
Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives

Abstract
Simple Summary
Abstract
1. Introduction
2. FLT3 Mutations in AML
2.1. General Aspects of FLT3 Mutations and Treatment
2.2. Therapeutic Implications of Distinct FLT3 Mutations
2.3. Maintenance Treatment in FLT3 Mutated AML beyond Midostaurin
2.4. Treatment of Relapsed AML with FLT3 Mutations
2.5. FLT3 Mutations in AML Patients Not Eligible for Intensive Treatment
3. Inhibitors of IDH1 and IDH2
4. Epigenetic Treatment of AML
The Role of Venetoclax in HMA-Based Treatment
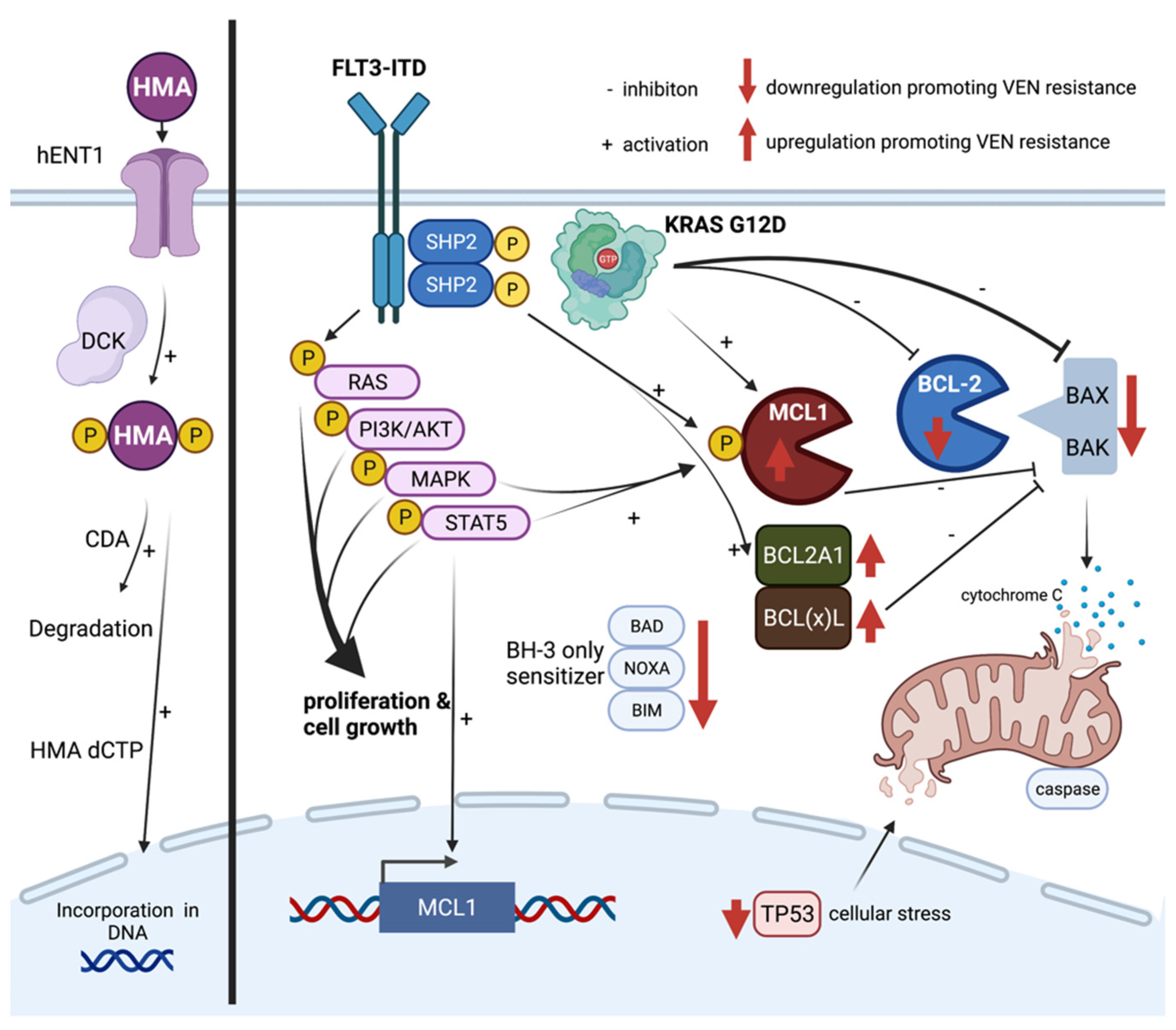
5. Mechanisms of Resistance towards HMA and Venetoclax
5.1. Hypomethylating Agents (HMAs)
5.2. Venetoclax
6. Potential Strategies to Overcome Venetoclax Resistance in AML
7. Inhibition of Hedgehog, Menin, or MDM2
7.1. Glasdegib
7.2. Menin
7.3. MDM2
8. Other Targeted Therapies of AML
9. Immunotherapy of AML
9.1. Gemtuzumab Ozogamicin
9.2. BiTEs and Bispecific Antibodies
9.3. CD33
9.4. CD123
9.5. FLT3 and CLL-1
9.6. TIM-3-Directed Treatment
9.7. Checkpoint Inhibitors
9.8. CAR-T Cell Approaches in AML
9.9. Novel Promising Targets: CD47 and CD70
9.9.1. CD47
9.9.2. CD70
10. Effective Targeting of Leukemic Stem Cells
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Bhatt, V.; Bixby, D.; Blum, W.; Coutre, S.E.; et al. Acute Myeloid Leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2017, 15, 926–957. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Voso, M.T.; Larson, R.A.; Jones, D.; Marcucci, G.; Prior, T.; Krauter, J.; Heuser, M.; Lavorgna, S.; Nomdedeu, J.; Geyer, S.M.; et al. Midostaurin in patients with acute myeloid leukemia and FLT3-TKD mutations: A subanalysis from the RATIFY trial. Blood Adv.. 2020, 4, 4945–4954. [Google Scholar] [CrossRef]
- Bornhauser, M.; Illmer, T.; Schaich, M.; Soucek, S.; Ehninger, G.; Thiede, C. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood 2007, 109, 2264–2265. [Google Scholar] [CrossRef]
- Oran, B.; Cortes, J.; Beitinjaneh, A.; Chen, H.C.; de Lima, M.; Patel, K.; Ravandi, F.; Wang, X.; Brandt, M.; Andersson, B.S.; et al. Allogeneic Transplantation in First Remission Improves Outcomes Irrespective of FLT3-ITD Allelic Ratio in FLT3-ITD-Positive Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. 2016, 22, 1218–1226. [Google Scholar] [CrossRef]
- Breitenbuecher, F.; Schnittger, S.; Grundler, R.; Markova, B.; Carius, B.; Brecht, A.; Duyster, J.; Haferlach, T.; Huber, C.; Fischer, T. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood 2009, 113, 4074–4077. [Google Scholar] [CrossRef]
- Kayser, S.; Schlenk, R.F.; Londono, M.C.; Breitenbuecher, F.; Wittke, K.; Du, J.; Groner, S.; Spath, D.; Krauter, J.; Ganser, A.; et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009, 114, 2386–2392. [Google Scholar] [CrossRef]
- Breitenbuecher, F.; Markova, B.; Kasper, S.; Carius, B.; Stauder, T.; Böhmer, F.D.; Masson, K.; Rönnstrand, L.; Huber, C.; Kindler, T.; et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood 2009, 113, 4063–4073. [Google Scholar] [CrossRef]
- Rucker, F.G.; Du, L.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3-Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Xuan, L.; Wang, Y.; Huang, F.; Fan, Z.; Xu, Y.; Sun, J.; Xu, N.; Deng, L.; Li, X.; Liang, X.; et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: An open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020, 21, 1201–1212. [Google Scholar] [CrossRef]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Müller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018, 24, 282–291. [Google Scholar] [CrossRef]
- Shih, L.Y.; Huang, C.F.; Wu, J.H.; Lin, T.L.; Dunn, P.; Wang, P.N.; Kuo, M.C.; Lai, C.L.; Hsu, H.C. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: A comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood 2002, 100, 2387–2392. [Google Scholar] [CrossRef]
- Kottaridis, P.D.; Gale, R.E.; Langabeer, S.E.; Frew, M.E.; Bowen, D.T.; Linch, D.C. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: Implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood 2002, 100, 2393–2398. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Kramer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Hosono, N.; Montesinos, P.; Podoltsev, N.A.; Martinelli, G.; Smith, C.C.; Levis, M.; Röllig, C.; Groß-Langenhoff, M.; et al. Clinical Outcomes in Patients with Relapsed/Refractory Acute Myeloid Leukemia Treated with Gilteritinib Who Received Prior Midostaurin or Sorafenib. Blood 2020, 136, 22–23. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Aldoss, I.; Zhang, J.; Mei, M.; Al Malki, M.M.; Arslan, S.; Ngo, D.; Aribi, A.; Ali, H.; Sandhu, K.; Salhotra, A.; et al. Venetoclax and hypomethylating agents in FLT3-mutated acute myeloid leukemia. Am. J. Hematol. 2020, 95, 1193–1199. [Google Scholar] [CrossRef]
- Mali, R.S.; Zhang, Q.; DeFilippis, R.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2020. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.M.; Muftuoglu, M.; Kadia, T.M.; Konopleva, M.; Borthakur, G.; Dinardo, C.D.; Pemmaraju, N.; Short, N.J.; Alvarado, Y.; et al. Quizartinib with decitabine and venetoclax (triplet) is highly active in patients with FLT3-ITD mutated acute myeloid leukemia (AML). J. Clin. Oncol. 2021, 39, e19019. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Ravandi, F.; Agresta, S.; Konopleva, M.; Takahashi, K.; Kadia, T.; Routbort, M.; Patel, K.P.; Mark, B.; Pierce, S.; et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am. J. Hematol. 2015, 90, 732–736. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Ward, P.S.; Lu, C.; Cross, J.R.; Abdel-Wahab, O.; Levine, R.L.; Schwartz, G.K.; Thompson, C.B. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 2013, 288, 3804–3815. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Dunlap, J.B.; Leonard, J.; Rosenberg, M.; Cook, R.; Press, R.; Fan, G.; Raess, P.W.; Druker, B.J.; Traer, E. The combination of NPM1, DNMT3A, and IDH1/2 mutations leads to inferior overall survival in AML. Am. J. Hematol. 2019, 94, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; Investigators, A.C.S. Differentiation Syndrome Associated With Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Fathi, A.T. Targeting IDH Mutations in AML: Wielding the Double-edged Sword of Differentiation. Curr. Cancer Drug Targets 2020, 20, 490–500. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.; Almstedt, M.; Lubbert, M. Epigenetic treatment of hematopoietic malignancies: In vivo targets of demethylating agents. Semin. Oncol. 2005, 32, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.P.; Chou, W.C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Kubasch, A.S.; Platzbecker, U. Beyond the Edge of Hypomethylating Agents: Novel Combination Strategies for Older Adults with Advanced MDS and AML. Cancers 2018, 10, 158. [Google Scholar] [CrossRef]
- Wei, A.H.; Dohner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef]
- Laille, E.; Shi, T.; Garcia-Manero, G.; Cogle, C.R.; Gore, S.D.; Hetzer, J.; Kumar, K.; Skikne, B.; MacBeth, K.J. Pharmacokinetics and Pharmacodynamics with Extended Dosing of CC-486 in Patients with Hematologic Malignancies. PLoS ONE 2015, 10, e0135520. [Google Scholar] [CrossRef]
- Oliva, E.N. CC-486 Reduces Hospitalization and Associated Estimated Costs in Patients with Acute Myeloid Leukemia (AML) in First Remission after Intensive Chemotherapy: Results from the QUAZAR AML-001 Maintenance Trial. Blood 2020, 126, 0006-4971. [Google Scholar] [CrossRef]
- de Lima, M.; Oran, B.; Champlin, R.E.; Papadopoulos, E.B.; Giralt, S.A.; Scott, B.L.; William, B.M.; Hetzer, J.; Laille, E.; Hubbell, B.; et al. CC-486 Maintenance after Stem Cell Transplantation in Patients with Acute Myeloid Leukemia or Myelodysplastic Syndromes. Biol. Blood Marrow Transplant. 2018, 24, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.; Czibere, A.; Platzbecker, U.; Bug, G.; Uharek, L.; Luft, T.; Giagounidis, A.; Zohren, F.; Bruns, I.; Wolschke, C.; et al. Azacitidine and donor lymphocyte infusions as first salvage therapy for relapse of AML or MDS after allogeneic stem cell transplantation. Leukemia 2013, 27, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.; Rautenberg, C. Treatment of MDS, AML and CMML Relapse after Allogeneic Blood Stem Cell Transplantation with Azacitidine, Lenalidomide and Donor Lymphocyte Infusions Results from the Second Interim Analysis of the Prospective Azalena-Trial (NCT02472691). Blood 2018, 132, 703. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- Aldoss, I.; Yang, D.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Mei, M.; Salhotra, A.; Khaled, S.; Nakamura, R.; et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica 2018, 103, e404–e407. [Google Scholar] [CrossRef]
- Morsia, E.; McCullough, K.; Joshi, M.; Cook, J.; Alkhateeb, H.B.; Al-Kali, A.; Begna, K.; Elliott, M.; Hogan, W.; Litzow, M.; et al. Venetoclax and hypomethylating agents in acute myeloid leukemia: Mayo Clinic series on 86 patients. Am. J. Hematol. 2020, 95, 1511–1521. [Google Scholar] [CrossRef]
- Tenold, M.E.; Moskoff, B.N.; Benjamin, D.J.; Hoeg, R.T.; Rosenberg, A.S.; Abedi, M.; Tuscano, J.M.; Jonas, B.A. Outcomes of Adults With Relapsed/Refractory Acute Myeloid Leukemia Treated With Venetoclax Plus Hypomethylating Agents at a Comprehensive Cancer Center. Front Oncol. 2021, 11, 649209. [Google Scholar] [CrossRef] [PubMed]
- Piccini, M.; Pilerci, S.; Merlini, M.; Grieco, P.; Scappini, B.; Bencini, S.; Peruzzi, B.; Caporale, R.; Signori, L.; Pancani, F.; et al. Venetoclax-Based Regimens for Relapsed/Refractory Acute Myeloid Leukemia in a Real-Life Setting: A Retrospective Single-Center Experience. J. Clin. Med. 2021, 10, 1684. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Rausch, C.R.; Benton, C.; Kadia, T.; Jain, N.; Pemmaraju, N.; Daver, N.; Covert, W.; Marx, K.R.; Mace, M.; et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol. 2018, 93, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Schuler, E.; Wagner-Drouet, E.M.; Ajib, S.; Bug, G.; Crysandt, M.; Dressler, S.; Hausmann, A.; Heidenreich, D.; Hirschbuhl, K.; Hoepting, M.; et al. Treatment of myeloid malignancies relapsing after allogeneic hematopoietic stem cell transplantation with venetoclax and hypomethylating agents-a retrospective multicenter analysis on behalf of the German Cooperative Transplant Study Group. Ann. Hematol. 2021, 100, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.; Danielson, N.; Sengsayadeth, S.; Rasche, A.; Culos, K.; Gatwood, K.; Wyatt, H.; Chinratanalab, W.; Dholaria, B.; Ferrell, P.B.; et al. The use of venetoclax-based salvage therapy for post-hematopoietic cell transplantation relapse of acute myeloid leukemia. Am. J. Hematol. 2020, 95, 1006–1014. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Dadwal, S.; Yang, D.; Mei, M.; Palmer, J.; Salhotra, A.; Al Malki, M.; Aribi, A.; Ali, H.; Khaled, S.; et al. Outcome of Allogeneic Hematopoietic Cell Transplantation after Venetoclax and Hypomethylating Agent Therapy for Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. 2020, 26, e322–e327. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Xiao, L.; Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax Combined With FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2021, 39, 2768–2778. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C., Jr.; Ferreira-Gonzalez, A.; Grant, S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef]
- Perl, A.D.N.; Pratz, K.; Dilley, K. Venetoclax in Combination with Gilteritinib in Patients with Relapsed/Refractory Acute Myeloid Leukemia: A Phase 1b Study. Blood 2019, 134, 3910. [Google Scholar] [CrossRef]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.P. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; D’Alo, F.; Zardo, G.; Voso, M.T.; Nervi, C. Epigenetic treatment of myelodysplastic syndromes and acute myeloid leukemias. Curr. Med. Chem. 2008, 15, 1274–1287. [Google Scholar] [CrossRef]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Hummel-Eisenbeiss, J.; Hascher, A.; Hals, P.A.; Sandvold, M.L.; Muller-Tidow, C.; Lyko, F.; Rius, M. The role of human equilibrative nucleoside transporter 1 on the cellular transport of the DNA methyltransferase inhibitors 5-azacytidine and CP-4200 in human leukemia cells. Mol. Pharmacol. 2013, 84, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Geng, S.; Weng, J.; Deng, C.; Lu, Z.; Luo, C.; Du, X. The hENT1 and DCK genes underlie the decitabine response in patients with myelodysplastic syndrome. Leuk Res. 2015, 39, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Zauri, M.; Berridge, G.; Thezenas, M.L.; Pugh, K.M.; Goldin, R.; Kessler, B.M.; Kriaucionis, S. CDA directs metabolism of epigenetic nucleosides revealing a therapeutic window in cancer. Nature 2015, 524, 114–118. [Google Scholar] [CrossRef]
- Coombs, C.C.; Sallman, D.A.; Devlin, S.M.; Dixit, S.; Mohanty, A.; Knapp, K.; Al Ali, N.H.; Lancet, J.E.; List, A.F.; Komrokji, R.S.; et al. Mutational correlates of response to hypomethylating agent therapy in acute myeloid leukemia. Haematologica 2016, 101, e457–e460. [Google Scholar] [CrossRef]
- Cedena, M.T.; Rapado, I.; Santos-Lozano, A.; Ayala, R.; Onecha, E.; Abaigar, M.; Such, E.; Ramos, F.; Cervera, J.; Diez-Campelo, M.; et al. Mutations in the DNA methylation pathway and number of driver mutations predict response to azacitidine in myelodysplastic syndromes. Oncotarget 2017, 8, 106948–106961. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef]
- Craddock, C.F.; Houlton, A.E.; Quek, L.S.; Ferguson, P.; Gbandi, E.; Roberts, C.; Metzner, M.; Garcia-Martin, N.; Kennedy, A.; Hamblin, A.; et al. Outcome of Azacitidine Therapy in Acute Myeloid Leukemia Is not Improved by Concurrent Vorinostat Therapy but Is Predicted by a Diagnostic Molecular Signature. Clin. Cancer Res. 2017, 23, 6430–6440. [Google Scholar] [CrossRef] [PubMed]
- Stasik, S.; Eckardt, J.N.; Kramer, M.; Rollig, C.; Kramer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brummendorf, T.H.; Naumann, R.; et al. Impact of PTPN11 mutations on clinical outcome analyzed in 1529 patients with acute myeloid leukemia. Blood Adv. 2021, 5, 3279–3289. [Google Scholar] [CrossRef]
- Middeke, J.M.; Teipel, R.; Rollig, C.; Stasik, S.; Zebisch, A.; Sill, H.; Kramer, M.; Scholl, S.; Hochhaus, A.; Jost, E.; et al. Decitabine treatment in 311 patients with acute myeloid leukemia: Outcome and impact of TP53 mutations—A registry based analysis. Leuk Lymphoma 2021, 62, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Leverson, J.D.; Sampath, D.; Souers, A.J.; Rosenberg, S.H.; Fairbrother, W.J.; Amiot, M.; Konopleva, M.; Letai, A. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov. 2017, 7, 1376–1393. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nakauchi, Y.; Kohnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, Q.; Dail, M.; Shi, C.; Cavazos, A.; Ruvolo, V.R.; Zhao, Y.; Kim, E.; Rahmani, M.; Mak, D.H.; et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica 2020, 105, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Mohi, M.G.; Neel, B.G. The role of Shp2 (PTPN11) in cancer. Curr. Opin. Genet. Dev. 2007, 17, 23–30. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hahlen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef]
- Nabinger, S.C.; Li, X.J.; Ramdas, B.; He, Y.; Zhang, X.; Zeng, L.; Richine, B.; Bowling, J.D.; Fukuda, S.; Goenka, S.; et al. The protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyperproliferation and malignant disease in vivo. Leukemia 2013, 27, 398–408. [Google Scholar] [CrossRef][Green Version]
- Chen, L.; Chen, W.; Mysliwski, M.; Serio, J.; Ropa, J.; Abulwerdi, F.A.; Chan, R.J.; Patel, J.P.; Tallman, M.S.; Paietta, E.; et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia 2015, 29, 1290–1300. [Google Scholar] [CrossRef]
- Yang, Z.; Li, Y.; Yin, F.; Chan, R.J. Activating PTPN11 mutants promote hematopoietic progenitor cell-cycle progression and survival. Exp. Hematol. 2008, 36, 1285–1296. [Google Scholar] [CrossRef]
- Zhu, X.Z.; Yu, Y.Z.; Fang, Y.M.; Liang, Y.; Lu, Q.H.; Xu, R.Z. Overexpression of Shp-2 is associated with the unlimited growth and apoptosis resistance of p210 bcr-abl-mediated chronic myeloid leukemia. Zhonghua Yi Xue Za Zhi 2005, 85, 1903–1906. [Google Scholar]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic Biomarkers Of Sensitivity and Resistance to Venetoclax Monotherapy in Patients With Relapsed Acute Myeloid Leukemia. Am. J. Hematol. 2018. [Google Scholar] [CrossRef]
- Li, L.; Modi, H.; McDonald, T.; Rossi, J.; Yee, J.K.; Bhatia, R. A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood 2011, 118, 1504–1515. [Google Scholar] [CrossRef]
- Singh Mali, R.L.E.A. FLT3-ITD Activation Mediates Resistance to the BCL-2 Selective Antagonist, Venetoclax, in FLT3-ITD Mutant AML Models. Blood 2017, 130, 1348. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol. Life. Sci. 2009, 66, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, D.A.; Niu, X.; Pan, Y.; Zhao, J.; Liu, S.; Edwards, H.; Chen, K.; Lin, H.; Taub, J.W.; Ge, Y. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct. Target Ther. 2017, 2, 17012. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Li, Z.; He, S.; Look, A.T. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 2019, 33, 262–266. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Wei, A.H.; Huang, D.C. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood 2021, 138, 1120–1136. [Google Scholar] [CrossRef]
- Kasper, S.; Breitenbuecher, F.; Heidel, F.; Hoffarth, S.; Markova, B.; Schuler, M.; Fischer, T. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012, 2, e60. [Google Scholar] [CrossRef]
- Chen, J.; Jin, S.; Abraham, V.; Huang, X.; Liu, B.; Mitten, M.J.; Nimmer, P.; Lin, X.; Smith, M.; Shen, Y.; et al. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Edwards, H.; Taub, J.W.; Ge, Y. Evaluating venetoclax and its potential in treatment-naive acute myeloid leukemia. Cancer Manag. Res. 2019, 11, 3197–3213. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef] [PubMed]
- Bogenberger, J.; Whatcott, C.; Hansen, N.; Delman, D.; Shi, C.X.; Kim, W.; Haws, H.; Soh, K.; Lee, Y.S.; Peterson, P.; et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017, 8, 107206–107222. [Google Scholar] [CrossRef]
- Daver, N.G.; Pollyea, D.A.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.; Fenaux, P.; Assouline, S.; Vey, N.; Olin, R.; Roboz, G.J.; et al. Safety, Efficacy, Pharmacokinetic (PK) and Biomarker Analyses of BCL2 Inhibitor Venetoclax (Ven) Plus MDM2 Inhibitor Idasanutlin (idasa) in Patients (pts) with Relapsed or Refractory (R/R) AML: A Phase Ib, Non-Randomized, Open-Label Study. Blood 2018, 132, 767. [Google Scholar] [CrossRef]
- Lainez-González, D.; Serrano-López, J.; Alonso-Domínguez, J.M. Understanding the Hedgehog Signaling Pathway in Acute Myeloid Leukemia Stem Cells: A Necessary Step toward a Cure. Biology 2021, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Sun, Z.; Ding, B.; Jiang, X.; Wang, Z.; Zhu, Y.; Meng, F. Suppressing Hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia. Onco Targets Ther. 2019, 12, 7477–7488. [Google Scholar] [CrossRef] [PubMed]
- Tibes, R.; Al-Kali, A.; Oliver, G.R.; Delman, D.H.; Hansen, N.; Bhagavatula, K.; Mohan, J.; Rakhshan, F.; Wood, T.; Foran, J.M.; et al. The Hedgehog pathway as targetable vulnerability with 5-azacytidine in myelodysplastic syndrome and acute myeloid leukemia. J Hematol. Oncol. 2015, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef]
- Schoch, C.; Schnittger, S.; Klaus, M.; Kern, W.; Hiddemann, W.; Haferlach, T. AML with 11q23/MLL abnormalities as defined by the WHO classification: Incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003, 102, 2395–2402. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Ding, L.W.; Tan, K.T.; Chien, W.; Mayakonda, A.; Lin, D.C.; Loh, X.Y.; Xiao, J.F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [Google Scholar] [CrossRef]
- Matkar, S.S.; Thiel, A.T.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402. [Google Scholar] [CrossRef]
- Wang, E.S.; Altman, J.K.; Pettit, K.; De Botton, S.; Walter, R.P.; Fenaux, P.; Burrows, F.; Tomkinson, B.E.; Martell, B.; Fathi, A.T. Preliminary Data on a Phase 1/2A First in Human Study of the Menin-KMT2A (MLL) Inhibitor KO-539 in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Blood 2020, 136, 7–8. [Google Scholar] [CrossRef]
- McGeehan, J. A first-in-class Menin-MLL1 antagonist for the treatment of MLL-r and NPM1 mutant leukemias. In Proceedings of the AACR Annual Meeting, Los Angeles, CA, USA, 22–24 June 2020. [Google Scholar]
- Morgado, E.; Albouhair, S.p.; Lavau, C. Flt3 is dispensable to the Hoxa9/Meis1 leukemogenic cooperation. Blood 2007, 109, 4020–4022. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dzama, M.M.; Steiner, M.; Rausch, J.; Sasca, D.; Schönfeld, J.; Kunz, K.; Taubert, M.C.; McGeehan, G.M.; Chen, C.W.; Mupo, A.; et al. Synergistic Targeting of FLT3 Mutations in AML via Combined Menin-MLL and FLT3 Inhibition. Blood 2020, 136, 2442–2456. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Garcia, J.S.; Jonas, B.A.; Kelly, K.R.; Assouline, S.; Brandwein, J.M.; Fenaux, P.; Olin, R.L.; Martinelli, G.; Paolini, S.; et al. Updated Results from the Venetoclax (Ven) in Combination with Idasanutlin (Idasa) Arm of a Phase 1b Trial in Elderly Patients (Pts) with Relapsed or Refractory (R/R) AML Ineligible for Cytotoxic Chemotherapy. Blood 2019, 134, 229. [Google Scholar] [CrossRef]
- Sallman, D.A.; Borate, U.; Cull, E.H.; Donnellan, W.B.; Komrokji, R.S.; Steidl, U.G.; Corvez, M.M.; Payton, M.; Annis, D.A.; Pinchasik, D.; et al. Phase 1/1b Study of the Stapled Peptide ALRN-6924, a Dual Inhibitor of MDMX and MDM2, As Monotherapy or in Combination with Cytarabine for the Treatment of Relapsed/Refractory AML and Advanced MDS with TP53 Wild-Type. Blood 2018, 132, 4066. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Ridinger, M.; Lin, T.L.; Becker, P.S.; Schiller, G.J.; Patel, P.A.; Spira, A.I.; Tsai, M.L.; Samuelsz, E.; Silberman, S.L.; et al. A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 6132–6140. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Menghrajani, K.; Cai, S.F.; Devlin, S.M.; Armstrong, S.A.; Piekartz, R.; Rudek, M.; Stein, E.M. A Phase Ib/II Study of the Histone Methyltransferase Inhibitor Pinometostat in Combination with Azacitidine in Patients with 11q23-Rearranged Acute Myeloid Leukemia. Blood 2019, 134, 2655. [Google Scholar] [CrossRef]
- Borthakur, G.; Odenike, O.; Aldoss, I.; Rizzieri, D.A.; Prebet, T.; Chen, C.; Popovic, R.; Modi, D.A.; Joshi, R.H.; Wolff, J.E.; et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, B.; Zhao, Q.; Mims, A.S.; Vasu, S.; Behbehani, G.K.; Larkin, K.; Blachly, J.S.; Blum, W.; Klisovic, R.B.; Ruppert, A.S.; et al. Selinexor in combination with decitabine in patients with acute myeloid leukemia: Results from a phase 1 study. Leuk Lymphoma 2020, 61, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Lee, D.J.; Frattini, M.; Fine, G.D.; Costas, J.; Kolibaba, K.; Anthony, S.P.; Bearss, D.; Smith, B.D. Phase I Study of Alvocidib Followed by 7+3 (Cytarabine + Daunorubicin) in Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef]
- Lamba, J.K.; Chauhan, L.; Shin, M.; Loken, M.R.; Pollard, J.A.; Wang, Y.C.; Ries, R.E.; Aplenc, R.; Hirsch, B.A.; Raimondi, S.C.; et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2017, 35, 2674–2682. [Google Scholar] [CrossRef]
- Gale, R.E.; Popa, T.; Wright, M.; Khan, N.; Freeman, S.D.; Burnett, A.K.; Russell, N.H.; Hills, R.K.; Linch, D.C. No evidence that CD33 splicing SNP impacts the response to GO in younger adults with AML treated on UK MRC/NCRI trials. Blood 2018, 131, 468–471. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Stasi, R.; Salih, H.R.; Selleslag, D.; Muus, P.; Fabritiis, P.D.; Venditti, A.; Ho, A.D.; Lübbert, M.; et al. Sequential Combination of Gemtuzumab Ozogamicin and Standard Chemotherapy in Older Patients With Newly Diagnosed Acute Myeloid Leukemia: Results of a Randomized Phase III Trial by the EORTC and GIMEMA Consortium (AML-17). J. Clin. Oncol. 2013, 31, 4424–4430. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Paschka, P.; Krzykalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T.; et al. Gemtuzumab Ozogamicin in NPM1-Mutated Acute Myeloid Leukemia: Early Results From the Prospective Randomized AMLSG 09-09 Phase III Study. J. Clin. Oncol. 2020, 38, 623–632. [Google Scholar] [CrossRef]
- Kapp-Schwoerer, S.; Weber, D.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Krönke, J.; Theis, F.; Rücker, F.G.; Teleanu, M.V.; Panina, E.; et al. Impact of gemtuzumab ozogamicin on MRD and relapse risk in patients with NPM1-mutated AML: Results from the AMLSG 09-09 trial. Blood 2020, 136, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Heidel, F.; Tenzer, S.; Radsak, M.P.; Solem, F.K.; Britten, C.M.; Huber, C.; Fischer, T.; Wölfel, T. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood 2007, 109, 2985–2988. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Brauchle, B.; Goldstein, R.L.; Karbowski, C.M.; Henn, A.; Li, C.-M.; Bücklein, V.L.; Krupka, C.; Boyle, M.C.; Koppikar, P.; Haubner, S.; et al. Characterization of a Novel FLT3 BiTE Molecule for the Treatment of Acute Myeloid Leukemia. Mol. Cancer Ther. 2020, 19, 1875–1888. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Böhmer, S.A.; Koch, S.; Müller, J.P.; Blei, L.; Cornils, H.; Bauer, R.; Korasikha, S.; Thiede, C.; Böhmer, F.D. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood 2009, 113, 3568–3576. [Google Scholar] [CrossRef]
- van Loo, P.F.; Hangalapura, B.N.; Thordardottir, S.; Gibbins, J.D.; Veninga, H.; Hendriks, L.J.A.; Kramer, A.; Roovers, R.C.; Leenders, M.; de Kruif, J.; et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opin. Biol. Ther. 2019, 19, 721–733. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Wang, M.; Zhang, L.; Yu, L. One Stone, Two Birds: The Roles of Tim-3 in Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 1098. [Google Scholar] [CrossRef]
- Gonçalves Silva, I.; Rüegg, L.; Gibbs, B.F.; Bardelli, M.; Fruehwirth, A.; Varani, L.; Berger, S.M.; Fasler-Kan, E.; Sumbayev, V.V. The immune receptor Tim-3 acts as a trafficker in a Tim-3/galectin-9 autocrine loop in human myeloid leukemia cells. Oncoimmunology 2016, 5, e1195535. [Google Scholar] [CrossRef]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem. Cell 2015, 17, 341–352. [Google Scholar] [CrossRef]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. Blood 2020, 136, 1–2. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Vereecque, R.; Saudemont, A.; Quesnel, B. Cytosine arabinoside induces costimulatory molecule expression in acute myeloid leukemia cells. Leukemia 2004, 18, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519.e505. [Google Scholar] [CrossRef]
- Liu, F.; Cao, Y.; Pinz, K.; Ma, Y.; Wada, M.; Chen, K.; Ma, G.; Shen, J.; Tse, C.O.; Su, Y.; et al. First-in-Human CLL1-CD33 Compound CAR T Cell Therapy Induces Complete Remission in Patients with Refractory Acute Myeloid Leukemia: Update on Phase 1 Clinical Trial. Blood 2018, 132, 901. [Google Scholar] [CrossRef]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Sallman, D.A.; Malki, M.A.; Asch, A.S.; Lee, D.J.; Kambhampati, S.; Donnellan, W.B.; Bradley, T.J.; Vyas, P.; Jeyakumar, D.; Marcucci, G.; et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: Phase Ib results. J. Clin. Oncol. 2020, 38, 7507. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Daver, N.G.; Xu, J.; Chao, M.; Chung, T.; Tan, A.; Wang, V.; Wei, A.; Vyas, P.; Sallman, D.A. Magrolimab + azacitidine versus azacitidine + placebo in untreated higher risk (HR) myelodysplastic syndrome (MDS): The phase 3, randomized, ENHANCE study. J. Clin. Oncol. 2021, 39, TPS7055. [Google Scholar] [CrossRef]
- Riether, C.; Schürch, C.M.; Bührer, E.D.; Hinterbrandner, M.; Huguenin, A.-L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J. Exp. Med. 2016, 214, 359–380. [Google Scholar] [CrossRef]
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nature Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef]
- Ochsenbein, A.F.; Pabst, T.; Höpner, S.; Bacher, V.U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.; Francisco, D.; et al. Targeting CD70 with Cusatuzumab Eliminates Acute Myeloid Leukemia Stem Cells in Humans. Blood 2019, 134, 234. [Google Scholar] [CrossRef]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Müller-Tidow, C.; et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef] [PubMed]
- van Gils, N.; Denkers, F.; Smit, L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front Oncol. 2021, 11, 659253. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef]
- Arnone, M.; Konantz, M.; Hanns, P.; Paczulla Stanger, A.M.; Bertels, S.; Godavarthy, P.S.; Christopeit, M.; Lengerke, C. Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity. Cancers 2020, 12, 3742. [Google Scholar] [CrossRef]
- Jung, N.; Dai, B.; Gentles, A.; Majeti, R.; Feinberg, A. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nature Commun. 2015, 6, 8489. [Google Scholar] [CrossRef]
- Ng, S.W.K.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Uy, G.L.; Rettig, M.P.; Stone, R.M.; Konopleva, M.Y.; Andreeff, M.; McFarland, K.; Shannon, W.; Fletcher, T.R.; Reineck, T.; Eades, W.; et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 2017, 7, e542. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Yu, C.; Liu, Y.; Hu, C.; Ma, R.; Lu, X.; Ji, P.; Chen, J.; Mizukawa, B.; Huang, Y.; et al. FOXM1 regulates leukemia stem cell quiescence and survival in MLL-rearranged AML. Nature Commun. 2020, 11, 928. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.M.; Xiang, M.; Heppler, L.N.; Tošić, I.; Jiang, K.; Munoz, J.O.; Gaikwad, A.S.; Horton, T.M.; Long, X.; Narayanan, P.; et al. Atovaquone is active against AML by upregulating the integrated stress pathway and suppressing oxidative phosphorylation. Blood Adv.. 2019, 3, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Munoz-Sagredo, L.; Streule, K.; Muschong, P.; Bayer, E.; Walter, R.J.; Gutjahr, J.C.; Greil, R.; Concha, M.L.; Muller-Tidow, C.; et al. CD44 loss of function sensitizes AML cells to the BCL-2 inhibitor venetoclax by decreasing CXCL12-driven survival cues. Blood 2021, 138, 1067–1080. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target Molecule | Target Function | Rationale | Compounds in Development | Clinical Trials | Ref. |
|---|---|---|---|---|---|
| Polo-like kinase 1 | G2->M entrance interaction with PI3K/mTOR | high expression of PLK1 in AML cells compared to CD34 progenitor cells | Onvansertib | NCT03303339: Phase 1b/2 study/45 pts. Onvansertib plus DEC or LDAC CR 9 pts. (20%), 4 pts with durable response of at least 9 months; 2 pts. proceeded to alloHSCT | [142] |
| TP53mutant | important tumor suppressor | poor prognosis of TP53mut AML restoration of TP53 function synergistic effect with AZA | APR-246 (Eprenetapopt) | NCT03072043: Phase 1b/2 study/55 pts., including 11 r/r AML pts. APR-246 plus AZA ORR 7 pts. (64%), CR 4 pts. (36%) | [143] |
| DOT1L | H3K79 methyltransferase | DOT1L plays a central role in leukemogenesis of AML with KMT2Ar AML | EPZ-5676 (Pinometostat) | Phase 1 study/43 pts. CR 2 pts., resolution of extramedullary AML 2 pts., signs of differentiation 9 pts. | [144] |
| BET | important epigenetic regulator | BRD4 overexpression in AML pivotal role in LSC transcriptional programs | Mivebresib (ABBV-075) pan-BET inhibitor | NCT02391480: Phase 1 study/44 pts. Mivebresib mono (MIV) or combination therapy with VEN (MIV-VEN) response MIV-VEN (30 pts.): CR 2 pts., PR 2 pts., MLFS 2 pts. | [145] |
| XPO1 | nuclear transport protein | high expression of XPO1 in LSC modulation of protein shuttling | Selinexor (KPT-330) | NCT02093403: Phase 1 study/25 pts. Selinexor plus DEC ORR 40%, median PFS 5.9 months PFS responders 11.8 months | [146] |
| CDK9 | regulation of RNA polymerase II activity | inhibition of transcription of genes involved in proliferation and survival (e.g., suppression of MCL-1) | Alvocidib (flavopiridol) | NCT03298984: Phase 1 study/32 pts. ND AML/Alvocidib -> 7 + 3 ORR 75%, CR 69% | [147] |
| Target Molecule | Expression LSC/HSC | Drug | Mechanism of Action | Treatment Schedule | First Results | Clinical Trial | Ref. |
|---|---|---|---|---|---|---|---|
| CD33 (Siglec-3) | +/+ | AMG330 | CD33xCD3 BiTE | continuous infusion D1-D28 2 weeks off | 55 pts. in 16 dose cohorts: AMG330-related AEs: 49/55 pts. (89%) CRS: 67%, grade ≥ 3: 13% Nausea: 11/55 (20%) Evaluable response in 42 pts. (17%) CRc: 7 pts. | NCT02520427 | [160] |
| AMG673 | CD33xCD3 BiTE half-life- extended | Infusion @D1 and D5 every 14 days | 30 pts. in 10 dose cohorts: CRS: 15/30 pts. (50%) TRSAE: 11/30 (37%) blast reduction > 50% in 6/27 pts.; CRi 1 pts. | NCT03224819 | [161] | ||
| CD123 (IL3Rα) | ++/(+) | Flotetuzumab (MGD006) | bispecific DART CD123xCD3 | two schedules: 7-day CIV 4 days on/3 days off CIV | 42 pts. dose-escalation/46 pts.@RP2D CRS: 81%, grade ≥ 3: 7% Nausea: 26% CRc @RP2D: 30% | NCT02152956 | [162] |
| Vibecotamab (XmAb14045) | Bispecific Antibody CD123xCD3 | weekly infusion | CRS 62/106 pts. (58%) no TLS ORR@>0.75 µg/kg 7/51 pts. (14%) CRc 5/51 pts. (10%) | NCT02730312 | [163] |
| Clinical Trial | Molecular Stratification | Study Population | Checkpoint Inhibitor | Combination Therapy | Trial Phase |
|---|---|---|---|---|---|
| NCT03730012 | FLT3 mutation | r/r AML | Atezolizumab | Gilteritinib | Phase 1/2 |
| NCT04044209 | IDH1 mutation | r/r AML | Nivolumab | Ivosidenib | Phase 2 |
| NCT04277442 | TP53 mutation | 1L AML | Nivolumab | Decitabine and Venetoclax | Phase 1 |
| NCT03066648 | none | r/r AML or 1L AML | PDR001 | Sabatolimab (MBG453) or DEC | Randomized Phase 1 |
| NCT03922477 | none | r/r AML | Atezolizumab | Magrolimab (Hu5F9-G4) | Phase 1 |
| NCT02890329 | none | r/r AML | Ipilimumab | Decitabine | Phase 1 |
| NCT02397720 | none | r/r AML or 1L AML | Nivolumab ± Ipilimumab | AZA | Non-randomized Phase 1 |
| NCT02464657 | none | 1L AML | Nivolumab | Idarubicin and Cytarabine | Phase 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleischmann, M.; Schnetzke, U.; Hochhaus, A.; Scholl, S. Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives. Cancers 2021, 13, 5722. https://doi.org/10.3390/cancers13225722
Fleischmann M, Schnetzke U, Hochhaus A, Scholl S. Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives. Cancers. 2021; 13(22):5722. https://doi.org/10.3390/cancers13225722
Chicago/Turabian StyleFleischmann, Maximilian, Ulf Schnetzke, Andreas Hochhaus, and Sebastian Scholl. 2021. "Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives" Cancers 13, no. 22: 5722. https://doi.org/10.3390/cancers13225722
APA StyleFleischmann, M., Schnetzke, U., Hochhaus, A., & Scholl, S. (2021). Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives. Cancers, 13(22), 5722. https://doi.org/10.3390/cancers13225722