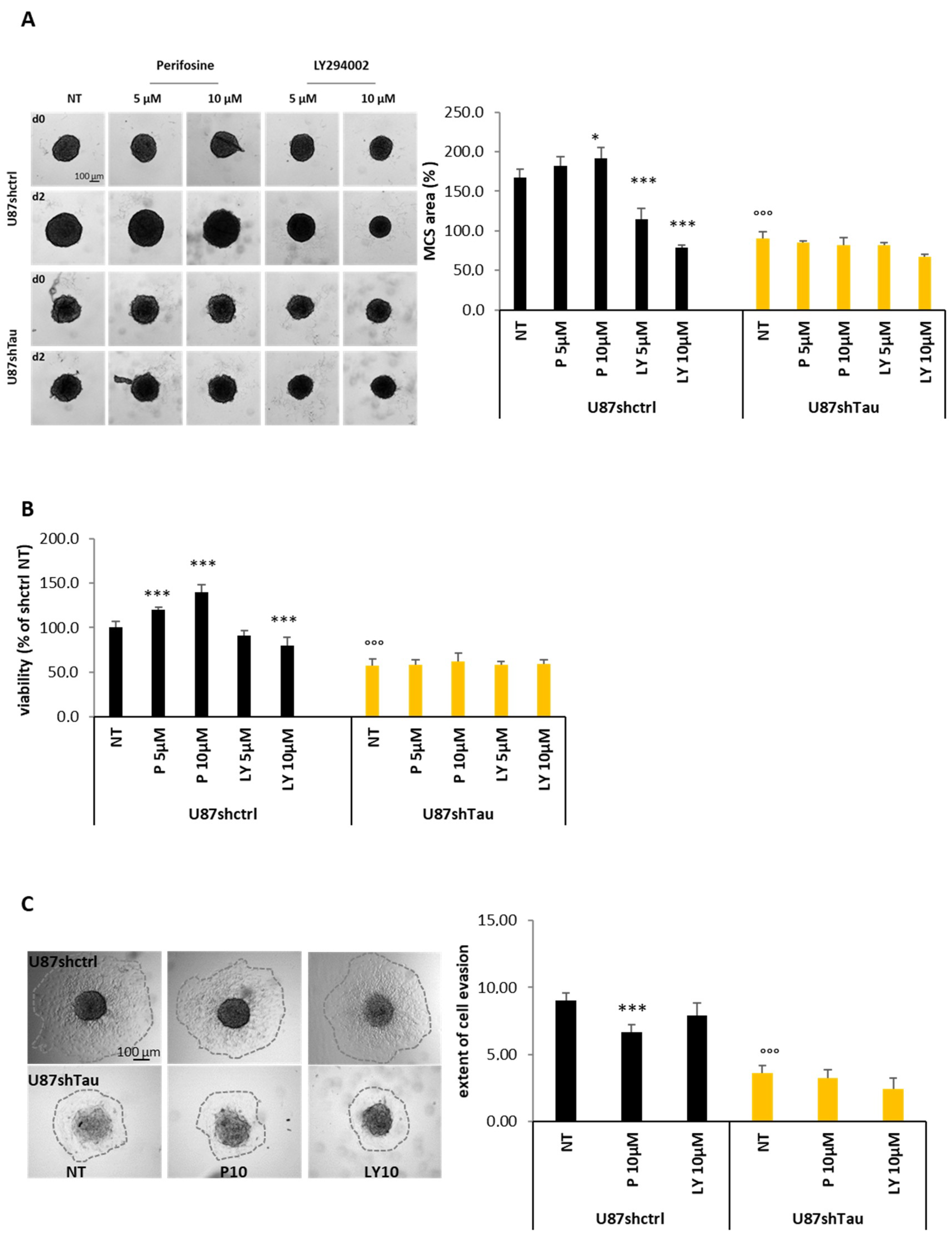
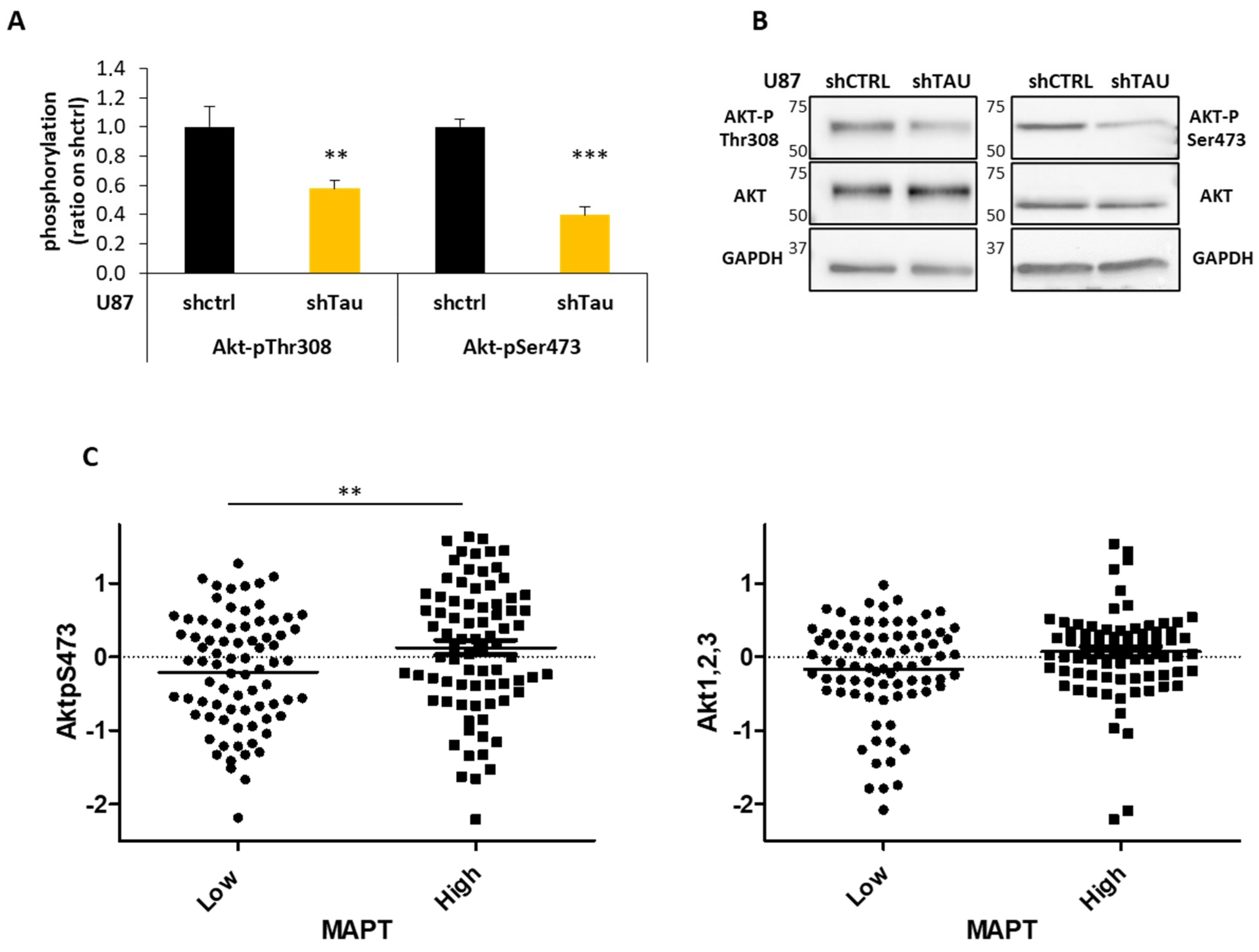
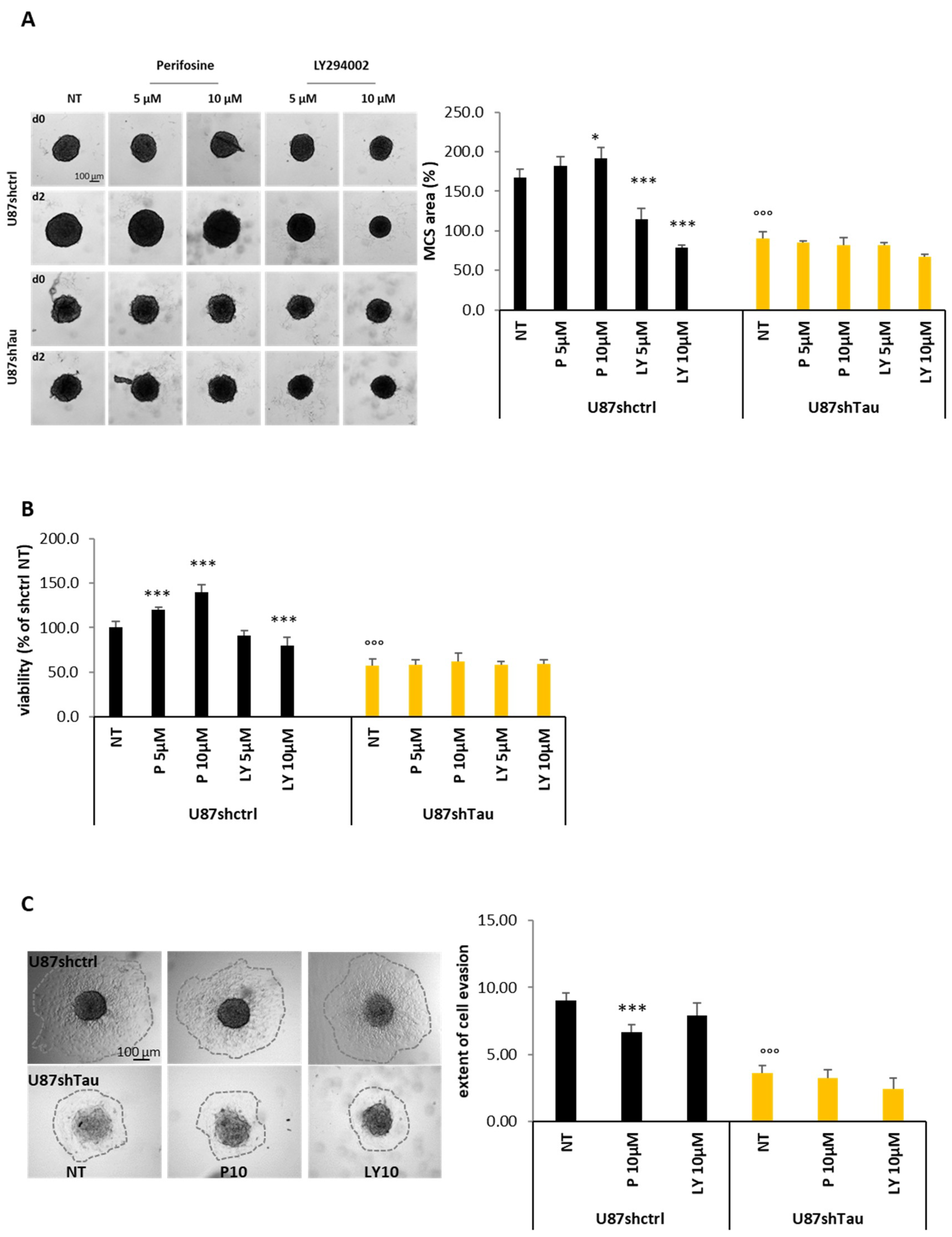
3.4. Tau Regulates Multicellular Spheroid Organization, Growth, and Migration via the PI3K-Akt Signalling Pathway
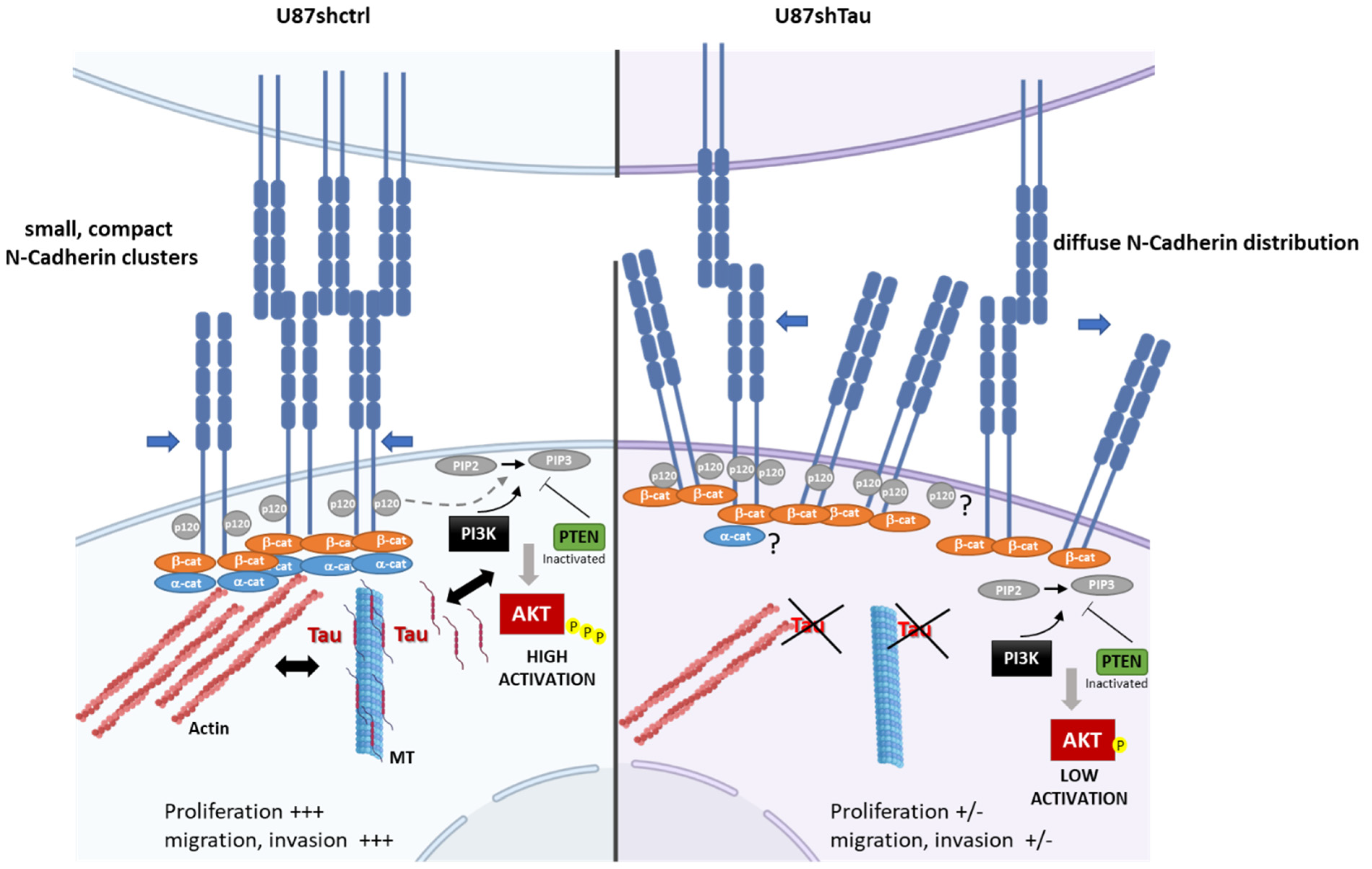
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Figarella-Branger, D.; Chappe, C.; Padovani, L.; Mercurio, S.; Colin, C.; Forest, F.; Bouvier, C. Glial and glioneuronal tumors in adults and children: Main genetic alterations and towards a histomolecular classification. Bull. Cancer 2013, 100, 715–726. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncol 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1198. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, A.; Honoré, S.; Mohan, R.; Berges, R.; Akhmanova, A.; Braguer, D. Epothilone B inhibits migration of glioblastoma cells by inducing microtubule catastrophes and affecting EB1 accumulation at microtubule plus ends. Biochem. Pharmacol. 2012, 84, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Honore, S.; Pasquier, E.; Braguer, D. Understanding microtubule dynamics for improved cancer therapy. Cell Mol. Life Sci. 2005, 62, 3039–3056. [Google Scholar] [CrossRef]
- Pasquier, E.; Kavallaris, M. Microtubules: A dynamic target in cancer therapy. IUBMB Life 2008, 60, 165–170. [Google Scholar] [CrossRef]
- Breuzard, G.; Hubert, P.; Nouar, R.; De Bessa, T.; Devred, F.; Barbier, P.; Sturgis, J.N.; Peyrot, V. Molecular mechanisms of Tau binding to microtubules and its role in microtubule dynamics in live cells. J. Cell Sci. 2013, 126, 2810–2819. [Google Scholar] [CrossRef] [Green Version]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of tau as a microtubule-associated protein: Structural and functional aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, P.O.; La Rocca, R.; Malesinski, S.; Devred, F. Characterization of microtubule-associated proteins (MAPs) and tubulin interactions by isothermal titration calorimetry (ITC). Methods Mol. Biol. 2019, 1964, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Barbolina, M.V. Dichotomous role of microtubule associated protein tau as a biomarker of response to and a target for increasing efficacy of taxane treatment in cancers of epithelial origin. Pharmacol. Res. 2021, 168, 105585. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, C.; Gurard-Levin, Z.A.; Andre, F.; Pusztai, L.; Rouzier, R. Predictive and prognostic value of the tauprotein in breast cancer. Anticancer Res. 2015, 35, 5179–5184. [Google Scholar] [PubMed]
- Yang, J.; Yu, Y.; Liu, W.; Li, Z.; Wei, Z.; Jiang, R. Microtubule-associated protein tau is associated with the resistance to docetaxel in prostate cancer cell lines. Res. Rep. Urol. 2017, 9, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.; Redaelli, V.; Perego, P.; Ferrari, R.; Giaccone, G.; Tagliavini, F. Tau mutations as a novel risk factor for cancer-response. Cancer Res. 2018, 78, 6525. [Google Scholar] [CrossRef] [Green Version]
- Smoter, M.; Bodnar, L.; Grala, B.; Stec, R.; Zieniuk, K.; Kozlowski, W.; Szczylik, C. Tau protein as a potential predictive marker in epithelial ovarian cancer patients treated with paclitaxel/platinum first-line chemotherapy. J. Exp. Clin. Cancer Res. 2013, 32, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, C.; Grell, J.; Hube-Magg, C.; Kluth, M.; Lang, D.; Simon, R.; Höflmayer, D.; Minner, S.; Burandt, E.; Clauditz, T.S.; et al. Aberrant expression of the microtubule-associated protein tau is an independent prognostic feature in prostate cancer. BMC Cancer 2019, 19, 193. [Google Scholar] [CrossRef] [Green Version]
- Papin, S.; Paganetti, P. Emerging evidences for an implication of the neurodegeneration-associated protein tau in cancer. Brain Sci. 2020, 10, 862. [Google Scholar] [CrossRef]
- Baquero, M.T.; Lostritto, K.; Gustavson, M.D.; Bassi, K.A.; Appia, F.; Camp, R.L.; Molinaro, A.M.; Harris, L.N.; Rimm, D.L. Evaluation of prognostic and predictive value of microtubule associated protein tau in two independent cohorts. Breast Cancer Res. 2011, 13, R85. [Google Scholar] [CrossRef] [Green Version]
- Gargini, R.; Segura-Collar, B.; Sánchez-Gómez, P. Novel functions of the neurodegenerative-related gene tau in cancer. Front. Aging Neurosci. 2019, 11, 231. [Google Scholar] [CrossRef] [Green Version]
- Breuzard, G.; Pagano, A.; Bastonero, S.; Malesinski, S.; Parat, F.; Barbier, P.; Peyrot, V.; Kovacic, H. Tau regulates the microtubule-dependent migration of glioblastoma cells via the Rho-ROCK signaling pathway. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Matrone, M.A.; Whipple, R.A.; Thompson, K.; Cho, E.H.; Vitolo, M.I.; Balzer, E.M.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Tan, M.; et al. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene 2010, 29, 3217–3227. [Google Scholar] [CrossRef] [Green Version]
- Silvani, G.; Romanov, V.; Cox, C.D.; Martinac, B. Biomechanical Characterization of Endothelial Cells Exposed to Shear Stress Using Acoustic Force Spectroscopy. Front. Bioeng. Biotechnol. 2021, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Chocry, M.; Leloup, L.; Kovacic, H. Reversion of resistance to oxaliplatin by inhibition of p38 MAPK in colorectal cancer cell lines: Involvement of the calpain/Nox1 pathway. Oncotarget 2017, 8, 103710–103730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berges, R.; Baeza-Kallee, N.; Tabouret, E.; Chinot, O.; Petit, M.; Kruczynski, A.; Figarella-Branger, D.; Honore, S.; Braguer, D. End-binding 1 protein overexpression correlates with glioblastoma progression and sensitizes to Vinca-alkaloids in vitro and in vivo. Oncotarget 2014, 5, 12769–12787. [Google Scholar] [CrossRef] [Green Version]
- Denicolaï, E.; Baeza-Kallee, N.; Tchoghandjian, A.; Carré, M.; Colin, C.; Jiglaire, C.J.; Mercurio, S.; Beclin, C.; Figarella-Branger, D. Proscillaridin A is cytotoxic for glioblastoma cell lines and controls tumor xenograft growth in vivo. Oncotarget 2014, 5, 10934–10948. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Zhang, S.; Li, C.; Zhou, C.; Li, D.; Liu, P.; Huang, M.; Shen, X. Cryptotanshinone inhibits human glioma cell proliferation in vitro and in vivo through SHP-2-dependent inhibition of STAT3 activation. Cell Death Dis. 2017, 8, e2767. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.; Broom, B.M.; Verhaak, R.G.W.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S.F. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genomics 2008, 1, 52. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in multicellular spheroids formation. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef]
- Sokolova, V.; Mekky, G.; van der Meer, S.B.; Seeds, M.C.; Atala, A.J.; Epple, M. Transport of ultrasmall gold nanoparticles (2 nm) across the blood-brain barrier in a six-cell brain spheroid model. Sci. Rep. 2020, 10, 18033. [Google Scholar] [CrossRef]
- Mège, R.M.; Ishiyama, N. Integration of cadherin adhesion and cytoskeleton at adherens junctions. Cold Spring Harb. Perspect. Biol. 2017, 9, a028738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mège, R.-M.; Gavard, J.; Lambert, M. Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 2006, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Groen, R.W.J.; de Rooij, M.F.M.; Kocemba, K.A.; Reijmers, R.M.; de Haan-Kramer, A.; Overdijk, M.B.; Aalders, L.; Rozemuller, H.; Martens, A.C.M.; Bergsagel, P.L.; et al. N-cadherin-mediated interaction with multiple myeloma cells inhibits osteoblast differentiation. Haematologica 2011, 96, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtidis, A.; Lu, R.; Pence, L.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef] [PubMed]
- De Bessa, T.C.; Pagano, A.; Moretti, A.I.S.; Oliveira, P.V.S.; Mendonça, S.A.; Kovacic, H.; Laurindo, F.R.M. Subverted regulation of Nox1 NADPH oxidase-dependent oxidant generation by protein disulfide isomerase A1 in colon carcinoma cells with overactivated KRas. Cell Death Dis. 2019, 10, 143. [Google Scholar] [CrossRef]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [Green Version]
- Snyder, H.M.; Ahles, T.; Calderwood, S.; Carrillo, M.C.; Chen, H.; Chang, C.-C.H.; Craft, S.; De Jager, P.; Driver, J.A.; Fillit, H.; et al. Exploring the nexus of Alzheimer’s disease and related dementias with cancer and cancer therapies: A convening of the Alzheimer’s Association & Alzheimer’s Drug Discovery Foundation. Alzheimers Dement. 2017, 13, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yu, J.-T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Zaman, S.; Chobrutskiy, B.I.; Sikaria, D.; Blanck, G. MAPT (Tau) expression is a biomarker for an increased rate of survival for low-grade glioma. Oncol. Rep. 2019, 41, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Gargini, R.; Segura-Collar, B.; Herránz, B.; García-Escudero, V.; Romero-Bravo, A.; Núñez, F.J.; García-Pérez, D.; Gutiérrez-Guamán, J.; Ayuso-Sacido, A.; Seoane, J.; et al. The IDH-TAU-EGFR triad defines the neovascular landscape of diffuse gliomas. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Pan, Y.; Pan, Y.; Cheng, Y.; Yang, F.; Yao, Z.; Wang, O. Knockdown of LncRNA MAPT-AS1 inhibites proliferation and migration and sensitizes cancer cells to paclitaxel by regulating MAPT expression in ER-negative breast cancers. Cell Biosci. 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Sekino, Y.; Babasaki, T.; Goto, K.; Inoue, S.; Hayashi, T.; Teishima, J.; Sakamoto, N.; Sentani, K.; Oue, N.; et al. Microtubule-associated protein tau (MAPT) is a promising independent prognostic marker and tumor suppressive protein in clear cell renal cell carcinoma. Urol. Oncol. 2020, 38, 605.e9–605.e17. [Google Scholar] [CrossRef]
- Yamauchi, A.; Kobayashi, A.; Oikiri, H.; Yokoyama, Y. Functional role of the Tau protein in epithelial ovarian cancer cells. Reprod. Med. Biol. 2017, 16, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.; Dalprà, L.; Crosti, F.; Lissoni, S.; Sciacca, F.L.; Catania, M.; Mangieri, M.; Giaccone, G.; Croci, D.; Tagliavini, F. A new function of microtubule-associated protein tau: Involvement in chromosome stability. Cell Cycle 2008, 7, 1788–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Muzi, P.; Mattei, V.; Bologna, M.; Angelucci, A. Tau oligomers accumulation sensitizes prostate cancer cells to docetaxel treatment. J. Cancer Res. Clin. Oncol. 2021, 147, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- Panopoulos, A.; Howell, M.; Fotedar, R.; Margolis, R.L. Glioblastoma motility occurs in the absence of actin polymer. Mol. Biol. Cell 2011, 22, 2212–2220. [Google Scholar] [CrossRef]
- Torres-Cruz, F.M.; Rodríguez-Cruz, F.; Escobar-Herrera, J.; Barragán-Andrade, N.; Basurto-Islas, G.; Ripova, D.; Ávila, J.; Garcia-Sierra, F. Expression of tau produces aberrant plasma membrane blebbing in glial cells through rhoa-rock-dependent F-actin remodeling. J. Alzheimer’s Dis. 2016, 52, 463–482. [Google Scholar] [CrossRef]
- Lin, R.-Z.; Chou, L.-F.; Chien, C.-C.M.; Chang, H.-Y. Dynamic analysis of hepatoma spheroid formation: Roles of E-cadherin and beta1-integrin. Cell Tissue Res. 2006, 324, 411–422. [Google Scholar] [CrossRef]
- Smyrek, I.; Mathew, B.; Fischer, S.C.; Lissek, S.M.; Becker, S.; Stelzer, E.H.K. E-cadherin, actin, microtubules and FAK dominate different spheroid formation phases and important elements of tissue integrity. Biol. Open 2019, 8, bio037051. [Google Scholar] [CrossRef] [Green Version]
- Muniz-Talavera, H.; Schmidt, J.V. The mouse Jhy gene regulates ependymal cell differentiation and ciliogenesis. PLoS ONE 2017, 12, e0184957. [Google Scholar] [CrossRef] [Green Version]
- Shimazui, T.; Schalken, J.A.; Kawai, K.; Kawamoto, R.; van Bockhoven, A.; Oosterwijk, E.; Akaza, H. Role of complex cadherins in cell-cell adhesion evaluated by spheroid formation in renal cell carcinoma cell lines. Oncol. Rep. 2004, 11, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Mary, S.; Charrasse, S.; Meriane, M.; Comunale, F.; Travo, P.; Blangy, A.; Gauthier-Rouvière, C. Biogenesis of N-cadherin-dependent cell-cell contacts in living fibroblasts is a microtubule-dependent kinesin-driven mechanism. MBoC 2002, 13, 285–301. [Google Scholar] [CrossRef] [Green Version]
- De Santis, G.; Miotti, S.; Mazzi, M.; Canevari, S.; Tomassetti, A. E-cadherin directly contributes to PI3K/AKT activation by engaging the PI3K-p85 regulatory subunit to adherens junctions of ovarian carcinoma cells. Oncogene 2009, 28, 1206–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekino, Y.; Han, X.; Babasaki, T.; Goto, K.; Inoue, S.; Hayashi, T.; Teishima, J.; Shiota, M.; Takeshima, Y.; Yasui, W.; et al. Microtubule-associated protein tau (MAPT) promotes bicalutamide resistance and is associated with survival in prostate cancer. Urol. Oncol. 2020, 38, 795.e1–795.e8. [Google Scholar] [CrossRef]
- Furnari, F.B.; Lin, H.; Huang, H.-J.S.; Cavenee, W.K. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-H.; Huang, Y.-F.; Chen, C.-C.; Chou, C.-Y. Akt inhibitor SC66 promotes cell sensitivity to cisplatin in chemoresistant ovarian cancer cells through inhibition of COL11A1 expression. Cell Death Dis. 2019, 10, 322. [Google Scholar] [CrossRef]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Phyu, S.M.; Tseng, C.-C.; Fleming, I.N.; Smith, T.A.D. Probing the PI3K/Akt/mTor pathway using 31P-NMR spectroscopy: Routes to glycogen synthase kinase 3. Sci. Rep. 2016, 6, 36544. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shemezis, J.R.; McQuinn, E.R.; Wang, J.; Sverdlov, M.; Chenn, A. AKT activation by N-cadherin regulates beta-catenin signaling and neuronal differentiation during cortical development. Neural. Dev. 2013, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful partners in cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, K.; Mahajan, N.P. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111 (Pt 21), 3167–3177. [Google Scholar] [CrossRef]
- Souter, S.; Lee, G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J. Cell Biochem. 2009, 108, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.-B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmström, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xun, G.; Hu, W.; Li, B. PTEN loss promotes oncogenic function of STMN1 via PI3K/AKT pathway in lung cancer. Sci. Rep. 2021, 11, 14318. [Google Scholar] [CrossRef]
- Massacesi, C.; Di Tomaso, E.; Urban, P.; Germa, C.; Quadt, C.; Trandafir, L.; Aimone, P.; Fretault, N.; Dharan, B.; Tavorath, R.; et al. PI3K inhibitors as new cancer therapeutics: Implications for clinical trial design. Onco Targets Ther. 2016, 9, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [Green Version]
- Colardo, M.; Segatto, M.; Di Bartolomeo, S. Targeting RTK-PI3K-mTOR axis in gliomas: An update. Int. J. Mol. Sci. 2021, 22, 4899. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, A.; Breuzard, G.; Parat, F.; Tchoghandjian, A.; Figarella-Branger, D.; De Bessa, T.C.; Garrouste, F.; Douence, A.; Barbier, P.; Kovacic, H. Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis. Cancers 2021, 13, 5818. https://doi.org/10.3390/cancers13225818
Pagano A, Breuzard G, Parat F, Tchoghandjian A, Figarella-Branger D, De Bessa TC, Garrouste F, Douence A, Barbier P, Kovacic H. Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis. Cancers. 2021; 13(22):5818. https://doi.org/10.3390/cancers13225818
Chicago/Turabian StylePagano, Alessandra, Gilles Breuzard, Fabrice Parat, Aurélie Tchoghandjian, Dominique Figarella-Branger, Tiphany Coralie De Bessa, Françoise Garrouste, Alexis Douence, Pascale Barbier, and Hervé Kovacic. 2021. "Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis" Cancers 13, no. 22: 5818. https://doi.org/10.3390/cancers13225818
APA StylePagano, A., Breuzard, G., Parat, F., Tchoghandjian, A., Figarella-Branger, D., De Bessa, T. C., Garrouste, F., Douence, A., Barbier, P., & Kovacic, H. (2021). Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis. Cancers, 13(22), 5818. https://doi.org/10.3390/cancers13225818