
Glycyrrhizin as a Nitric Oxide Regulator in Cancer Chemotherapy




Abstract
:Simple Summary
Abstract
1. Introduction
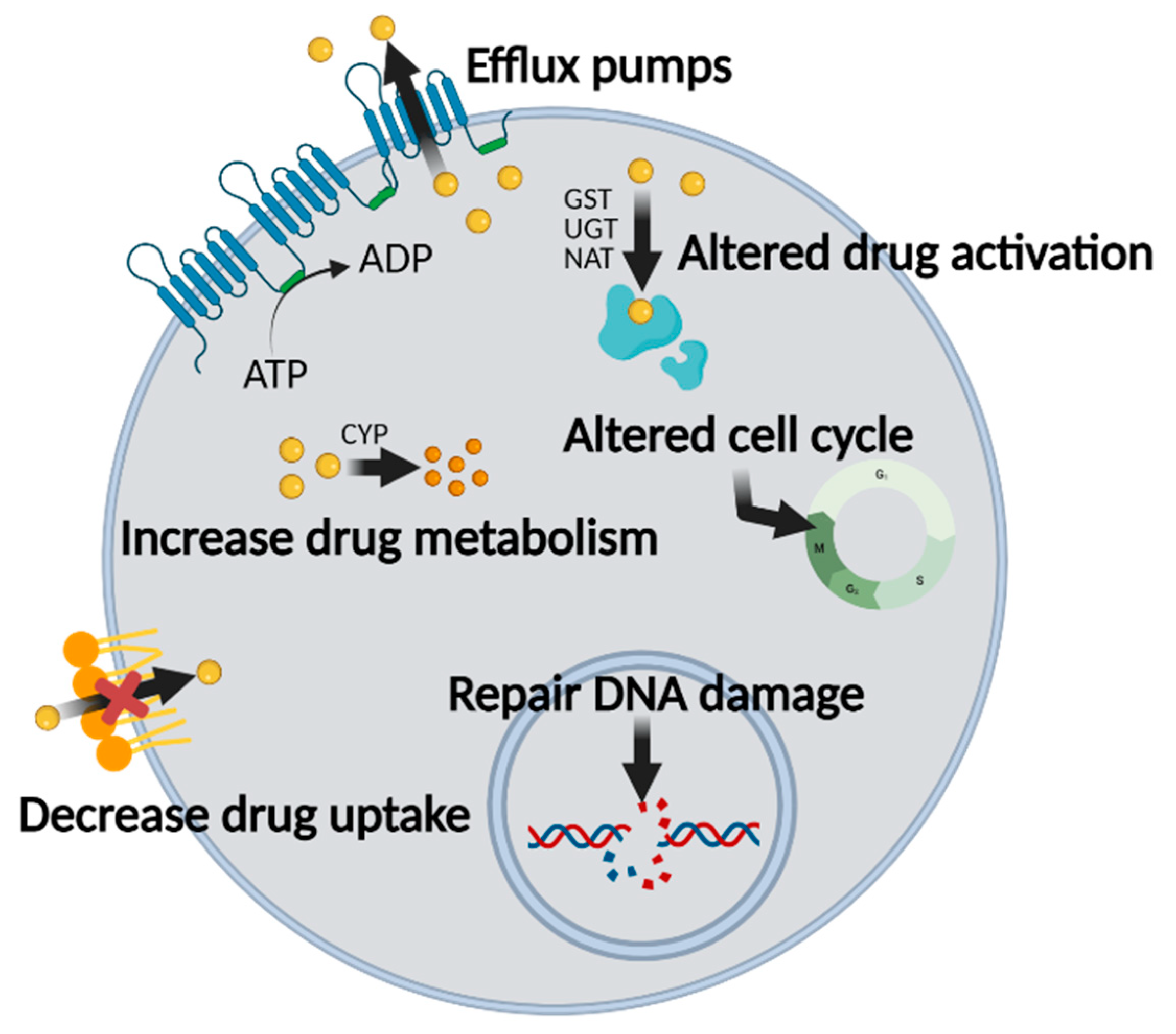
2. Multidrug Resistance in Cancer Chemotherapy
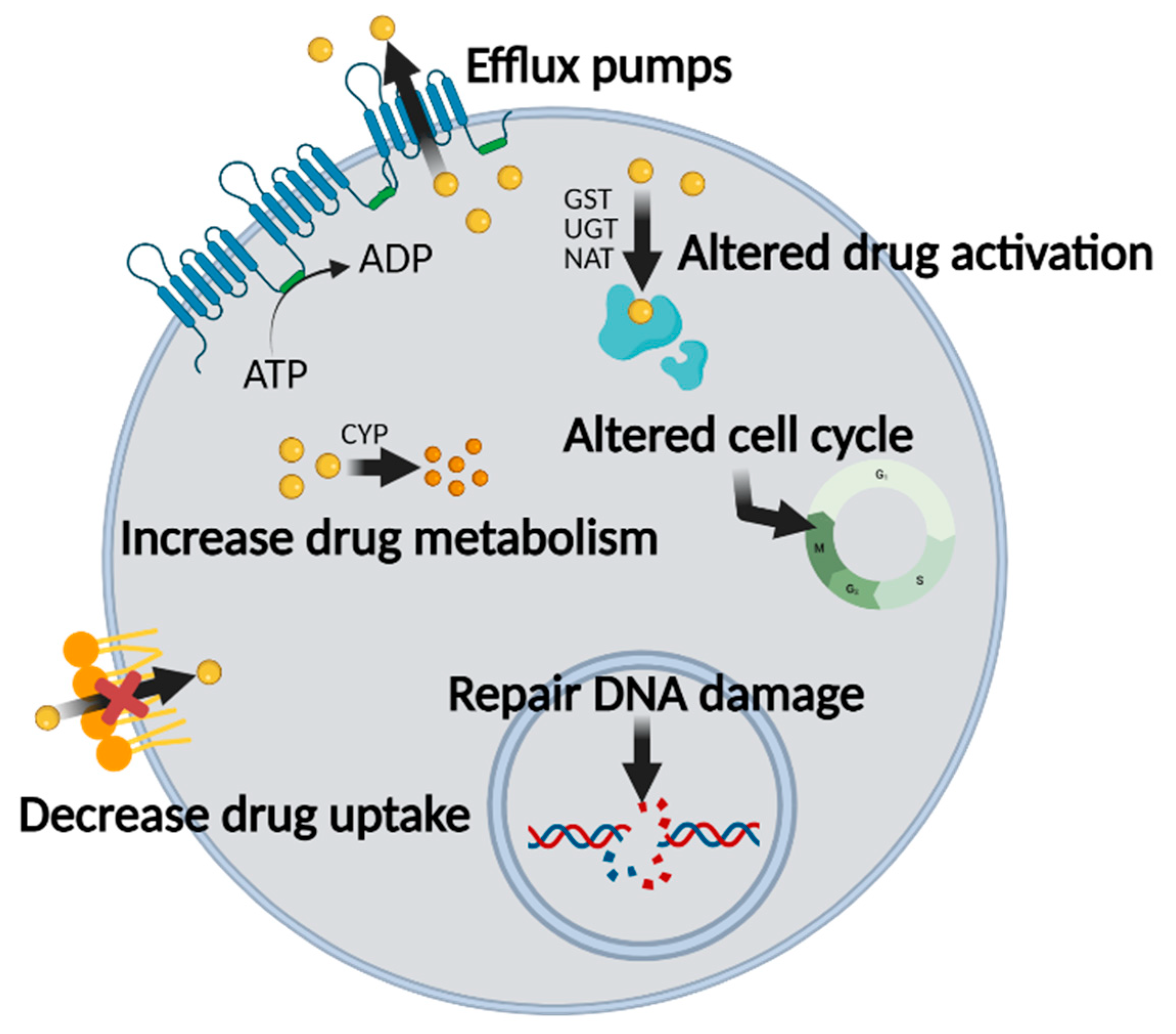
2.1. Mechanisms of MDR in Chemotherapy
2.2. ABC Transporters Are Involved in MDR
2.3. Strategies to Overcome MDR
2.3.1. P-gp Inhibitors
2.3.2. MRP1 Inhibitors
2.3.3. BCRP Inhibitors
3. Physiology of Nitric Oxide
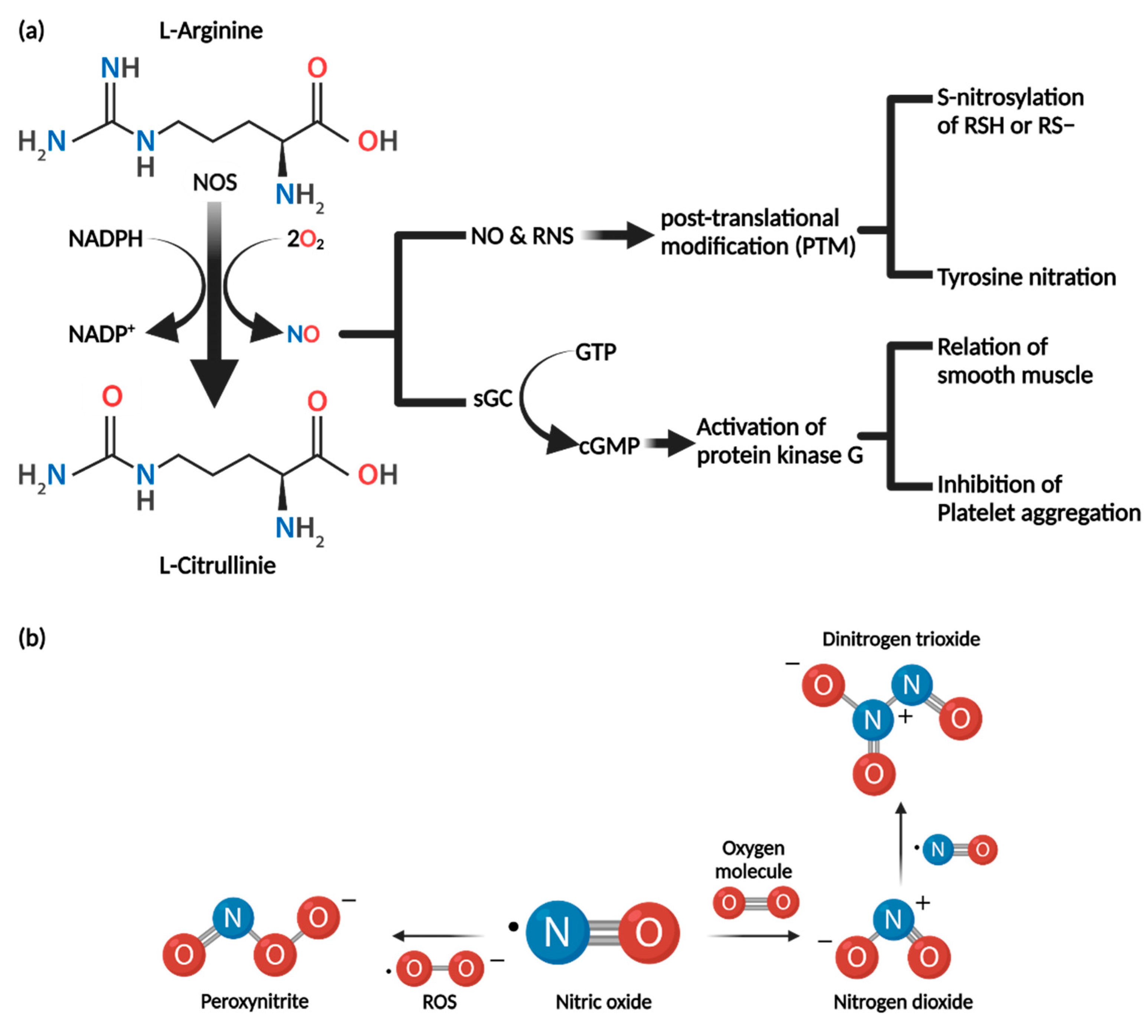
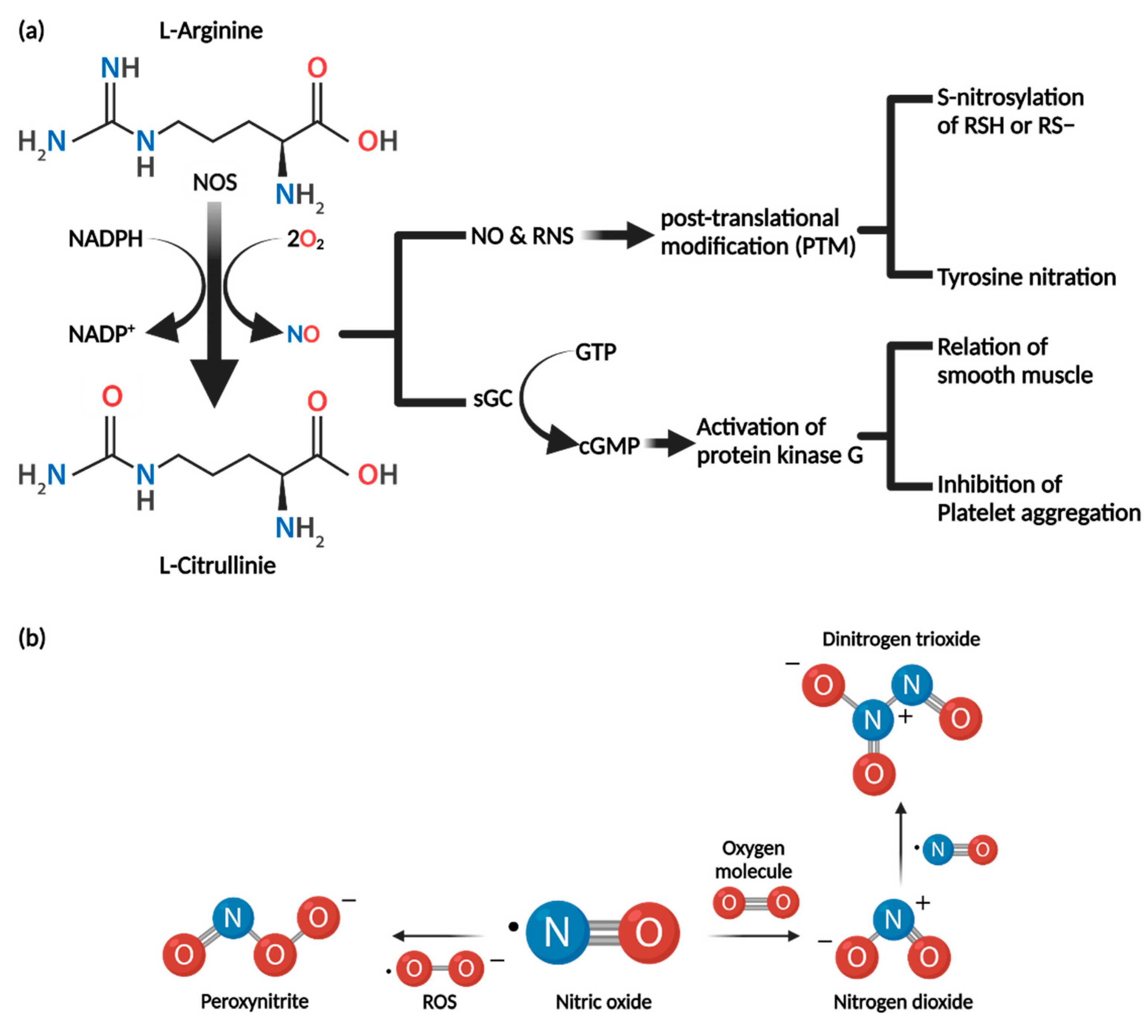
3.1. Synthesis of Nitric Oxide
3.2. Biochemical Properties of Nitric Oxide
3.3. Nitric Oxide Mechanism of Action
3.3.1. cGMP-Dependent Pathway
3.3.2. cGMP-Independent Pathway
4. Nitric Oxide and Cancer
4.1. Cancer-Promoting Role of NO
4.2. Cancer-Inhibiting Role of NO
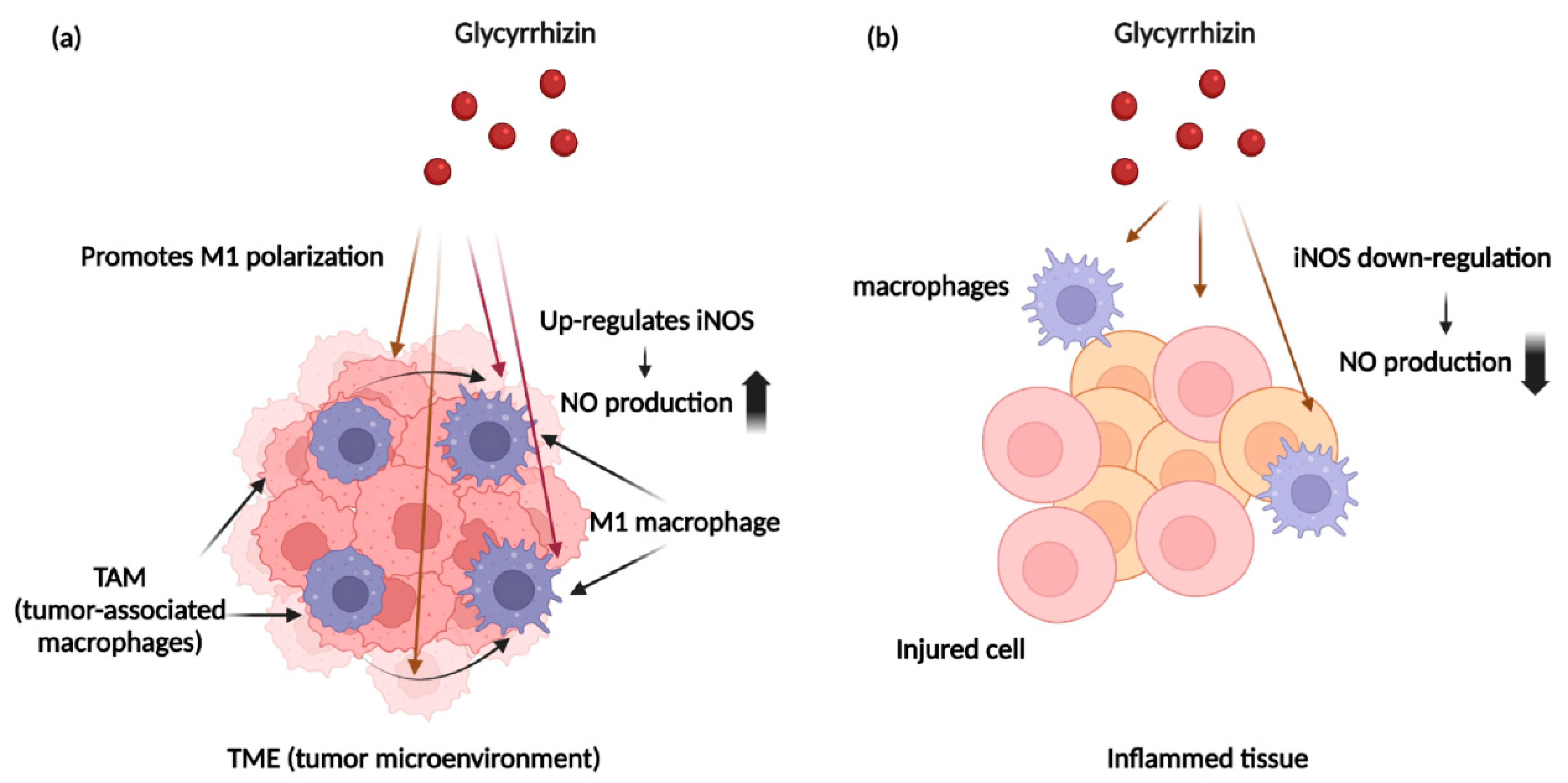
4.3. NO and Immune Cells within Cancer Tissues
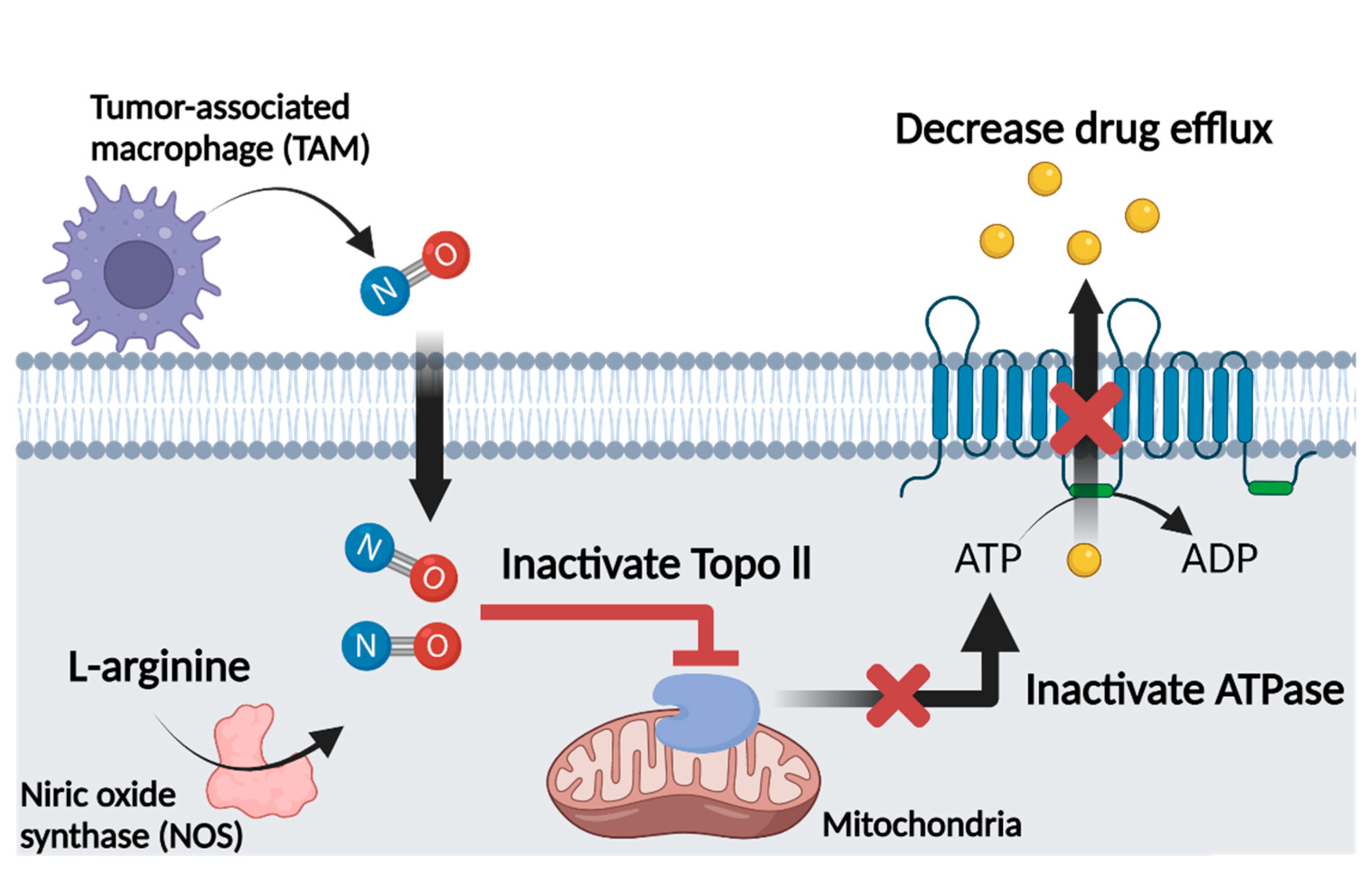
4.4. Nitric Oxide and Multidrug Resistance
4.5. NO-Donors
5. Glycyrrhizin as an Anti-Cancer Therapeutic Agent and Nitric Oxide Regulator
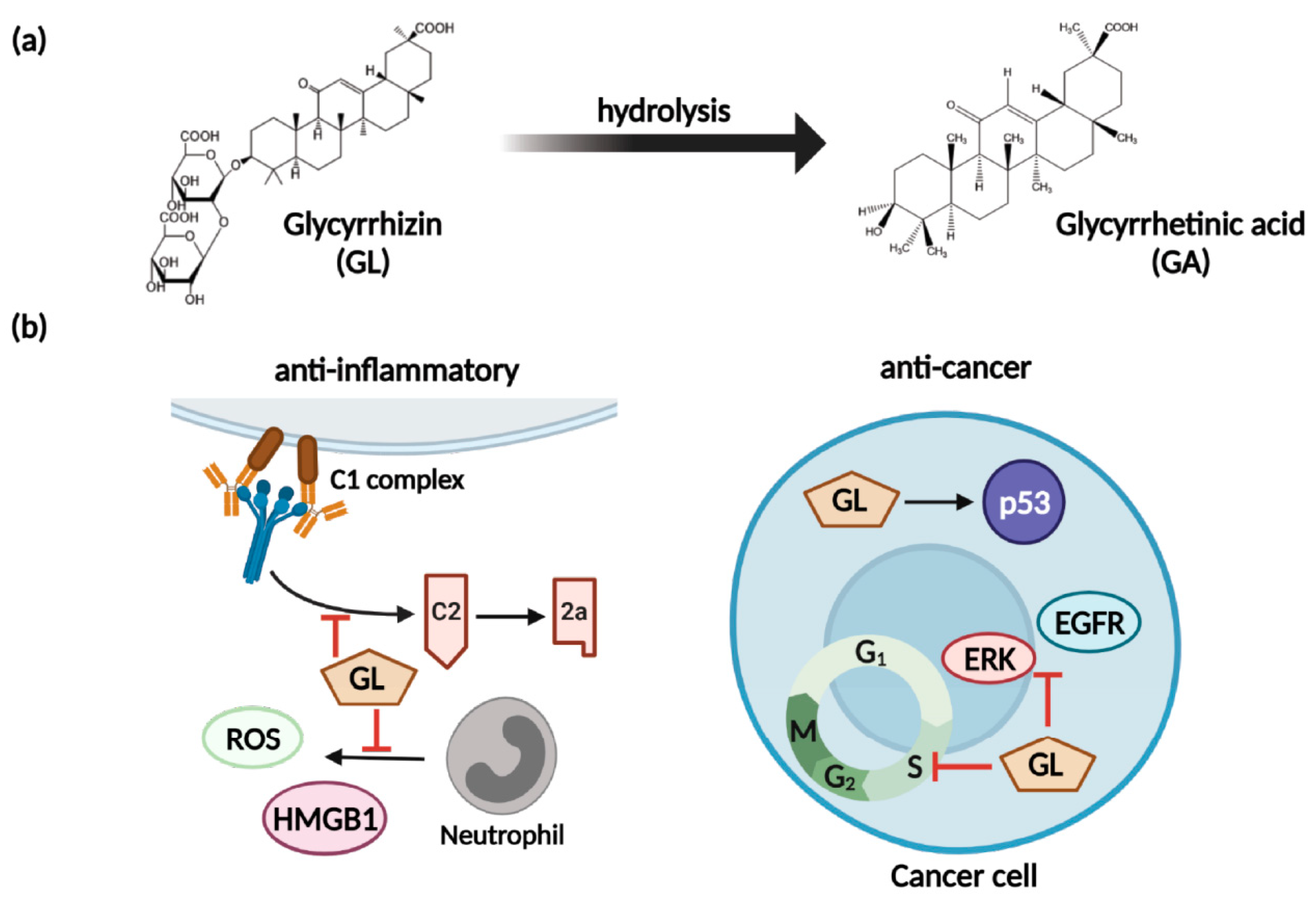
5.1. Glycyrrhizin and Nitric Oxide
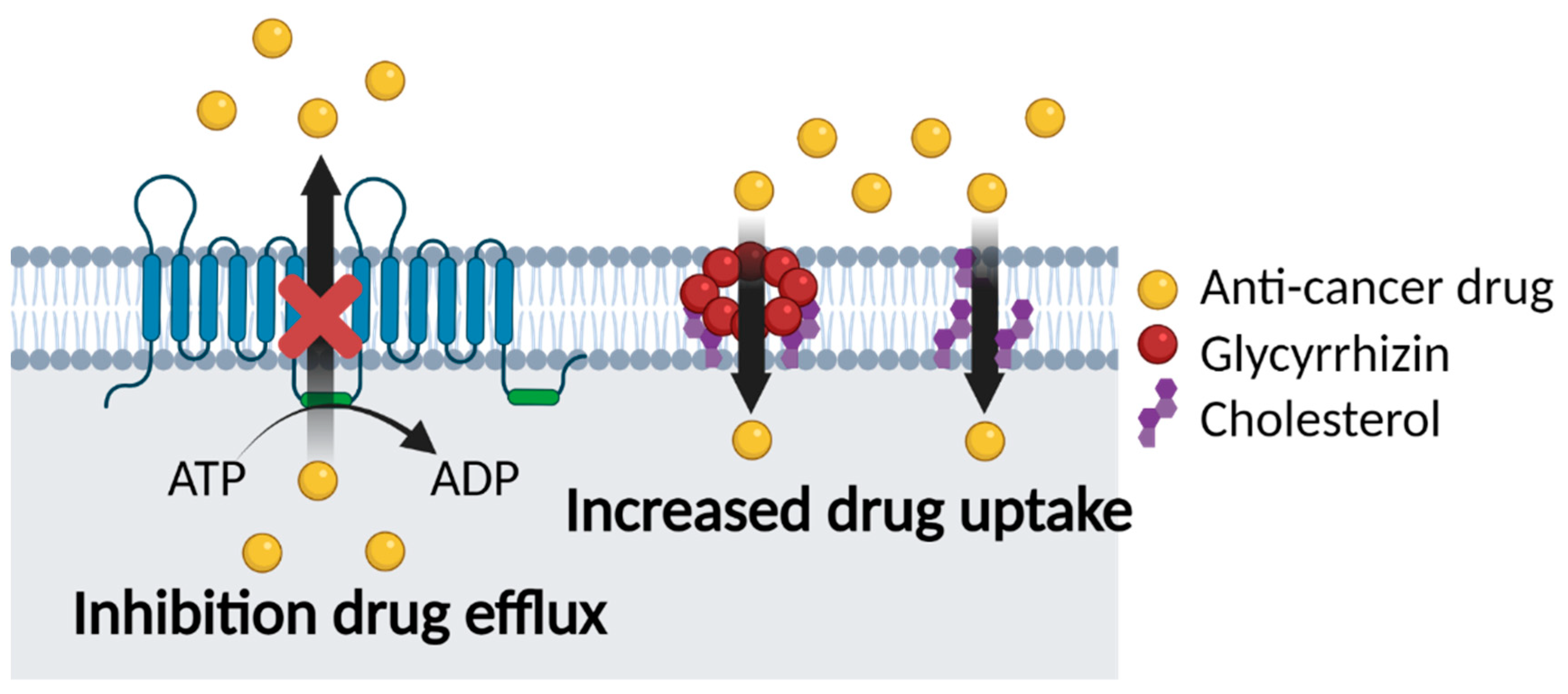
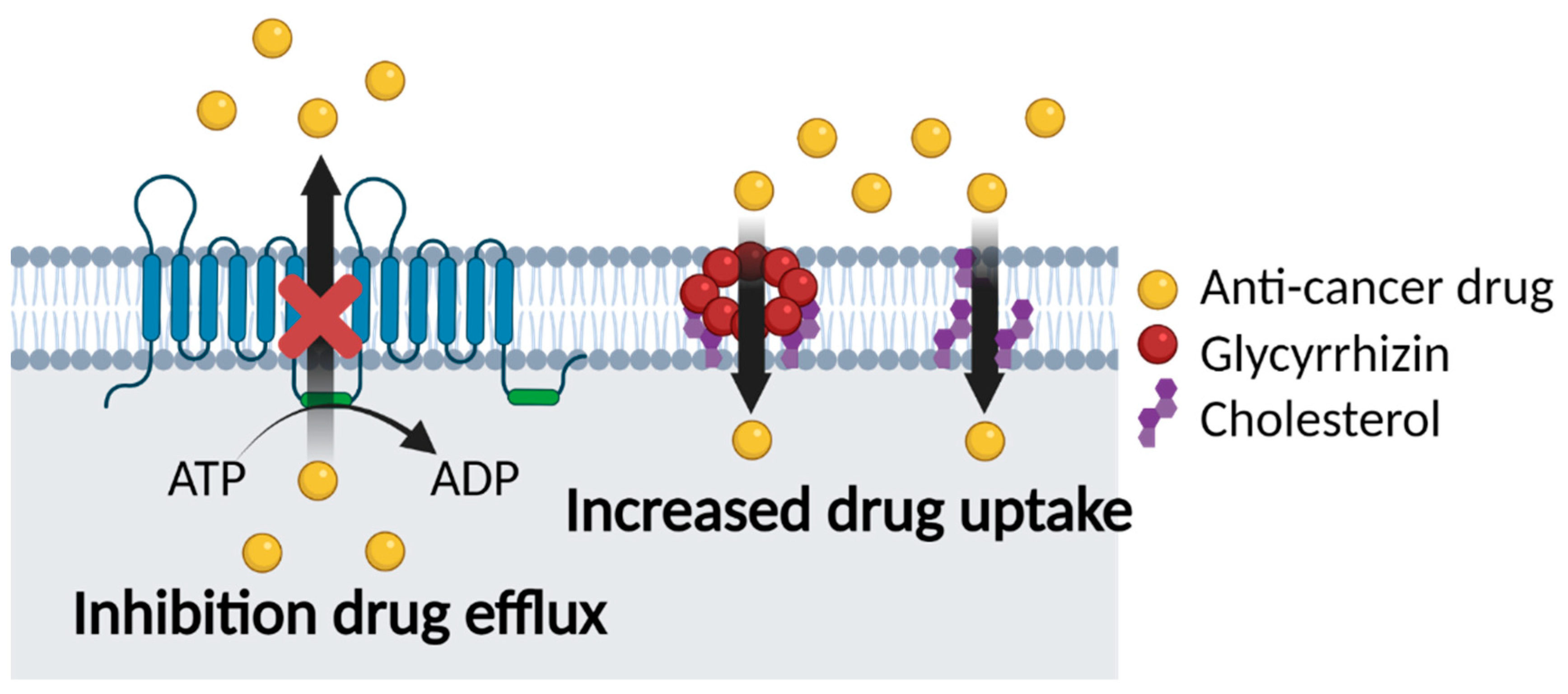
5.2. Glycyrrhizin and Multidrug Resistance
5.2.1. Co-Delivery of GL and Anti-Cancer Drugs
5.2.2. GL Carriers for Cancer Therapy
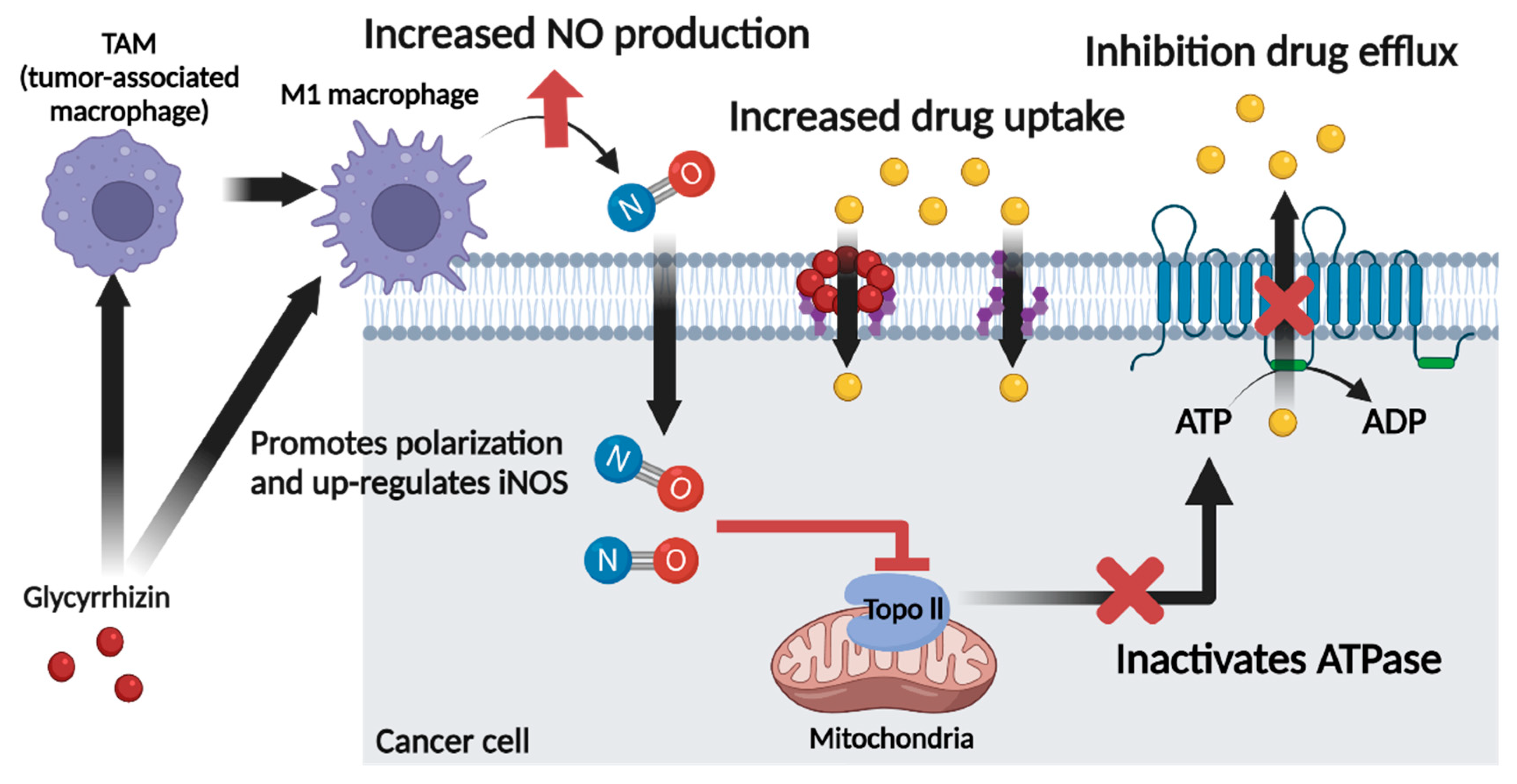
6. Role of Glycyrrhizin as a Nitric Oxide Regulator in Cancer Chemotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chabner, B.A.; Roberts, T.G. Timeline—Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.H.; Ling, T.L.; Cao, H. Current status of surgical treatment of gastric cancer in the era of minimally invasive surgery in China: Opportunity and challenge. Int. J. Surg. 2016, 28, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, P. What is cancer chemotherapy? Acta Oncol. 2001, 40, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Yu, A.M. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Shroff, E.H.; Liu, J.; Chandel, N.S. Nitric Oxide Induces Cell Death by Regulating Anti-Apoptotic BCL-2 Family Members. PLoS ONE 2009, 4, e7059. [Google Scholar] [CrossRef] [Green Version]
- Caneba, C.A.; Yang, L.; Baddour, J.; Curtis, R.; Win, J.; Hartig, S.; Marini, J.; Nagrath, D. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014, 5, e1302. [Google Scholar] [CrossRef]
- Kim, J.; Yung, B.C.; Kim, W.J.; Chen, X. Combination of nitric oxide and drug delivery systems: Tools for overcoming drug resistance in chemotherapy. J. Control. Release 2017, 263, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Jordan, B.F.; Gallez, B.; Feron, O. Nitric oxide delivery to cancer: Why and how? Eur. J. Cancer 2009, 45, 1352–1369. [Google Scholar] [CrossRef]
- Sinha, B.K.; Bortner, C.D.; Mason, R.P.; Cannon, R.E. Nitric oxide reverses drug resistance by inhibiting ATPase activity of p-glycoprotein in human multi-drug resistant cancer cells. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2806–2814. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Radzievski, R.; Xie, J. Free Radical: Nitric Oxide in Cancer Therapy. Int. J. Anal. Med. Chem. 2018, 1, 1–4. [Google Scholar]
- Kato, T.; Horie, N.; Hashimoto, K.; Satoh, K.; Shimoyama, T.; Kaneko, T.; Kusama, K.; Sakagami, H. Bimodal effect of glycyrrhizin on macrophage nitric oxide and prostaglandin E2 production. In Vivo 2008, 22, 583–586. [Google Scholar] [PubMed]
- Yi, H.; Nakashima, I.; Isobe, K. Enhancement of nitric oxide production from activated macrophages by glycyrrhizin. Am. J. Chin. Med. 1996, 24, 271–278. [Google Scholar] [CrossRef]
- Jeong, H.G.; Kim, J.Y. Induction of inducible nitric oxide synthase expression by 18 beta-glycyrrhetinic acid in macrophages. FEBS Lett. 2002, 513, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Sinha, B.K.; Kumar, A.; Mason, R.P. Nitric oxide inhibits ATPase activity and induces resistance to topoisomerase II-poisons in human MCF-7 breast tumor cells. Biochem. Biophys. Rep. 2017, 10, 252–259. [Google Scholar] [CrossRef]
- Selyutina, O.Y.; Polyakov, N.E.; Korneev, D.V.; Zaitsev, B.N. Influence of glycyrrhizin on permeability and elasticity of cell membrane: Perspectives for drugs delivery. Drug Deliv. 2016, 23, 858–865. [Google Scholar] [CrossRef]
- Selyutina, O.Y.; Apanasenko, I.E.; Kim, A.V.; Shelepova, E.A.; Khalikov, S.S.; Polyakov, N.E. Spectroscopic and molecular dynamics characterization of glycyrrhizin membrane-modifying activity. Colloids Surf. B Biointerfaces 2016, 147, 459–466. [Google Scholar] [CrossRef]
- Chai, S.; To, K.K.; Lin, G. Circumvention of multi-drug resistance of cancer cells by Chinese herbal medicines. Chin. Med. 2010, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Zamora, P.; Feliu, J.; Gonzalez Baron, M. Classification of anticancer drugs—A new system based on therapeutic targets. Cancer Treat. Rev. 2003, 29, 515–523. [Google Scholar] [CrossRef]
- He, L.; Vasiliou, K.; Nebert, D.W. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genom. 2009, 3, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Lemstrova, R.; Soucek, P.; Melichar, B.; Mohelnikova-Duchonova, B. Role of solute carrier transporters in pancreatic cancer: A review. Pharmacogenomics 2014, 15, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.J.; Xu, J.; Liang, C.; Meng, Q.C.; Hua, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.J.; Shi, S. Emerging roles of the solute carrier family in pancreatic cancer. Clin. Transl. Med. 2021, 11, e356. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Trouillas, P.; Gasparovic, A.C.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Update 2020, 49, 100670. [Google Scholar] [CrossRef]
- Van Vlerken, L.E.; Duan, Z.F.; Seiden, M.V.; Amiji, M.M. Modulation of intracellular ceramide using polymeric nanoparticles to overcome multidrug resistance in cancer. Cancer Res. 2007, 67, 4843–4850. [Google Scholar] [CrossRef] [Green Version]
- Gouaze-Andersson, V.; Yu, J.Y.; Kreitenberg, A.J.; Bielawska, A.; Giuliano, A.E.; Cabot, M.C. Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim. Biophys. Acta 2007, 1771, 1407–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.M.; Reisfeld, B.; Mayeno, A.N. Cytochromes p450: A structure-based summary of biotransformations using representative substrates. Drug Metab. Rev. 2008, 40, 1–100. [Google Scholar] [CrossRef]
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: Implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.L.; Jun, M.; Jeong, W.S. Phytochemical and Over-The-Counter Drug Interactions: Involvement of Phase I and II Drug-Metabolizing Enzymes and Phase III Transporters. J. Med. Food 2021, 24, 786–805. [Google Scholar] [CrossRef]
- Ghezzi, P.; Di Sirnplicio, P. Glutathionylation pathways in drug response. Curr. Opin. Pharmacol. 2007, 7, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Hemmerich, S.; Verdugo, D.; Rath, V.L. Strategies for drug discovery by targeting sulfation pathways. Drug Discov. Today 2004, 9, 967–975. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase Ii Drug Metabolizing Enzymes. Biomed. Pap. 2010, 154, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanwar, J.; Das, S.; Fatima, Z.; Hameed, S. Multidrug Resistance: An Emerging Crisis. Interdiscip. Perspect. Infect. Dis. 2014, 2014, 541340. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Linton, K.J. Structure and function of ABC transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- El-Awady, R.; Saleh, E.; Hashim, A.; Soliman, N.; Dallah, A.; Elrasheed, A.; Elakraa, G. The Role of Eukaryotic and Prokaryotic ABC Transporter Family in Failure of Chemotherapy. Front. Pharmacol. 2016, 7, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.W.; Ruefli, A.A.; Smyth, M.J. Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem. Sci. 2000, 25, 1–6. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Zhang, J. Multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphism: From discovery to clinical application. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011, 36, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P.C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharm. 2005, 204, 216–237. [Google Scholar] [CrossRef]
- Westover, D.; Li, F.Z. New trends for overcoming ABCG2/BCRP-mediated resistance to cancer therapies. J. Exp. Clin. Cancer Res. 2015, 34, 159. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Ross, D.D. Breast cancer resistance protein (BCRP/ABCG2): Its role in multidrug resistance and regulation of its gene expression. Chin. J. Cancer 2012, 31, 73–99. [Google Scholar] [CrossRef] [Green Version]
- Lage, H. Therapeutic potential of RNA interference in drug-resistant cancers. Future Oncol. 2009, 5, 169–185. [Google Scholar] [CrossRef]
- Green, W.J.F.; James, P.A.; Ratan, H.L. Potential Use of RNA Interference as Therapeutic Strategy in Urologic Cancer. Urology 2011, 78, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.W.; Mumper, R.J. Nanomedicinal strategies to treat multidrug-resistant tumors: Current progress. Nanomedicine 2010, 5, 597–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadali, F.; Pourfathollah, A.A.; Alimoghaddam, K.; Nikougoftar, M.; Rostami, S.; Dizaji, A.; Azizi, E.; Zomorodipour, A.; Ghavamzadeh, A. Multidrug resistance inhibition by antisense oligonucleotide against MDR1/mRNA in P-glycoprotein expressing leukemic cells. Hematology 2007, 12, 393–401. [Google Scholar] [CrossRef]
- Tran, V.H.; Marks, D.; Duke, R.K.; Bebawy, M.; Duke, C.C.; Roufogalis, B.D. Modulation of P-glycoprotein-Mediated Anticancer Drug Accumulation, Cytotoxicity, and ATPase Activity by Flavonoid Interactions. Nutr. Cancer 2011, 63, 435–443. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.L. Development of Fourth Generation ABC Inhibitors from Natural Products: A Novel Approach to Overcome Cancer Multidrug Resistance. Anti-Cancer Agents Med. Chem. 2015, 15, 605–615. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Guns, E.S.; Denyssevych, T.; Dixon, R.; Bally, M.B.; Mayer, L. Drug interaction studies between paclitaxel (Taxol) and OC144-093—A new modulator of MDR in cancer chemotherapy. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 119–126. [Google Scholar] [CrossRef]
- Sinha, B.K.; Perera, L.; Cannon, R.E. NCX-4040, a Unique Nitric Oxide Donor, Induces Reversal of Drug-Resistance in Both ABCB1-and ABCG2-Expressing Multidrug Human Cancer Cells. Cancers 2021, 13, 1680. [Google Scholar] [CrossRef] [PubMed]
- Bajelan, E.; Haeri, A.; Vali, A.M.; Ostad, S.N.; Dadashzadeh, S. Co-delivery of Doxorubicin and PSC 833 (Valspodar) by Stealth Nanoliposomes for Efficient Overcoming of Multidrug Resistance. J. Pharm. Pharm. Sci. 2012, 15, 568–582. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, L.; Gao, H.; Liu, Y.; Zhang, Q.; Ran, R.; Zhang, Z.; He, Q. Co-delivery of doxorubicin and P-gp inhibitor by a reduction-sensitive liposome to overcome multidrug resistance, enhance anti-tumor efficiency and reduce toxicity. Drug Deliv. 2016, 23, 1130–1143. [Google Scholar] [CrossRef]
- Li, N.; Zhang, P.; Huang, C.; Song, Y.; Garg, S.; Luan, Y. Co-delivery of doxorubicin hydrochloride and verapamil hydrochloride by pH-sensitive polymersomes for the reversal of multidrug resistance. RSC Adv. 2015, 5, 77986–77995. [Google Scholar] [CrossRef]
- Gao, W.; Lin, Z.; Chen, M.; Yang, X.; Zhang, X.; Lan, Y.; Zhang, Q.; Cui, Z. The co-delivery of a low-dose P-glycoprotein inhibitor with doxorubicin sterically stabilized liposomes against breast cancer with low P-glycoprotein expression. Int. J. Nanomed. 2014, 9, 3425. [Google Scholar] [CrossRef] [Green Version]
- Tonbul, H.; Sahin, A.; Tavukcuoglu, E.; Esendagli, G.; Capan, Y. Combination drug delivery with actively-targeted PLGA nanoparticles to overcome multidrug resistance in breast cancer. J. Drug Deliv. Sci. Techol. 2019, 54, 101380. [Google Scholar] [CrossRef]
- Wang, H.Y.; Li, F.; Du, C.A.; Wang, H.X.; Mahato, R.I.; Huang, Y.Z. Doxorubicin and Lapatinib Combination Nanomedicine for Treating Resistant Breast Cancer. Mol. Pharm. 2014, 11, 2600–2611. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Shi, H.; Qiao, M.X.; Zhang, Z.H.; Yang, W.T.; Dong, L.Y.; Xie, F.C.; Zhao, C.P.; Kang, L. pH-sensitive micelles for the intracellular co-delivery of curcumin and Pluronic L61 unimers for synergistic reversal effect of multidrug resistance. Sci. Rep. 2021, 11, 18061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Cheng, D.; Weichel, A.K.; Osipo, C.; Wing, L.K.; Chen, B.; Louis, T.E.; Jordan, V.C. Cooperative effect of gefitinib and fumitremorgin c on cell growth and chemosensitivity in estrogen receptor alpha negative fulvestrant-resistant MCF-7 cells. Int. J. Oncol. 2006, 29, 1237–1246. [Google Scholar]
- Wu, C.-P.; Hsiao, S.-H.; Huang, Y.-H.; Hung, L.-C.; Yu, Y.-J.; Chang, Y.-T.; Hung, T.-H.; Wu, Y.-S. Sitravatinib Sensitizes ABCB1- and ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Drugs. Cancers 2020, 12, 195. [Google Scholar] [CrossRef] [Green Version]
- Ravar, F.; Saadat, E.; Kelishadi, P.D.; Dorkoosh, F.A. Liposomal formulation for co-delivery of paclitaxel and lapatinib, preparation, characterization and optimization. J. Liposome Res. 2016, 26, 175–187. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, Y.; Chen, Y.; Yu, S.; Hao, J.; Luo, J.; Sha, X.; Fang, X. Enhanced antitumor efficacy by paclitaxel-loaded pluronic P123/F127 mixed micelles against non-small cell lung cancer based on passive tumor targeting and modulation of drug resistance. Eur. J. Pharm. Biopharm. 2010, 75, 341–353. [Google Scholar] [CrossRef]
- Shou, J.W.; You, L.K.; Yao, J.L.; Xie, J.S.; Jing, J.; Jing, Z.; Jiang, L.M.; Sui, X.B.; Pan, H.M.; Han, W.D. Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer Lett. 2020, 493, 13–15. [Google Scholar] [CrossRef]
- Patil, Y.; Sadhukha, T.; Ma, L.N.; Panyam, J. Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J. Control. Release 2009, 136, 21–29. [Google Scholar] [CrossRef]
- Limtrakul, P.; Chearwae, W.; Shukla, S.; Phisalphong, C.; Ambudkar, S.V. Modulation of function of three ABC drug transporters, P-glycoprotein (ABCB1), mitoxantrone resistance protein (ABCG2) and multidrug resistance protein 1 (ABCC1) by tetrahydrocurcumin, a major metabolite of curcumin. Mol. Cell. Biochem. 2007, 296, 85–95. [Google Scholar] [CrossRef]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kogel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H.M. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef] [Green Version]
- Kopanitsa, L.; Kopanitsa, M.V.; Safitri, D.; Ladds, G.; Bailey, D.S. Suppression of Proliferation of Human Glioblastoma Cells by Combined Phosphodiesterase and Multidrug Resistance-Associated Protein 1 Inhibition. Int. J. Mol. Sci. 2021, 22, 9665. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.A.; Dunn, M.J.; Sorokin, A. Regulation of MDR-1 (P-glycoprotein) by cyclooxygenase-2. J. Biol. Chem. 2002, 277, 38915–38920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Selvarajan, K.; Hasan, M.R.; Chan, A.P.; Jin, C.; Kim, J.; Chan, S.K.; Le, N.D.; Kim, Y.B.; Tai, I.T. Inhibition of COX-2 in colon cancer modulates tumor growth and MDR-1 expression to enhance tumor regression in therapy-refractory cancers in vivo. Neoplasia 2012, 14, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef]
- Lingineni, K.; Belekar, V.; Tangadpalliwar, S.R.; Garg, P. The role of multidrug resistance protein (MRP-1) as an active efflux transporter on blood-brain barrier (BBB) permeability. Mol. Divers. 2017, 21, 355–365. [Google Scholar] [CrossRef]
- Muntane, J.; la Mata, M.D. Nitric oxide and cancer. World J. Hepatol. 2010, 2, 337–344. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Ali, A. Biochemistry of nitric oxide. Indian J. Clin. Biochem. 2011, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta, S. Nitric oxide for cancer therapy. Future Sci. OA 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. [Google Scholar] [CrossRef]
- Xu, W.M.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.P.; McAndrew, J.; Sellak, H.; White, C.R.; Jo, H.J.; Freeman, B.A.; Darley-Usmar, V.M. Biological aspects of reactive nitrogen species. Biochim. Biophys. Acta-Bioenerg. 1999, 1411, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Fessenden, J.D.; Schacht, J. The nitric oxide/cyclic GMP pathway: A potential major regulator of cochlear physiology. Hear. Res. 1998, 118, 168–176. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-Dependent Protein Kinases and cGMP Phosphodiesterases in Nitric Oxide and cGMP Action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Shahani, N.; Sawa, A. Protein S-nitrosylation: Role for nitric oxide signaling in neuronal death. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 736–742. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Thomas, D.D.; Donzelli, S.; Espey, M.G.; Roberts, D.D.; Wink, D.A.; Isenberg, J.S. The biphasic nature of nitric oxide responses in tumor biology. Antioxid. Redox Signal. 2006, 8, 1329–1337. [Google Scholar] [CrossRef]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Szeliga, M.; Albrecht, J. Roles of nitric oxide and polyamines in brain tumor growth. Adv. Med. Sci. 2021, 66, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Billiar, T.R.; Talanian, R.V.; Kim, Y.M. Nitric Oxide Reversibly Inhibits Seven Members of the Caspase Family via S-Nitrosylation. Biochem. Biophys. Res. Commun. 1997, 240, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.M.; Pae, H.O.; Jang, S.I.; Kim, Y.M.; Chung, H.T. Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. J. Biochem. Mol. Biol. 2002, 35, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Knethen, A.; Brune, B. Cyclooxygenase-2: An essential regulator of NO-mediated apoptosis. FASEB J. 1997, 11, 887–895. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Peñarando, J.; Aranda, E.; Rodríguez-Ariza, A. Immunomodulatory roles of nitric oxide in cancer: Tumor microenvironment says “NO” to antitumor immune response. Transl. Res. 2019, 210, 99–108. [Google Scholar] [CrossRef]
- Korde, S.; Sridharan, G.; Gadbail, A.; Poornima, V. Nitric oxide and oral cancer: A review. Oral Oncol. 2012, 48, 475–483. [Google Scholar] [CrossRef]
- Fahey, J.M.; Girotti, A.W. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: Role of nitric oxide. Nitric Oxide-Biol. Chem. 2015, 49, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; FelleyBosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messmer, U.K.; Ankarcrona, M.; Nicotera, P.; Brune, B. P53 Expression in Nitric Oxide-Induced Apoptosis. FEBS Lett. 1994, 355, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Boyd, C.S.; Cadenas, E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol. Chem. 2002, 383, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Glockzin, S.; von Knethen, A.; Scheffner, M.; Brune, B. Activation of the cell death program by nitric oxide involves inhibition of the proteasome. J. Biol. Chem. 1999, 274, 19581–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.M.; Liu, L.Z.; Smith, G.C.M.; Charles, I.G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat. Cell Biol. 2000, 2, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Baritaki, S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: Downregulation of the NF-kappa B/Snail/YY1/RKIP circuitry. Nitric Oxide-Biol. Chem. 2011, 24, 1–7. [Google Scholar] [CrossRef]
- Rosselli, M.; Keller, P.J.; Dubey, R.K. Role of nitric oxide in the biology, physiology and pathophysiology of reproduction. Hum. Reprod. Update 1998, 4, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Vedenko, A.; Panara, K.; Goldstein, G.; Ramasamy, R.; Arora, H. Tumor Microenvironment and Nitric Oxide: Concepts and Mechanisms. Adv. Exp. Med. Biol. 2020, 1277, 143–158. [Google Scholar] [CrossRef]
- Weigert, A.; Brune, B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide-Biol. Chem. 2008, 19, 95–102. [Google Scholar] [CrossRef]
- Lin, Y.X.; Xu, J.X.; Lan, H.Y. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.L.; Wang, B.K.; Xu, X.; Du, W.; Li, W.F.; Wang, Y.M. Glycyrrhizic Acid Promotes M1 Macrophage Polarization in Murine Bone Marrow-Derived Macrophages Associated with the Activation of JNK and NF-kappa B. Mediat. Inflamm. 2015, 2015, 372931. [Google Scholar] [CrossRef] [Green Version]
- Hasinoff, B.B.; Wu, X.; Krokhin, O.V.; Ens, W.; Standing, K.G.; Nitiss, J.L.; Sivaram, T.; Giorgianni, A.; Yang, S.H.; Jiang, Y.; et al. Biochemical and proteomics approaches to characterize topoisomerase II alpha cysteines and DNA as targets responsible for cisplatin-induced inhibition of topoisomerase II alpha. Mol. Pharmacol. 2005, 67, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Kumar, A.; Kumari, A.; Tokar, E.J.; Waalkes, M.P.; Bortner, C.D.; Williams, J.; Ehrenshaft, M.; Mason, R.P.; Sinha, B.K. Nitric Oxide Down-Regulates Topoisomerase I and Induces Camptothecin Resistance in Human Breast MCF-7 Tumor Cells. PLoS ONE 2015, 10, e0141897. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Miraglia, E.; Viarisio, D.; Costamagna, C.; Pescarmona, G.; Ghigo, D.; Bosia, A. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005, 65, 516–525. [Google Scholar]
- Sinha, B.K.; Perera, L.; Cannon, R.E. Reversal of drug resistance by JS-K and nitric oxide in ABCB1-and ABCG2-expressing multi-drug resistant human tumor cells. Biomed. Pharmacother. 2019, 120, 109468. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Bhattacharjee, S.; Chatterjee, S.; Jiang, J.J.; Motten, A.G.; Kumar, A.; Espey, M.G.; Mason, R.P. Role of Nitric Oxide in the Chemistry and Anticancer Activity of Etoposide (VP-16,213). Chem. Res. Toxicol. 2013, 26, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.Q.; Huang, Z.J.; Li, L.L. Advanced nitric oxide donors: Chemical structure of NO drugs, NO nanomedicines and biomedical applications. Nanoscale 2021, 13, 444–459. [Google Scholar] [CrossRef]
- Miller, M.R.; Megson, I.L. Review—Recent developments in nitric oxide donor drugs. Br. J. Pharmacol. 2007, 151, 305–321. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.G.; Xian, M.; Tang, X.P.; Wu, X.J.; Wen, Z.; Cai, T.W.; Janczuk, A.J. Nitric oxide donors: Chemical activities and biological applications. Chem. Rev. 2002, 102, 1091–1134. [Google Scholar] [CrossRef]
- Riccio, D.A.; Schoenfisch, M.H. Nitric oxide release: Part I. Macromolecular scaffolds. Chem. Soc. Rev. 2012, 41, 3731–3741. [Google Scholar] [CrossRef]
- Fei, Y.; Wu, J.B.; An, H.W.; Zhu, K.; Peng, B.; Cai, J.Q.; Zhang, Y.H.; Li, L.L.; Wang, H.; Huang, Z.J. Identification of New Nitric Oxide-Donating Peptides with Dual Biofilm Eradication and Antibacterial Activities for Intervention of Device-Related Infections. J. Med. Chem. 2020, 63, 9127–9135. [Google Scholar] [CrossRef]
- Rose, M.J.; Mascharak, P.K. Photoactive ruthenium nitrosyls: Effects of light and potential application as NO donors. Coord. Chem. Rev. 2008, 252, 2093–2114. [Google Scholar] [CrossRef] [Green Version]
- Fry, N.L.; Mascharak, P.K. Photoactive Ruthenium Nitrosyls as NO Donors: How To Sensitize Them toward Visible Light. Acc. Chem. Res. 2011, 44, 289–298. [Google Scholar] [CrossRef]
- Su, X.T.; Wu, L.; Hu, M.M.; Dong, W.X.; Xu, M.; Zhang, P. Glycyrrhizic acid: A promising carrier material for anticancer therapy. Biomed. Pharmacother. 2017, 95, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Sakamoto, M.; Matsushita, M.; Fujita, T.; Nishioka, K. Glycyrrhizin inhibits the lytic pathway of complement—Possible mechanism of its anti-inflammatory effect on liver cells in viral hepatitis. Microbiol. Immunol. 2000, 44, 799–804. [Google Scholar] [CrossRef]
- Kroes, B.H.; Beukelman, C.J.; vandenBerg, A.J.J.; Wolbink, G.J.; vanDijk, H.; Labadie, R.P. Inhibition of human complement by beta-glycyrrhetinic acid. Immunology 1997, 90, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Komura, J.; Asada, Y.; Niwa, Y. Mechanism of anti-inflammatory action of glycyrrhizin: Effect on neutrophil functions including reactive oxygen species generation. Planta Med. 1991, 57, 119–121. [Google Scholar] [CrossRef]
- Schleimer, R.P. Potential regulation of inflammation in the lung by local metabolism of hydrocortisone. Am. J. Respir. Cell Mol. Biol 1991, 4, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Teelucksingh, S.; Mackie, A.D.; Burt, D.; McIntyre, M.A.; Brett, L.; Edwards, C.R. Potentiation of hydrocortisone activity in skin by glycyrrhetinic acid. Lancet 1990, 335, 1060–1063. [Google Scholar] [CrossRef]
- Roohbakhsh, A.; Iranshahy, M.; Iranshahi, M. Glycyrrhetinic Acid and Its Derivatives: Anti-Cancer and Cancer Chemopreventive Properties, Mechanisms of Action and Structure-Cytotoxic Activity Relationship. Curr. Med. Chem. 2016, 23, 498–517. [Google Scholar] [CrossRef]
- He, S.Q.; Gao, M.; Fu, Y.F.; Zhang, Y.N. Glycyrrhizic acid inhibits leukemia cell growth and migration via blocking AKT/mTOR/STAT3 signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 5175–5181. [Google Scholar]
- Ma, Y.F.; Guo, N.N.; Chu, J.; Jin, S.; Yang, B.; Li, J.; Zhang, T.; Guo, J.T.; Chen, L.; Liang, C.Y.; et al. Glycyrrhizin treatment inhibits proliferation and invasive potential of lung cancer cells. Int. J. Clin. Exp. Med. 2016, 9, 10592–10596. [Google Scholar]
- Ibrahim, M.G.; EI-Senduny, F.F.; Youssef, M.M.; Elimam, D.M.; Bar, F.M.A.; Badria, F.A. Acetyl glycyrrhetinic acid methyl ester as a promising glycyrrhizin derivative against the breast cancer cells (MCF-7). J. Rep. Pharm. Sci. 2019, 8, 161–171. [Google Scholar] [CrossRef]
- Tsai, J.J.; Pan, P.J.; Hsu, F.T.; Chung, J.G.; Chiang, I.T. Glycyrrhizic Acid Modulates Apoptosis through Extrinsic/Intrinsic Pathways and Inhibits Protein Kinase B- and Extracellular Signal-Regulated Kinase-Mediated Metastatic Potential in Hepatocellular Carcinoma In Vitro and In Vivo. Am. J. Chin. Med. 2020, 48, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Davey, A.K.; Chen, Y.X.; Wang, J.P.; Liu, X.Q. Effects of diammonium glycyrrhizinate on the pharmacokinetics of aconitine in rats and the potential mechanism. Xenobiotica 2009, 39, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cui, Y.; Hao, K.J. Effects of glycyrrhizin on the pharmacokinetics of asiatic acid in rats and its potential mechanism. Pharm. Biol. 2018, 56, 119–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Takano, F. Nitric-Oxide Production in Mouse Peritoneal-Macrophages Enhanced with Glycyrrhizin. Biol. Pharm. Bull. 1994, 17, 759–761. [Google Scholar] [CrossRef] [Green Version]
- Tanemoto, R.; Okuyama, T.; Matsuo, H.; Okumura, T.; Ikeya, Y.; Nishizawa, M. The constituents of licorice (Glycyrrhiza uralensis) differentially suppress nitric oxide production in interleukin-1beta-treated hepatocytes. Biochem. Biophys. Rep. 2015, 2, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Lee, S.H.; Kim, J.Y.; Lee, W.S. Effects of glycyrrhizin on lipopolysaccharide-induced acute lung injury in a mouse model. J. Thorac. Dis. 2019, 11, 1287–1302. [Google Scholar] [CrossRef]
- Wang, H.L.; Li, Y.X.; Niu, Y.T.; Zheng, J.; Wu, J.; Shi, G.J.; Ma, L.; Niu, Y.; Sun, T.; Yu, J.Q. Observing Anti-inflammatory and Anti-nociceptive Activities of Glycyrrhizin Through Regulating COX-2 and Pro-inflammatory Cytokines Expressions in Mice. Inflammation 2015, 38, 2269–2278. [Google Scholar] [CrossRef]
- Selyutina, O.Y.; Apanasenko, I.E.; Polyakov, N.E. Membrane-modifying activity of glycyrrhizic acid. Russ. Chem. Bull. 2015, 64, 1555–1559. [Google Scholar] [CrossRef]
- Zhou, J.-X.; Wink, M. Reversal of Multidrug Resistance in Human Colon Cancer and Human Leukemia Cells by Three Plant Extracts and Their Major Secondary Metabolites. Medicines 2018, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, T.; Nakahashi, Y.; Hachimine, D.; Seki, T.; Okazaki, K. The combination of glycyrrhizin and lamivudine can reverse the cisplatin resistance in hepatocellular carcinoma cells through inhibition of multidrug resistance-associated proteins. Int. J. Oncol. 2007, 31, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Nabekura, T.; Yamaki, T.; Ueno, K.; Kitagawa, S. Inhibition of P-glycoprotein and multidrug resistance protein 1 by dietary phytochemicals. Cancer Chemother. Pharm. 2008, 62, 867–873. [Google Scholar] [CrossRef]
- Wang, Q.S.; Gao, L.N.; Zhu, X.N.; Zhang, Y.; Zhang, C.N.; Xu, D.; Cui, Y.L. Co-delivery of glycyrrhizin and doxorubicin by alginate nanogel particles attenuates the activation of macrophage and enhances the therapeutic efficacy for hepatocellular carcinoma. Theranostics 2019, 9, 6239–6255. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Lan, Y.; Cao, M.; Ma, X.Q.; Cao, A.C.; Sun, Y.; Yang, J.H.; Li, L.; Liu, Y.H. Glycyrrhetinic acid-conjugated polymeric prodrug micelles co-delivered with doxorubicin as combination therapy treatment for liver cancer. Colloids Surf. B Biointerfaces 2019, 175, 106–115. [Google Scholar] [CrossRef]
- He, S.S.; Lin, Q.; Qu, M.K.; Wang, L.Y.; Deng, L.; Xiao, L.Y.; Zhang, Z.R.; Zhang, L. Liver-Targeted Co-delivery of Entecavir and Glycyrrhetinic Acid Based on Albumin Nanoparticle To Enhance the Accumulation of Entecavir. Mol. Pharm. 2018, 15, 3953–3961. [Google Scholar] [CrossRef] [PubMed]
- Trucillo, P. Drug Carriers: Classification, Administration, Release Profiles, and Industrial Approach. Processes 2021, 9, 470. [Google Scholar] [CrossRef]
- Sparreboom, A.; van Asperen, J.; Mayer, U.; Schinkel, A.H.; Smit, J.W.; Meijer, D.K.F.; Borst, P.; Nooijen, W.J.; Beijnen, J.H.; van Tellingen, O. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 1997, 94, 2031–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.H.; Zhang, Q.; Liang, Q.Y.; Wang, S.Q.; Zhao, B.X.; Wang, Y.T.; Cai, Y.; Li, G.F. Bioavailability enhancement of paclitaxel via a novel oral drug delivery system: Paclitaxel-loaded glycyrrhizic acid micelles. Molecules 2015, 20, 4337–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.H.; Feng, Z.M.; Wang, H.; Su, C.; Lu, Z.H.; Yu, J.B.; Dushkin, A.V.; Su, W.K. Preparation of camptothecin micelles self-assembled from disodium glycyrrhizin and tannic acid with enhanced antitumor activity. Eur. J. Pharm. Biopharm. 2021, 164, 75–85. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cell Type | Anti-Cancer Drug | ABC Transporter | Inhibitor | References |
|---|---|---|---|---|
| Breast | Doxorubicin | P-gp | PSC-833 | [59] |
| Doxorubicin | P-gp | Verapamil | [60,61] | |
| Doxorubicin | P-gp | Cyclosporine A | [62] | |
| Paclitaxel | P-gp | Elacridar | [63] | |
| Doxorubicin | BCRP | Lapatinib | [64] | |
| Doxorubicin | BCRP | Pluronic L61 | [65] | |
| Gefitinib | BCRP | Fumitremorgin C | [66] | |
| Paclitaxel | BCRP | Sitravatinib | [67] | |
| Paclitaxel | BCRP | Lapatinib | [68] | |
| Lung | Paclitaxel | P-gp | Pluronic P123/F127 | [69] |
| Gefitinib | P-gp | Cyclosporine A | [70] | |
| Ovarian | Paclitaxel | P-gp | Tariquidar | [71] |
| Paclitaxel | P-gp, MRP1 | Curcumin | [72] | |
| Brain | Vincristine | MRP1 | Reversan | [73] |
| PF-2545920 | MRP1 | Reversan | [74] |
| Physiological Function | Mechanisms of Action | References |
|---|---|---|
| Cell proliferation | Phosphorylation of proteins in the ERK and AKT pathways | [91,97] |
| Phosphorylation of proteins in the JNK/SAPK pathway | ||
| Phosphorylation of proteins in the P38K pathway | ||
| Anti-apoptotic effect | Inactivation of caspases 1–4 and 6–8 | [79,95,97] |
| Inactivates p53 protein by inducing GC to AT mutation | ||
| Activation of cyclooxygenase-2 | ||
| Induction of Hsp 70 and Hsp 32 | ||
| Inhibition of ceramide production | ||
| Cell survival | PI3K/AKT pathway upregulation | [83,91,97,98] |
| β-Catenin downregulation | ||
| Angiogenesis | Stabilization of HIF1a | [91,92,93,95,97,99] |
| Induction of VEGF secretion | ||
| Downregulation of Angiostatin and Thrombospondin-1 | ||
| Induction of interleukin-8 secretion | ||
| Migration and invasion | Upregulation of α2β1 integrin | [97,100] |
| Upregulation of matrix metalloproteinases (MMPs) |
| Physiological Function | Mechanisms of Action | References |
|---|---|---|
| Apoptosis | Upregulation of tumor-suppressor p53 protein | [101,102,103,104,105] |
| Induction of the BCL-2 regulated apoptotic pathway | ||
| Release of cytochrome C | ||
| Induction of proteosomal degradation of anti-apoptotic proteins | ||
| Anti-invasion and Anti-metastatic effects | Downregulation of E-selectine | [97,107,108] |
| Inhibition of platelet aggregation | ||
| Inhibition of the NF-κB/Snail/YY1/Raf-1 kinase inhibitor protein (RKIP) circuitry |
| Class of NO Donor | Type | NO-Producing Mechanism | References |
|---|---|---|---|
| Organic nitrates | Glyceryl trinitrate (nitroglycerin) Isosorbide dinitrate Nicorandi Nipradilol Pentaerythrityl tetranitrate | Can be metabolized by specific enzymes such as mitochondrial aldehyde dehydrogenase (mtADH) | [118,119,120] |
| N-Diazeniumdiolates (NONOates) | PYPRO/NO DEA/NO DETA/NO PROLI/NO JS-K | Produces two NO molecules; release of NO occurs spontaneously and does not require specific metabolism | [118,119,120] |
| S-Nitrosothiols (RSNO) | S-nitrosoglutathione S-nitroso-N-acetylpenicillamine S-nitroso-N-valerylpenicillamine | S-NO bond cleavage promotes release of NO | [118,119,120] |
| Nitrobenzenes | PhNO2 -1 PhNO2 -2 | Nitro groups are converted into nitroso groups by light irradiation, leading to cleavage of oxygen and nitrogen bonds to produce NO | [118,120,121] |
| Furoxans | Furoxans-1 Furoxans-2 Furoxans-3 | NO produced in the presence of sulfhydryl compounds | [118,122] |
| Metal nitrosyl compounds | Ru-NO Fe-NO Sodium nitroprusside dihydrate | Photoelectron release from the π orbital of the metal ion to the π antibonding orbital of NO is promoted by light irradiation. As a result, NO is released rapidly from metal nitrosyl compounds | [118,123,124] |
| Cancer Cell Type | Anti-Cancer Drug | Delivery Method | Reference |
|---|---|---|---|
| Colon | Doxorubicin | KB-C2 cells were treated with GL (100 μM) and doxorubicin (5 ng/mL) | [146] |
| Aconitine | Oral administration of GL (50 mg/kg) and aconitine (0.2 mg/kg) | [137] | |
| Asiatic acid | Oral administration of GL (100 mg/kg) and asiatic acid (20 mg/kg) | [138] | |
| Liver | Cisplatin | Hepatocellular carcinoma cells were treated with GL (100 μg/mL) and cisplatin (10 ng/mL) | [145] |
| Doxorubicin | Alginate nanogel particle | [147] | |
| Polymeric prodrug micellar carrier based on polyethylene glycol-derivatized GA | [148] | ||
| Entecavir | Albumin nanoparticle | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Park, S.C.; Lee, D.Y. Glycyrrhizin as a Nitric Oxide Regulator in Cancer Chemotherapy. Cancers 2021, 13, 5762. https://doi.org/10.3390/cancers13225762
Kim M, Park SC, Lee DY. Glycyrrhizin as a Nitric Oxide Regulator in Cancer Chemotherapy. Cancers. 2021; 13(22):5762. https://doi.org/10.3390/cancers13225762
Chicago/Turabian StyleKim, Minsu, Seok Chan Park, and Dong Yun Lee. 2021. "Glycyrrhizin as a Nitric Oxide Regulator in Cancer Chemotherapy" Cancers 13, no. 22: 5762. https://doi.org/10.3390/cancers13225762
APA StyleKim, M., Park, S. C., & Lee, D. Y. (2021). Glycyrrhizin as a Nitric Oxide Regulator in Cancer Chemotherapy. Cancers, 13(22), 5762. https://doi.org/10.3390/cancers13225762