
The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Metabolic Reprogramming in Cancers: The Warburg Effect
2.1. Warburg Effect, a Major Feature of Metabolic Reprogramming
2.2. Warburg Effect: Release of Lactate, a Fuel for Normoxic Cancer Cells
2.3. Lactates: A Fuel for Cancer Cells to Initiate Angiogenesis
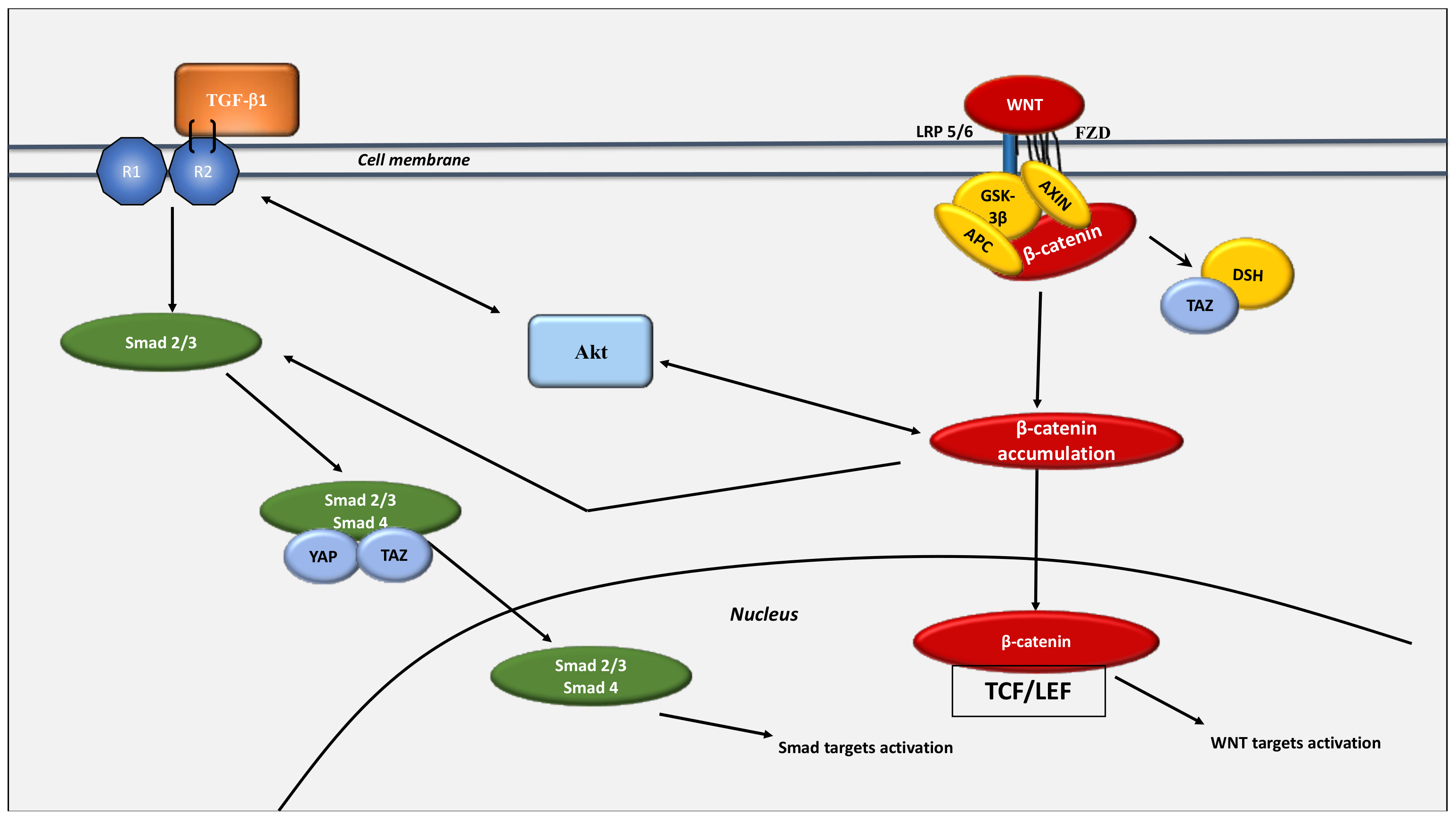
3. The Key Role of the WNT/β-Catenin Pathway in Cancer Development under Normoxic Conditions
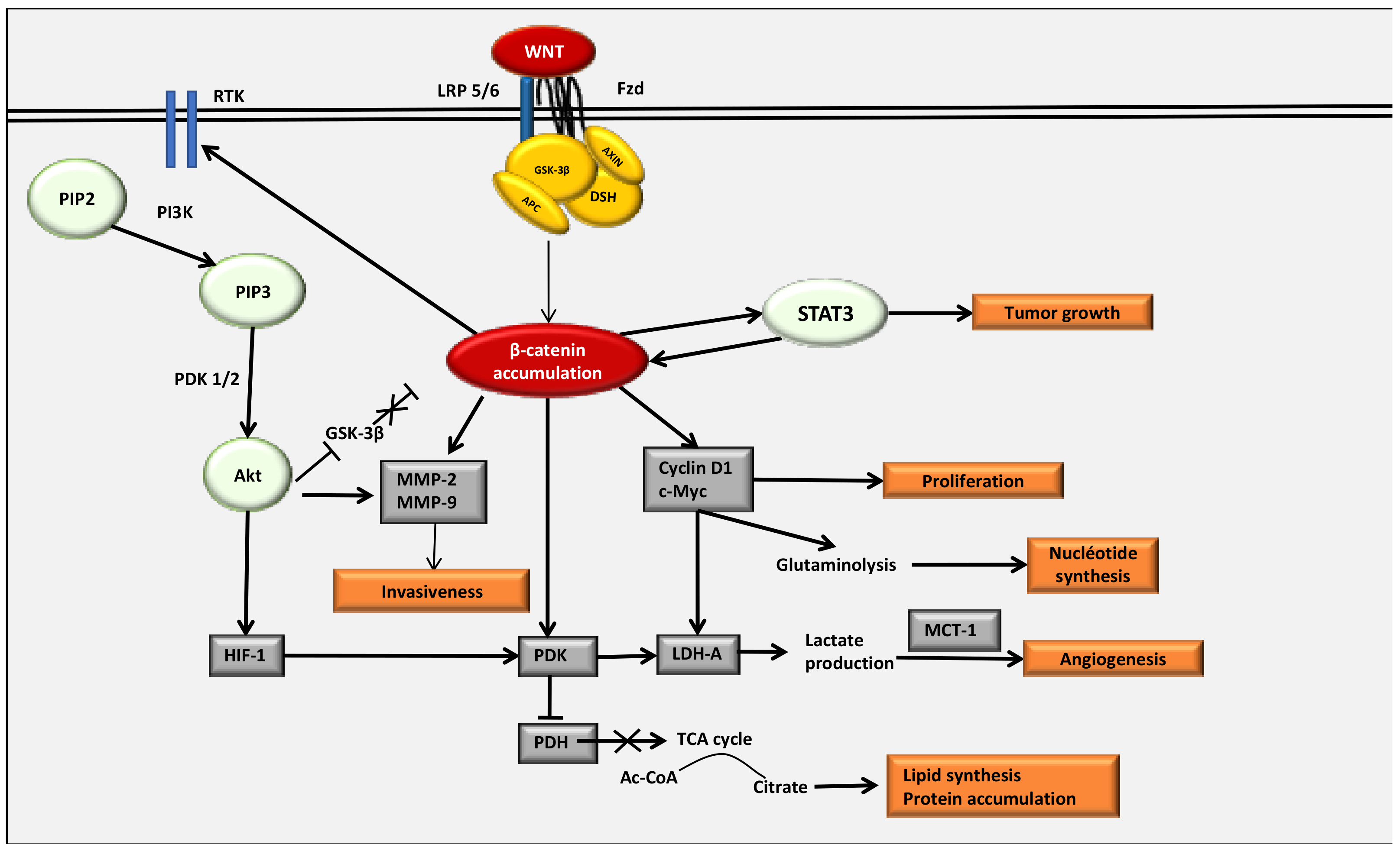
4. The WNT/β-Catenin Pathway Stimulates Cancer Metabolic Reprogramming under Normoxic Conditions
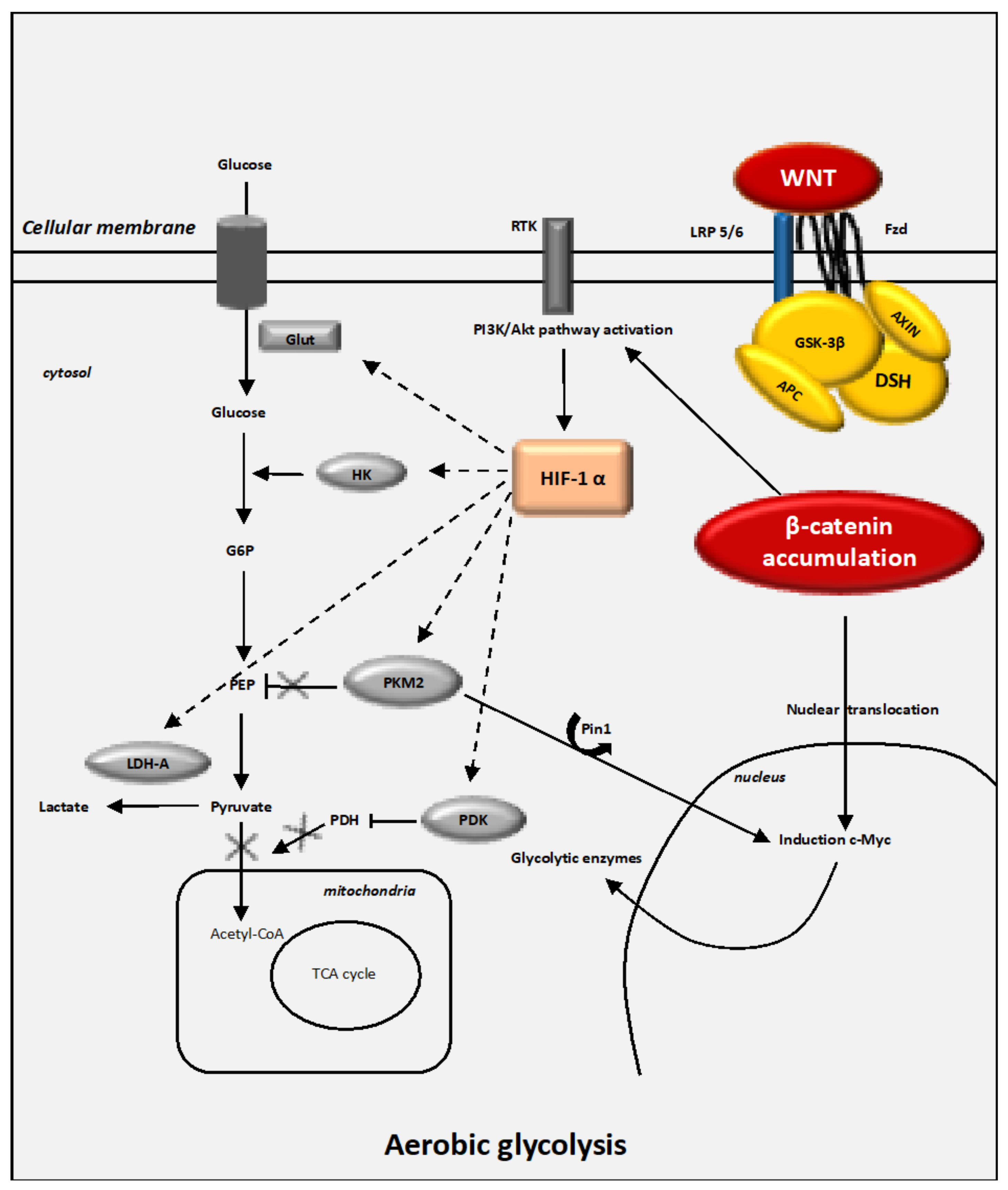
4.1. Interactions between the Canonical WNT/β-Catenin Pathway and the Warburg Effect
4.2. Interactions between the WNT/β-Catenin Pathway and STAT3 Pathway
4.3. Interaction between the WNT/β-Catenin Pathway and the PI3K/Akt Pathway
4.4. The Key Enzymes Involved in Metabolic Reprogramming in Cancer Cells
4.4.1. HIF-1α
4.4.2. Glut-1 and HK2
4.4.3. PKM2
4.4.4. PDK-1
4.4.5. LDH-A
5. Definition of the Autophagy Mechanism
6. Autophagy in Cancer
7. Interplay between Glycolysis and Autophagy in the Cancer Process
7.1. Key Role of the WNT/β-Catenin Pathway in Cancer Development through Interaction with Autophagy
7.2. The Key Role of the WNT/β-Catenin Pathway in Cancer Development through Interaction with Glutaminolysis
8. Interaction between the WNT/β-Catenin Pathway and the Hippo Pathway: The Link between Glycolysis and Glutaminolysis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| CK1 | casein kinase 1 |
| COX-2 | Cyclooxygenase-2 |
| FZD | Frizzled |
| GSK-3β | Glycogen synthase kinase-3β |
| LRP 5/6 | Low-density lipoprotein receptor-related protein 5/6 |
| MAPK | Mitogen-activated protein kinases |
| NF-κB | nuclear factor κB |
| NOX | NADPH oxidase |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PI3K-Akt | Phosphatidylinositol 3-kinase-protein kinase B |
| ROS | reactive oxygen species |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TNF-α | tumor necrosis factor alpha. |
References
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory Signal Suppresses Proliferation and Shifts Macrophage Metabolism from Myc-Dependent to HIF1α-Dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors: Coupling Glucose Metabolism and Redox Regulation with Induction of the Breast Cancer Stem Cell Phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M.K. Autophagy in Lung Disease Pathogenesis and Therapeutics. Redox Biol. 2015, 4, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Fu, H.; Gao, H.; Qi, X.; Zhao, L.; Wu, D.; Bai, Y.; Li, H.; Liu, X.; Hu, J.; Shao, S. Aldolase A Promotes Proliferation and G1/S Transition via the EGFR/MAPK Pathway in Non-Small Cell Lung Cancer. Cancer Commun. 2018, 38, 18. [Google Scholar] [CrossRef]
- Liu, T.; Yin, H. PDK1 Promotes Tumor Cell Proliferation and Migration by Enhancing the Warburg Effect in Non-Small Cell Lung Cancer. Oncol. Rep. 2017, 37, 193–200. [Google Scholar] [CrossRef]
- Gong, T.; Cui, L.; Wang, H.; Wang, H.; Han, N. Knockdown of KLF5 Suppresses Hypoxia-Induced Resistance to Cisplatin in NSCLC Cells by Regulating HIF-1α-Dependent Glycolysis through Inactivation of the PI3K/Akt/MTOR Pathway. J. Transl. Med. 2018, 16, 164. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.; Du, L.; Xi, P.; Wang, Q.; Li, Y.; Liu, D. MiR-449a Suppresses LDHA-Mediated Glycolysis to Enhance the Sensitivity of Non-Small Cell Lung Cancer Cells to Ionizing Radiation. Oncol. Res. 2018, 26, 547–556. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Arelaki, S.; Koukourakis, M.I. Expression of Enzymes Related to Glucose Metabolism in Non-Small Cell Lung Cancer and Prognosis. Exp. Lung Res. 2017, 43, 167–174. [Google Scholar] [CrossRef]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Claes, V.; Vallée, A.; Hébert, J.-L. Thermodynamics in Cancers: Opposing Interactions between PPAR Gamma and the Canonical WNT/Beta-Catenin Pathway. Clin. Transl. Med. 2017, 6, 14. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Opposite Interplay Between the Canonical WNT/β-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018, 34, 573–588. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef]
- Vallée, A.; Guillevin, R.; Vallée, J.-N. Vasculogenesis and Angiogenesis Initiation under Normoxic Conditions through Wnt/β-Catenin Pathway in Gliomas. Rev. Neurosci. 2018, 29, 71–91. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-ΚB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and Downstream of Cancer Metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Zhou, W.; Liotta, L.A.; Petricoin, E.F. The Warburg Effect and Mass Spectrometry-Based Proteomic Analysis. Cancer Genom. Proteom. 2017, 14, 211–218. [Google Scholar] [CrossRef][Green Version]
- Vaupel, P. Hypoxia in Neoplastic Tissue. Microvasc. Res. 1977, 13, 399–408. [Google Scholar] [CrossRef]
- Vaupel, P.; Fortmeyer, H.P.; Runkel, S.; Kallinowski, F. Blood Flow, Oxygen Consumption, and Tissue Oxygenation of Human Breast Cancer Xenografts in Nude Rats. Cancer Res. 1987, 47, 3496–3503. [Google Scholar]
- Vaupel, P.; Mayer, A. Tumor Oxygenation Status: Facts and Fallacies. Adv. Exp. Med. Biol. 2017, 977, 91–99. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Availability, Not Respiratory Capacity Governs Oxygen Consumption of Solid Tumors. Int. J. Biochem. Cell Biol. 2012, 44, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Ralph, S.J.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. HIF-1alpha Modulates Energy Metabolism in Cancer Cells by Inducing over-Expression of Specific Glycolytic Isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.-H.; Park, J. Targeting Cancer Metabolism-Revisiting the Warburg Effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg Effect in Tumor Progression: Mitochondrial Oxidative Metabolism as an Anti-Metastasis Mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and Cancer: The Good, the Bad and the Importance of Context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Liao, Y. Cancer Metabolism as We Know It Today: A Prologue to a Special Issue of Cancer Metabolism. Genes Dis. 2017, 4, 4–6. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-Mediated Tumor Invasion: A Multidisciplinary Study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate Promotes Glioma Migration by TGF-Beta2-Dependent Regulation of Matrix Metalloproteinase-2. Neuro-Oncology 2009, 11, 368–380. [Google Scholar] [CrossRef]
- Seliger, C.; Leukel, P.; Moeckel, S.; Jachnik, B.; Lottaz, C.; Kreutz, M.; Brawanski, A.; Proescholdt, M.; Bogdahn, U.; Bosserhoff, A.-K.; et al. Lactate-Modulated Induction of THBS-1 Activates Transforming Growth Factor (TGF)-Beta2 and Migration of Glioma Cells in Vitro. PLoS ONE 2013, 8, e78935. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-Derived Lactic Acid Modulates Dendritic Cell Activation and Antigen Expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic Enzymes Can Modulate Cellular Life Span. Cancer Res. 2005, 65, 177–185. [Google Scholar]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-ΚB/IL-8 Pathway That Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef]
- Constant, J.S.; Feng, J.J.; Zabel, D.D.; Yuan, H.; Suh, D.Y.; Scheuenstuhl, H.; Hunt, T.K.; Hussain, M.Z. Lactate Elicits Vascular Endothelial Growth Factor from Macrophages: A Possible Alternative to Hypoxia. Wound Repair Regen. Off. Publ. Wound Health Soc. Eur. Tissue Repair Soc. 2000, 8, 353–360. [Google Scholar] [CrossRef]
- D’Arcangelo, D.; Facchiano, F.; Barlucchi, L.M.; Melillo, G.; Illi, B.; Testolin, L.; Gaetano, C.; Capogrossi, M.C. Acidosis Inhibits Endothelial Cell Apoptosis and Function and Induces Basic Fibroblast Growth Factor and Vascular Endothelial Growth Factor Expression. Circ. Res. 2000, 86, 312–318. [Google Scholar] [CrossRef]
- Goerges, A.L.; Nugent, M.A. PH Regulates Vascular Endothelial Growth Factor Binding to Fibronectin: A Mechanism for Control of Extracellular Matrix Storage and Release. J. Biol. Chem. 2004, 279, 2307–2315. [Google Scholar] [CrossRef]
- Jensen, J.A.; Hunt, T.K.; Scheuenstuhl, H.; Banda, M.J. Effect of Lactate, Pyruvate, and PH on Secretion of Angiogenesis and Mitogenesis Factors by Macrophages. Lab. Investig. J. Tech. Methods Pathol. 1986, 54, 574–578. [Google Scholar]
- Sattler, U.G.A.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic Metabolism and Tumour Response to Fractionated Irradiation. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic Extracellular PH Induces Vascular Endothelial Growth Factor (VEGF) in Human Glioblastoma Cells via ERK1/2 MAPK Signaling Pathway: Mechanism of Low PH-Induced VEGF. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef]
- Zadeh, G.; Koushan, K.; Pillo, L.; Shannon, P.; Guha, A. Role of Ang1 and Its Interaction with VEGF-A in Astrocytomas. J. Neuropathol. Exp. Neurol. 2004, 63, 978–989. [Google Scholar] [CrossRef]
- Griffiths, J.R. Are Cancer Cells Acidic? Br. J. Cancer 1991, 64, 425–427. [Google Scholar] [CrossRef]
- Raghunand, N.; Gatenby, R.A.; Gillies, R.J. Microenvironmental and Cellular Consequences of Altered Blood Flow in Tumours. Br. J. Radiol. 2003, 76, S11–S22. [Google Scholar] [CrossRef] [PubMed]
- Wike-Hooley, J.L.; Haveman, J.; Reinhold, H.S. The Relevance of Tumour PH to the Treatment of Malignant Disease. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1984, 2, 343–366. [Google Scholar] [CrossRef]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of Vascular Endothelial Growth Factor Expression by Acidosis in Human Cancer Cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and Acidosis Independently Up-Regulate Vascular Endothelial Growth Factor Transcription in Brain Tumors in Vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Chang, L.K.; Garcia-Cardena, G.; Farnebo, F.; Fannon, M.; Chen, E.J.; Butterfield, C.; Moses, M.A.; Mulligan, R.C.; Folkman, J.; Kaipainen, A. Dose-Dependent Response of FGF-2 for Lymphangiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 11658–11663. [Google Scholar] [CrossRef] [PubMed]
- Eichten, A.; Hyun, W.C.; Coussens, L.M. Distinctive Features of Angiogenesis and Lymphangiogenesis Determine Their Functionality during de Novo Tumor Development. Cancer Res. 2007, 67, 5211–5220. [Google Scholar] [CrossRef]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically Derived Lactate Stimulates Revascularization and Tissue Repair via Redox Mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Lactate Stimulates Vasculogenic Stem Cells via the Thioredoxin System and Engages an Autocrine Activation Loop Involving Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 2008, 28, 6248–6261. [Google Scholar] [CrossRef]
- Haaga, J.R.; Haaga, R. Acidic Lactate Sequentially Induced Lymphogenesis, Phlebogenesis, and Arteriogenesis (ALPHA) Hypothesis: Lactate-Triggered Glycolytic Vasculogenesis That Occurs in Normoxia or Hypoxia and Complements the Traditional Concept of Hypoxia-Based Vasculogenesis. Surgery 2013, 154, 632–637. [Google Scholar] [CrossRef]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-Inducible Factor 1 Activation by Aerobic Glycolysis Implicates the Warburg Effect in Carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible Inactivation of HIF-1 Prolyl Hydroxylases Allows Cell Metabolism to Control Basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. Pyruvate Dehydrogenase Complex Activity Controls Metabolic and Malignant Phenotype in Cancer Cells. J. Biol. Chem. 2008, 283, 22700–22708. [Google Scholar] [CrossRef]
- Mekhail, K.; Khacho, M.; Carrigan, A.; Hache, R.R.J.; Gunaratnam, L.; Lee, S. Regulation of Ubiquitin Ligase Dynamics by the Nucleolus. J. Cell Biol. 2005, 170, 733–744. [Google Scholar] [CrossRef]
- Kaur, B.; Tan, C.; Brat, D.J.; Post, D.E.; Van Meir, E.G. Genetic and Hypoxic Regulation of Angiogenesis in Gliomas. J. Neurooncol. 2004, 70, 229–243. [Google Scholar] [CrossRef]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma: A Population-Based Study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Parliament, M.B.; Allalunis-Turner, M.J.; Franko, A.J.; Olive, P.L.; Mandyam, R.; Santos, C.; Wolokoff, B. Vascular Endothelial Growth Factor Expression Is Independent of Hypoxia in Human Malignant Glioma Spheroids and Tumours. Br. J. Cancer 2000, 82, 635–641. [Google Scholar] [CrossRef][Green Version]
- Semenza, G.L. Defining the Role of Hypoxia-Inducible Factor 1 in Cancer Biology and Therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Zagzag, D.; Zhong, H.; Scalzitti, J.M.; Laughner, E.; Simons, J.W.; Semenza, G.L. Expression of Hypoxia-Inducible Factor 1alpha in Brain Tumors: Association with Angiogenesis, Invasion, and Progression. Cancer 2000, 88, 2606–2618. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and Cancer. J. Mol. Med. Berl. Ger. 2007, 85, 1301–1307. [Google Scholar] [CrossRef]
- Unruh, A.; Ressel, A.; Mohamed, H.G.; Johnson, R.S.; Nadrowitz, R.; Richter, E.; Katschinski, D.M.; Wenger, R.H. The Hypoxia-Inducible Factor-1 Alpha Is a Negative Factor for Tumor Therapy. Oncogene 2003, 22, 3213–3220. [Google Scholar] [CrossRef]
- Green, H.; Goldberg, B. Collagen and Cell Protein Synthesis by an Established Mammalian Fibroblast Line. Nature 1964, 204, 347–349. [Google Scholar] [CrossRef]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple Biological Activities of Lactic Acid in Cancer: Influences on Tumor Growth, Angiogenesis and Metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef]
- Polet, F.; Feron, O. Endothelial Cell Metabolism and Tumour Angiogenesis: Glucose and Glutamine as Essential Fuels and Lactate as the Driving Force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2016, 38, 119–133. [Google Scholar] [CrossRef]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate Activates HIF-1 in Oxidative but Not in Warburg-Phenotype Human Tumor Cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hussien, R.; Oommen, S.; Gohil, K.; Brooks, G.A. Lactate Sensitive Transcription Factor Network in L6 Cells: Activation of MCT1 and Mitochondrial Biogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the Lactate Transporter MCT1 in Endothelial Cells Inhibits Lactate-Induced HIF-1 Activation and Tumor Angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Stern, R. Hyaluronidases in Cancer Biology. Semin. Cancer Biol. 2008, 18, 275–280. [Google Scholar] [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and Motor of Tumor Malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef]
- Pardue, E.L.; Ibrahim, S.; Ramamurthi, A. Role of Hyaluronan in Angiogenesis and Its Utility to Angiogenic Tissue Engineering. Organogenesis 2008, 4, 203–214. [Google Scholar] [CrossRef]
- Schoenfelder, M.; Einspanier, R. Expression of Hyaluronan Synthases and Corresponding Hyaluronan Receptors Is Differentially Regulated during Oocyte Maturation in Cattle. Biol. Reprod. 2003, 69, 269–277. [Google Scholar] [CrossRef]
- Formby, B.; Stern, R. Lactate-Sensitive Response Elements in Genes Involved in Hyaluronan Catabolism. Biochem. Biophys. Res. Commun. 2003, 305, 203–208. [Google Scholar] [CrossRef]
- Stern, R.; Shuster, S.; Neudecker, B.A.; Formby, B. Lactate Stimulates Fibroblast Expression of Hyaluronan and CD44: The Warburg Effect Revisited. Exp. Cell Res. 2002, 276, 24–31. [Google Scholar] [CrossRef]
- West, D.C.; Hampson, I.N.; Arnold, F.; Kumar, S. Angiogenesis Induced by Degradation Products of Hyaluronic Acid. Science 1985, 228, 1324–1326. [Google Scholar] [CrossRef]
- Genasetti, A.; Vigetti, D.; Viola, M.; Karousou, E.; Moretto, P.; Rizzi, M.; Bartolini, B.; Clerici, M.; Pallotti, F.; De Luca, G.; et al. Hyaluronan and Human Endothelial Cell Behavior. Connect. Tissue Res. 2008, 49, 120–123. [Google Scholar] [CrossRef]
- Gao, F.; Yang, C.X.; Mo, W.; Liu, Y.W.; He, Y.Q. Hyaluronan Oligosaccharides Are Potential Stimulators to Angiogenesis via RHAMM Mediated Signal Pathway in Wound Healing. Clin. Investig. Med. Med. Clin. Exp. 2008, 31, E106–E116. [Google Scholar] [CrossRef]
- Ohno-Nakahara, M.; Honda, K.; Tanimoto, K.; Tanaka, N.; Doi, T.; Suzuki, A.; Yoneno, K.; Nakatani, Y.; Ueki, M.; Ohno, S.; et al. Induction of CD44 and MMP Expression by Hyaluronidase Treatment of Articular Chondrocytes. J. Biochem. 2004, 135, 567–575. [Google Scholar] [CrossRef]
- Zhang, Y.; Thant, A.A.; Machida, K.; Ichigotani, Y.; Naito, Y.; Hiraiwa, Y.; Senga, T.; Sohara, Y.; Matsuda, S.; Hamaguchi, M. Hyaluronan-CD44s Signaling Regulates Matrix Metalloproteinase-2 Secretion in a Human Lung Carcinoma Cell Line QG90. Cancer Res. 2002, 62, 3962–3965. [Google Scholar]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef]
- Oren, O.; Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef]
- Al-Harthi, L. Wnt/β-Catenin and Its Diverse Physiological Cell Signaling Pathways in Neurodegenerative and Neuropsychiatric Disorders. J. Neuroimmune Pharmacol. 2012, 7, 725–730. [Google Scholar] [CrossRef]
- Marchetti, B.; Pluchino, S. Wnt Your Brain Be Inflamed? Yes, It Wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Neurodegenerative Diseases: Interplay Between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromol. Med. 2018, 20, 174–204. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.-L. Circadian Rhythms, Wnt/Beta-Catenin Pathway and PPAR Alpha/Gamma Profiles in Diseases with Primary or Secondary Cardiac Dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Vallée, A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 100. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 Receptors for Wnt/β-Catenin Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome Proliferator-Activated Receptor Gamma Activation Can Regulate Beta-Catenin Levels via a Proteasome-Mediated and Adenomatous Polyposis Coli-Independent Pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [PubMed]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 Expression in Transgenic Mouse Models of Neurodegenerative Disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt Signaling: Role in Alzheimer Disease and Schizophrenia. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2012, 7, 788–807. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-Β1, Canonical WNT/β-Catenin Pathway and PPAR γ in Radiation-Induced Fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Hypothesis of Opposite Interplay Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr. Issues Mol. Biol. 2019, 31, 1–20. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-Catenin Is a Target for the Ubiquitin–Proteasome Pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef]
- Hur, E.-M.; Zhou, F.-Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef]
- Ambacher, K.K.; Pitzul, K.B.; Karajgikar, M.; Hamilton, A.; Ferguson, S.S.; Cregan, S.P. The JNK- and AKT/GSK3β- Signaling Pathways Converge to Regulate Puma Induction and Neuronal Apoptosis Induced by Trophic Factor Deprivation. PLoS ONE 2012, 7, e46885. [Google Scholar] [CrossRef]
- Orellana, A.M.M.; Vasconcelos, A.R.; Leite, J.A.; de Sá Lima, L.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-Related Neuroinflammation and Changes in AKT-GSK-3β and WNT/ β-CATENIN Signaling in Rat Hippocampus. Aging 2015, 7, 1094–1111. [Google Scholar] [CrossRef]
- Jha, M.K.; Suk, K. Pyruvate Dehydrogenase Kinase as a Potential Therapeutic Target for Malignant Gliomas. Brain Tumor Res. Treat. 2013, 1, 57–63. [Google Scholar] [CrossRef]
- Thompson, C.B. Wnt Meets Warburg: Another Piece in the Puzzle? EMBO J. 2014, 33, 1420–1422. [Google Scholar] [CrossRef]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt Signaling Directs a Metabolic Program of Glycolysis and Angiogenesis in Colon Cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Venneti, S.; Thompson, C.B. Metabolic Reprogramming in Brain Tumors. Annu. Rev. Pathol. 2017, 12, 515–545. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.R.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial Activation by Inhibition of PDKII Suppresses HIF1a Signaling and Angiogenesis in Cancer. Oncogene 2013, 32, 1638–1650. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Kim, J.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The Transcription Factor HIF-1alpha Plays a Critical Role in the Growth Factor-Dependent Regulation of Both Aerobic and Anaerobic Glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian Target of Rapamycin Up-Regulation of Pyruvate Kinase Isoenzyme Type M2 Is Critical for Aerobic Glycolysis and Tumor Growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.-H. Roles of P53, MYC and HIF-1 in Regulating Glycolysis-the Seventh Hallmark of Cancer. Cell. Mol. Life Sci. CMLS 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential Utilization of Ketone Bodies by Neurons and Glioma Cell Lines: A Rationale for Ketogenic Diet as Experimental Glioma Therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Arvelo, F.; Miccoli, L.; Apiou, F.; Dutrillaux, A.M.; Poisson, M.; Dutrillaux, B.; Poupon, M.F. High Glycolysis in Gliomas despite Low Hexokinase Transcription and Activity Correlated to Chromosome 10 Loss. Br. J. Cancer 1996, 74, 839–845. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of Cancer Stem Cell Metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Bouzier-Sore, A.K.; Canioni, P.; Merle, M. Effect of Exogenous Lactate on Rat Glioma Metabolism. J. Neurosci. Res. 2001, 65, 543–548. [Google Scholar] [CrossRef]
- Darnell, J.E. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Horvath, C.M. STAT Proteins and Transcriptional Responses to Extracellular Signals. Trends Biochem. Sci. 2000, 25, 496–502. [Google Scholar] [CrossRef]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive Activation of Stat3alpha in Brain Tumors: Localization to Tumor Endothelial Cells and Activation by the Endothelial Tyrosine Kinase Receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of P21WAF1/CIP1 and Cyclin D1 Expression by the Src Oncoprotein in Mouse Fibroblasts: Role of Activated STAT3 Signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.-H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.-W.; Ye, S.-K.; et al. STAT3 Is a Potential Modulator of HIF-1-Mediated VEGF Expression in Human Renal Carcinoma Cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1296–1298. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, C.; Zhang, W.; Zhang, G.; Zhao, X.; Yang, S.; Wang, Y.; Lu, N.; Zhu, H.; Xu, N. Beta-Catenin/TCF Pathway Upregulates STAT3 Expression in Human Esophageal Squamous Cell Carcinoma. Cancer Lett. 2008, 271, 85–97. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The Transcriptional Network for Mesenchymal Transformation of Brain Tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Ma, J.; Meng, Y.; Kwiatkowski, D.J.; Chen, X.; Peng, H.; Sun, Q.; Zha, X.; Wang, F.; Wang, Y.; Jing, Y.; et al. Mammalian Target of Rapamycin Regulates Murine and Human Cell Differentiation through STAT3/P63/Jagged/Notch Cascade. J. Clin. Investig. 2010, 120, 103–114. [Google Scholar] [CrossRef]
- Mazzone, M.; Selfors, L.M.; Albeck, J.; Overholtzer, M.; Sale, S.; Carroll, D.L.; Pandya, D.; Lu, Y.; Mills, G.B.; Aster, J.C.; et al. Dose-Dependent Induction of Distinct Phenotypic Responses to Notch Pathway Activation in Mammary Epithelial Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5012–5017. [Google Scholar] [CrossRef]
- Kang, S.-H.; Yu, M.O.; Park, K.-J.; Chi, S.-G.; Park, D.-H.; Chung, Y.-G. Activated STAT3 Regulates Hypoxia-Induced Angiogenesis and Cell Migration in Human Glioblastoma. Neurosurgery 2010, 67, 1386–1395. [Google Scholar] [CrossRef]
- Chakravarti, A.; Dicker, A.; Mehta, M. The Contribution of Epidermal Growth Factor Receptor (EGFR) Signaling Pathway to Radioresistance in Human Gliomas: A Review of Preclinical and Correlative Clinical Data. Int. J. Radiat. Oncol. 2004, 58, 927–931. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.Y.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR Signaling Pathway in Cancer Therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K.; Hart, J.R. PI3K and STAT3: A New Alliance. Cancer Discov. 2011, 1, 481–486. [Google Scholar] [CrossRef]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.-W. Mechanisms Regulating Glioma Invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sami, A.; Karsy, M. Targeting the PI3K/AKT/MTOR Signaling Pathway in Glioblastoma: Novel Therapeutic Agents and Advances in Understanding. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K Signalling: The Path to Discovery and Understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Williams, R.L. Synergy in Activating Class I PI3Ks. Trends Biochem. Sci. 2015, 40, 88–100. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Popescu, A.M.; Purcaru, S.O.; Alexandru, O.; Dricu, A. New Perspectives in Glioblastoma Antiangiogenic Therapy. Contemp. Oncol. Pozn. Pol. 2016, 20, 109–118. [Google Scholar] [CrossRef]
- Tan, X.; Apte, U.; Micsenyi, A.; Kotsagrelos, E.; Luo, J.-H.; Ranganathan, S.; Monga, D.K.; Bell, A.; Michalopoulos, G.K.; Monga, S.P.S. Epidermal Growth Factor Receptor: A Novel Target of the Wnt/Beta-Catenin Pathway in Liver. Gastroenterology 2005, 129, 285–302. [Google Scholar] [CrossRef]
- Mora, A.; Komander, D.; van Aalten, D.M.F.; Alessi, D.R. PDK1, the Master Regulator of AGC Kinase Signal Transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef]
- Loewith, R.; Hall, M.N. Target of Rapamycin (TOR) in Nutrient Signaling and Growth Control. Genetics 2011, 189, 1177–1201. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of MTORC1 by PI3K Signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for Direct Activation of MTORC2 Kinase Activity by Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef]
- Efeyan, A.; Sabatini, D.M. Nutrients and Growth Factors in MTORC1 Activation. Biochem. Soc. Trans. 2013, 41, 902–905. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific Activation of MTORC1 by Rheb G-Protein in Vitro Involves Enhanced Recruitment of Its Substrate Protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef]
- Tabancay, A.P.; Gau, C.-L.; Machado, I.M.P.; Uhlmann, E.J.; Gutmann, D.H.; Guo, L.; Tamanoi, F. Identification of Dominant Negative Mutants of Rheb GTPase and Their Use to Implicate the Involvement of Human Rheb in the Activation of P70S6K. J. Biol. Chem. 2003, 278, 39921–39930. [Google Scholar] [CrossRef]
- Tee, A.R.; Blenis, J.; Proud, C.G. Analysis of MTOR Signaling by the Small G-Proteins, Rheb and RhebL1. FEBS Lett. 2005, 579, 4763–4768. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Dodd, K.M.; Seymour, L.A.; Tee, A.R. Mammalian Target of Rapamycin Complex 1-Mediated Phosphorylation of Eukaryotic Initiation Factor 4E-Binding Protein 1 Requires Multiple Protein-Protein Interactions for Substrate Recognition. Cell. Signal. 2009, 21, 1073–1084. [Google Scholar] [CrossRef]
- Howell, J.J.; Manning, B.D. MTOR Couples Cellular Nutrient Sensing to Organismal Metabolic Homeostasis. Trends Endocrinol. Metab. TEM 2011, 22, 94–102. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Block, K.; Nayak, B.K.; Livi, C.B.; Venkatachalam, M.A.; Sudarshan, S. PI3K Regulation of the SKP-2/P27 Axis through MTORC2. Oncogene 2013, 32, 2027–2036. [Google Scholar] [CrossRef]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt Promotes Feedforward MTORC2 Activation through IKKα. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [PubMed]
- Fuhler, G.M.; Tyl, M.R.; Olthof, S.G.M.; Lyndsay Drayer, A.; Blom, N.; Vellenga, E. Distinct Roles of the MTOR Components Rictor and Raptor in MO7e Megakaryocytic Cells. Eur. J. Haematol. 2009, 83, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; van der Groep, P.; Kemming, D.; Shvarts, A.; Semenza, G.L.; Meijer, G.A.; van de Wiel, M.A.; Belien, J.a.M.; van Diest, P.J.; van der Wall, E. Up-Regulation of Gene Expression by Hypoxia Is Mediated Predominantly by Hypoxia-Inducible Factor 1 (HIF-1). J. Pathol. 2005, 206, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Berra, E.; Ginouvès, A.; Pouysségur, J. The Hypoxia-Inducible-Factor Hydroxylases Bring Fresh Air into Hypoxia Signalling. EMBO Rep. 2006, 7, 41–45. [Google Scholar] [CrossRef]
- Kaelin, W.G. The von Hippel-Lindau Tumor Suppressor Protein: Roles in Cancer and Oxygen Sensing. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 159–166. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, W.; Garcia-Prieto, C.; Huang, P. The Warburg Effect and Its Cancer Therapeutic Implications. J. Bioenerg. Biomembr. 2007, 39, 267–274. [Google Scholar] [CrossRef]
- Faure Vigny, H.; Heddi, A.; Giraud, S.; Chautard, D.; Stepien, G. Expression of Oxidative Phosphorylation Genes in Renal Tumors and Tumoral Cell Lines. Mol. Carcinog. 1996, 16, 165–172. [Google Scholar] [CrossRef]
- Unwin, R.D.; Craven, R.A.; Harnden, P.; Hanrahan, S.; Totty, N.; Knowles, M.; Eardley, I.; Selby, P.J.; Banks, R.E. Proteomic Changes in Renal Cancer and Co-Ordinate Demonstration of Both the Glycolytic and Mitochondrial Aspects of the Warburg Effect. Proteomics 2003, 3, 1620–1632. [Google Scholar] [CrossRef]
- Dekanty, A. The Insulin-PI3K/TOR Pathway Induces a HIF-Dependent Transcriptional Response in Drosophila by Promoting Nuclear Localization of HIF- /Sima. J. Cell Sci. 2005, 118, 5431–5441. [Google Scholar] [CrossRef]
- Shaw, R.J. Glucose Metabolism and Cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G. TSC2 Regulates VEGF through MTOR-Dependent and -Independent Pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of MTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-Inducible Factor 1alpha Is Regulated by the Mammalian Target of Rapamycin (MTOR) via an MTOR Signaling Motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef]
- Toschi, A.; Lee, E.; Gadir, N.; Ohh, M.; Foster, D.A. Differential Dependence of Hypoxia-Inducible Factors 1 Alpha and 2 Alpha on MTORC1 and MTORC2. J. Biol. Chem. 2008, 283, 34495–34499. [Google Scholar] [CrossRef]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 Blocks Both HIF-1 and VEGF Expression Induced by Multiple Oncogenic Growth Signaling Pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef]
- McEwen, B.S.; Reagan, L.P. Glucose Transporter Expression in the Central Nervous System: Relationship to Synaptic Function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Bos, R.; van Der Hoeven, J.J.M.; van Der Wall, E.; van Der Groep, P.; van Diest, P.J.; Comans, E.F.I.; Joshi, U.; Semenza, G.L.; Hoekstra, O.S.; Lammertsma, A.A.; et al. Biologic Correlates of (18)Fluorodeoxyglucose Uptake in Human Breast Cancer Measured by Positron Emission Tomography. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 379–387. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Pyruvate Kinase M2 Regulates Glucose Metabolism by Functioning as a Coactivator for Hypoxia-Inducible Factor 1 in Cancer Cells. Oncotarget 2011, 2, 551–556. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Pereira, K.M.A.; Chaves, F.N.; Viana, T.S.A.; Carvalho, F.S.R.; Costa, F.W.G.; Alves, A.P.N.N.; Sousa, F.B. Oxygen Metabolism in Oral Cancer: HIF and GLUTs (Review). Oncol. Lett. 2013, 6, 311–316. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the Facilitative Glucose Transporter GLUT1 Inhibits the Self-Renewal and Tumor-Initiating Capacity of Cancer Stem Cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-Dependent Phosphorylation and Nuclear Translocation of PKM2 Promotes the Warburg Effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose Catabolism in Cancer Cells: Identification and Characterization of a Marked Activation Response of the Type II Hexokinase Gene to Hypoxic Conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 Is a Key Mediator of Aerobic Glycolysis and Promotes Tumor Growth in Human Glioblastoma Multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Gershon, T.R.; Crowther, A.J.; Tikunov, A.; Garcia, I.; Annis, R.; Yuan, H.; Miller, C.R.; Macdonald, J.; Olson, J.; Deshmukh, M. Hexokinase-2-Mediated Aerobic Glycolysis Is Integral to Cerebellar Neurogenesis and Pathogenesis of Medulloblastoma. Cancer Metab. 2013, 1, 2. [Google Scholar] [CrossRef]
- Imamura, K.; Tanaka, T. Multimolecular Forms of Pyruvate Kinase from Rat and Other Mammalian Tissues. I. Electrophoretic Studies. J. Biochem. 1972, 71, 1043–1051. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. PKM2 Isoform-Specific Deletion Reveals a Differential Requirement for Pyruvate Kinase in Tumor Cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate Kinase Type M2: A Key Regulator of the Metabolic Budget System in Tumor Cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Tindale, L.; Cumming, R.C. Age-Dependent Metabolic Dysregulation in Cancer and Alzheimer’s Disease. Biogerontology 2014, 15, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.-G. Prevalent Overexpression of Prolyl Isomerase Pin1 in Human Cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 Regulates Turnover and Subcellular Localization of Beta-Catenin by Inhibiting Its Interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Roche, T.E.; Baker, J.C.; Yan, X.; Hiromasa, Y.; Gong, X.; Peng, T.; Dong, J.; Turkan, A.; Kasten, S.A. Distinct Regulatory Properties of Pyruvate Dehydrogenase Kinase and Phosphatase Isoforms. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 33–75. [Google Scholar]
- Burgner, J.W.; Ray, W.J. On the Origin of the Lactate Dehydrogenase Induced Rate Effect. Biochemistry 1984, 23, 3636–3648. [Google Scholar] [CrossRef]
- Kumar, V.B.S.; Viji, R.I.; Kiran, M.S.; Sudhakaran, P.R. Endothelial Cell Response to Lactate: Implication of PAR Modification of VEGF. J. Cell. Physiol. 2007, 211, 477–485. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; De Saedeleer, C.J.; Préat, V.; Thissen, J.-P.; Feron, O.; Sonveaux, P. Lactate Stimulates Angiogenesis and Accelerates the Healing of Superficial and Ischemic Wounds in Mice. Angiogenesis 2012, 15, 581–592. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Kim, J.; Dang, C.V. Cancer’s Molecular Sweet Tooth and the Warburg Effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic Regulation of Lactate Dehydrogenase A. Interaction between Hypoxia-Inducible Factor 1 and CAMP Response Elements. J. Biol. Chem. 1995, 270, 21021–21027. [Google Scholar] [CrossRef]
- Lewis, B.C.; Shim, H.; Li, Q.; Wu, C.S.; Lee, L.A.; Maity, A.; Dang, C.V. Identification of Putative C-Myc-Responsive Genes: Characterization of Rcl, a Novel Growth-Related Gene. Mol. Cell. Biol. 1997, 17, 4967–4978. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-Inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. C-Myc Transactivation of LDH-A: Implications for Tumor Metabolism and Growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef]
- Brooks, G.A. Lactate Shuttles in Nature. Biochem. Soc. Trans. 2002, 30, 258–264. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Bougioukas, G.; Didilis, V.; Gatter, K.C.; Harris, A.L. Tumour and Angiogenesis Research Group Lactate Dehydrogenase-5 (LDH-5) Overexpression in Non-Small-Cell Lung Cancer Tissues Is Linked to Tumour Hypoxia, Angiogenic Factor Production and Poor Prognosis. Br. J. Cancer 2003, 89, 877–885. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Trarbach, T.; Folprecht, G.; Shi, M.M.; Lebwohl, D.; Jalava, T.; Laurent, D.; et al. Prognostic and Predictive Role of Lactate Dehydrogenase 5 Expression in Colorectal Cancer Patients Treated with PTK787/ZK 222584 (Vatalanib) Antiangiogenic Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4892–4900. [Google Scholar] [CrossRef]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; Scheuenstuhl, H.; Königsrainer, A.; Hussain, M.Z.; Hunt, T.K. Lactate Stimulates Endothelial Cell Migration. Wound Repair Regen. Off. Publ. Wound Heal. Soc. Eur. Tissue Repair Soc. 2006, 14, 321–324. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Xu, L.; Fidler, I.J. Acidic PH-Induced Elevation in Interleukin 8 Expression by Human Ovarian Carcinoma Cells. Cancer Res. 2000, 60, 4610–4616. [Google Scholar] [PubMed]
- Brohée, L.; Peulen, O.; Nusgens, B.; Castronovo, V.; Thiry, M.; Colige, A.C.; Deroanne, C.F. Propranolol Sensitizes Prostate Cancer Cells to Glucose Metabolism Inhibition and Prevents Cancer Progression. Sci. Rep. 2018, 8, 7050. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dai, X.; Yao, Y.; Wang, J.; Yang, X.; Zhang, Y.; Yang, J.; Cao, R.; Wen, G.; Zhong, J. PRMT2β Suppresses Autophagy and Glycolysis Pathway in Human Breast Cancer MCF-7 Cell Lines. Acta Biochim. Biophys. Sin. 2019, 51, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt Signaling Pathway: A Focus on Wnt/β-Catenin Signaling. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and Regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Kohler, V.; Aufschnaiter, A.; Büttner, S. Closing the Gap: Membrane Contact Sites in the Regulation of Autophagy. Cells 2020, 9, 1184. [Google Scholar] [CrossRef]
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef]
- Alizadeh, J.; Shojaei, S.; Sepanjnia, A.; Hashemi, M.; Eftekharpour, E.; Ghavami, S. Simultaneous Detection of Autophagy and Epithelial to Mesenchymal Transition in the Non-Small Cell Lung Cancer Cells. Methods Mol. Biol. 2019, 1854, 87–103. [Google Scholar] [CrossRef]
- Brun, P.; Tarricone, E.; Di Stefano, A.; Mattiuzzo, E.; Mehrbod, P.; Ghavami, S.; Leonardi, A. The Regulatory Activity of Autophagy in Conjunctival Fibroblasts and Its Possible Role in Vernal Keratoconjunctivitis. J. Allergy Clin. Immunol. 2020, 146, 1210–1213.e9. [Google Scholar] [CrossRef]
- Wasik, A.M.; Grabarek, J.; Pantovic, A.; Cieślar-Pobuda, A.; Asgari, H.R.; Bundgaard-Nielsen, C.; Rafat, M.; Dixon, I.M.C.; Ghavami, S.; Łos, M.J. Reprogramming and Carcinogenesis--Parallels and Distinctions. Int. Rev. Cell Mol. Biol. 2014, 308, 167–203. [Google Scholar] [CrossRef]
- Kardideh, B.; Samimi, Z.; Norooznezhad, F.; Kiani, S.; Mansouri, K. Autophagy, Cancer and Angiogenesis: Where Is the Link? Cell Biosci. 2019, 9, 65. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and Apoptosis Dysfunction in Neurodegenerative Disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Rautou, P.-E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in Liver Diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and Aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar] [CrossRef]
- Ren, L.; Yang, H.; Cui, Y.; Xu, S.; Sun, F.; Tian, N.; Hua, J.; Peng, S. Autophagy Is Essential for the Differentiation of Porcine PSCs into Insulin-Producing Cells. Biochem. Biophys. Res. Commun. 2017, 488, 471–476. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; White, E. Role of Tumor and Host Autophagy in Cancer Metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-Deficient Mice Develop Multiple Liver Tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a Molecular Target for Cancer Treatment. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Nassour, J.; Radford, R.; Correia, A.; Fusté, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic Cell Death Restricts Chromosomal Instability during Replicative Crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic Degradation of the BCR-ABL Oncoprotein and Generation of Antileukemic Responses by Arsenic Trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kimmelman, A.C. Inhibition of Autophagy Attenuates Pancreatic Cancer Growth Independent of TP53/TRP53 Status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy Provides Metabolic Substrates to Maintain Energy Charge and Nucleotide Pools in Ras-Driven Lung Cancer Cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef]
- Akkoç, Y.; Berrak, Ö.; Arısan, E.D.; Obakan, P.; Çoker-Gürkan, A.; Palavan-Ünsal, N. Inhibition of PI3K Signaling Triggered Apoptotic Potential of Curcumin Which Is Hindered by Bcl-2 through Activation of Autophagy in MCF-7 Cells. Biomed. Pharmacother. 2015, 71, 161–171. [Google Scholar] [CrossRef]
- Chen, W.; Bai, Y.; Patel, C.; Geng, F. Autophagy Promotes Triple Negative Breast Cancer Metastasis via YAP Nuclear Localization. Biochem. Biophys. Res. Commun. 2019, 520, 263–268. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Aoki, S.; Hirao, T.; Morita, M.; Ito, K. Autophagy Is an Important Metabolic Pathway to Determine Leukemia Cell Survival Following Suppression of the Glycolytic Pathway. Biochem. Biophys. Res. Commun. 2016, 474, 188–192. [Google Scholar] [CrossRef]
- Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; Ge, P. RSL3 Induced Autophagic Death in Glioma Cells via Causing Glycolysis Dysfunction. Biochem. Biophys. Res. Commun. 2019, 518, 590–597. [Google Scholar] [CrossRef]
- Kim, J.H.; Nam, B.; Choi, Y.J.; Kim, S.Y.; Lee, J.-E.; Sung, K.J.; Kim, W.S.; Choi, C.-M.; Chang, E.-J.; Koh, J.S.; et al. Enhanced Glycolysis Supports Cell Survival in EGFR-Mutant Lung Adenocarcinoma by Inhibiting Autophagy-Mediated EGFR Degradation. Cancer Res. 2018, 78, 4482–4496. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Zhang, M.; Cong, Q.; Zhang, M.-X.; Zhang, M.-Y.; Lu, Y.-Y.; Xu, C.-J. Hexokinase 2 Confers Resistance to Cisplatin in Ovarian Cancer Cells by Enhancing Cisplatin-Induced Autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Takai, T.; Tsujino, T.; Yoshikawa, Y.; Inamoto, T.; Sugito, N.; Kuranaga, Y.; Heishima, K.; Soga, T.; Hayashi, K.; Miyata, K.; et al. Synthetic MiR-143 Exhibited an Anti-Cancer Effect via the Downregulation of K-RAS Networks of Renal Cell Cancer Cells In Vitro and In Vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1017–1027. [Google Scholar] [CrossRef]
- Wu, X.; Won, H.; Rubinsztein, D.C. Autophagy and Mammalian Development. Biochem. Soc. Trans. 2013, 41, 1489–1494. [Google Scholar] [CrossRef]
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.M.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal β-Catenin Degradation Regulates Wnt-Autophagy-P62 Crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef]
- Tao, H.; Chen, F.; Liu, H.; Hu, Y.; Wang, Y.; Li, H. Wnt/β-Catenin Signaling Pathway Activation Reverses Gemcitabine Resistance by Attenuating Beclin1-Mediated Autophagy in the MG63 Human Osteosarcoma Cell Line. Mol. Med. Rep. 2017, 16, 1701–1706. [Google Scholar] [CrossRef]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy Negatively Regulates Wnt Signalling by Promoting Dishevelled Degradation. Nat. Cell Biol. 2010, 12, 781–790. [Google Scholar] [CrossRef]
- Hoverter, N.P.; Waterman, M.L. A Wnt-Fall for Gene Regulation: Repression. Sci. Signal. 2008, 1, pe43. [Google Scholar] [CrossRef]
- Nàger, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 Signaling Upregulates SQSTM1 and Sensitizes Glioblastoma Cells to Autophagy Blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-H.; Bang, S.; Kang, K.S.; Kang, D.; Chung, J. ULK1 Negatively Regulates Wnt Signaling by Phosphorylating Dishevelled. Biochem. Biophys. Res. Commun. 2019, 508, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Colella, B.; Faienza, F.; Carinci, M.; D’Alessandro, G.; Catalano, M.; Santoro, A.; Cecconi, F.; Limatola, C.; Di Bartolomeo, S. Autophagy Induction Impairs Wnt/β-Catenin Signalling through β-Catenin Relocalisation in Glioblastoma Cells. Cell. Signal. 2019, 53, 357–364. [Google Scholar] [CrossRef]
- Zhong, Z.; Yu, J.; Virshup, D.M.; Madan, B. Wnts and the Hallmarks of Cancer. Cancer Metastasis Rev. 2020, 39, 625–645. [Google Scholar] [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and Its Interactions with the PI3K/AKT/MTOR Signalling Network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Buttrick, G.J.; Wakefield, J.G. PI3-K and GSK-3: Akt-Ing Together with Microtubules. Cell Cycle Georget. Tex 2008, 7, 2621–2625. [Google Scholar] [CrossRef]
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer Is a Mediator of MTORC1 and GSK3-TIP60 Signaling in Regulation of Autophagosome Maturation and Lipid Metabolism. Mol. Cell 2019, 73, 788–802.e7. [Google Scholar] [CrossRef]
- Aznar, N.; Sun, N.; Dunkel, Y.; Ear, J.; Buschman, M.D.; Ghosh, P. A Daple-Akt Feed-Forward Loop Enhances Noncanonical Wnt Signals by Compartmentalizing β-Catenin. Mol. Biol. Cell 2017, 28, 3709–3723. [Google Scholar] [CrossRef]
- Pérez-Plasencia, C.; López-Urrutia, E.; García-Castillo, V.; Trujano-Camacho, S.; López-Camarillo, C.; Campos-Parra, A.D. Interplay Between Autophagy and Wnt/β-Catenin Signaling in Cancer: Therapeutic Potential Through Drug Repositioning. Front. Oncol. 2020, 10, 1037. [Google Scholar] [CrossRef]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy Promotes Metastasis and Glycolysis by Upregulating MCT1 Expression and Wnt/β-Catenin Signaling Pathway Activation in Hepatocellular Carcinoma Cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef]
- Jing, Q.; Li, G.; Chen, X.; Liu, C.; Lu, S.; Zheng, H.; Ma, H.; Qin, Y.; Zhang, D.; Zhang, S.; et al. Wnt3a Promotes Radioresistance via Autophagy in Squamous Cell Carcinoma of the Head and Neck. J. Cell. Mol. Med. 2019, 23, 4711–4722. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic Shifts toward Glutamine Regulate Tumor Growth, Invasion and Bioenergetics in Ovarian Cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 Controls Glutamine Uptake and Tumour Growth in Triple-Negative Basal-like Breast Cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]
- Kung, H.-N.; Marks, J.R.; Chi, J.-T. Glutamine Synthetase Is a Genetic Determinant of Cell Type-Specific Glutamine Independence in Breast Epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V.; et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine Supports Pancreatic Cancer Growth through a KRAS-Regulated Metabolic Pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Li, C.; Zhang, G.; Zhao, L.; Ma, Z.; Chen, H. Metabolic Reprogramming in Cancer Cells: Glycolysis, Glutaminolysis, and Bcl-2 Proteins as Novel Therapeutic Targets for Cancer. World J. Surg. Oncol. 2016, 14, 15. [Google Scholar] [CrossRef]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Lévy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New Targets of Beta-Catenin Signaling in the Liver Are Involved in the Glutamine Metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc Suppression of MiR-23a/b Enhances Mitochondrial Glutaminase Expression and Glutamine Metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Wu, H.; Li, Z.; Yang, P.; Zhang, L.; Fan, Y.; Li, Z. PKM2 Depletion Induces the Compensation of Glutaminolysis through β-Catenin/c-Myc Pathway in Tumor Cells. Cell. Signal. 2014, 26, 2397–2405. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High Resistance to Cisplatin in Human Ovarian Cancer Cell Lines Is Associated with Marked Increase of Glutathione Synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the XCT Subunit of System Xc(-) and Thereby Promotes Tumor Growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.-P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of Stem-like Cancer Cells by Glutamine through β-Catenin Pathway Mediated by Redox Signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef]
- Mihm, S.; Galter, D.; Dröge, W. Modulation of Transcription Factor NF Kappa B Activity by Intracellular Glutathione Levels and by Variations of the Extracellular Cysteine Supply. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 246–252. [Google Scholar] [CrossRef]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of Glutathione Promotes Apoptosis in Triple-Negative Breast Cancer Cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 2803–2813. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as Mediator of Wnt Signaling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, N.H.; Cho, E.S.; Yang, J.H.; Cha, Y.H.; Kang, H.E.; Yun, J.S.; Cho, S.B.; Lee, S.-H.; Paclikova, P.; et al. Dishevelled Has a YAP Nuclear Export Function in a Tumor Suppressor Context-Dependent Manner. Nat. Commun. 2018, 9, 2301. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Konsavage, W.M.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/β-Catenin Signaling Regulates Yes-Associated Protein (YAP) Gene Expression in Colorectal Carcinoma Cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M.; Garmaroudi, F.S.; Luo, Z.; McManus, B.M. Wnt3a Induces Myofibroblast Differentiation by Upregulating TGF-β Signaling through SMAD2 in a β-Catenin-Dependent Manner. PLoS ONE 2011, 6, e19809. [Google Scholar] [CrossRef]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ Controls Smad Nucleocytoplasmic Shuttling and Regulates Human Embryonic Stem-Cell Self-Renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. Yap Reprograms Glutamine Metabolism to Increase Nucleotide Biosynthesis and Enable Liver Growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The Role of YAP/TAZ Activity in Cancer Metabolic Reprogramming. Mol. Cancer 2018, 17, 134. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.-Y.; et al. Vascular Stiffness Mechanoactivates YAP/TAZ-Dependent Glutaminolysis to Drive Pulmonary Hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK Modulates Hippo Pathway Activity to Regulate Energy Homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef]
- Song, L.; Tang, H.; Liao, W.; Luo, X.; Li, Y.; Chen, T.; Zhang, X. FOXC2 Positively Regulates YAP Signaling and Promotes the Glycolysis of Nasopharyngeal Carcinoma. Exp. Cell Res. 2017, 357, 17–24. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions. Cancers 2021, 13, 5557. https://doi.org/10.3390/cancers13215557
Vallée A, Lecarpentier Y, Vallée J-N. The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions. Cancers. 2021; 13(21):5557. https://doi.org/10.3390/cancers13215557
Chicago/Turabian StyleVallée, Alexandre, Yves Lecarpentier, and Jean-Noël Vallée. 2021. "The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions" Cancers 13, no. 21: 5557. https://doi.org/10.3390/cancers13215557
APA StyleVallée, A., Lecarpentier, Y., & Vallée, J.-N. (2021). The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions. Cancers, 13(21), 5557. https://doi.org/10.3390/cancers13215557