
Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase

 , ,
, ,
Abstract
Simple Summary
Abstract

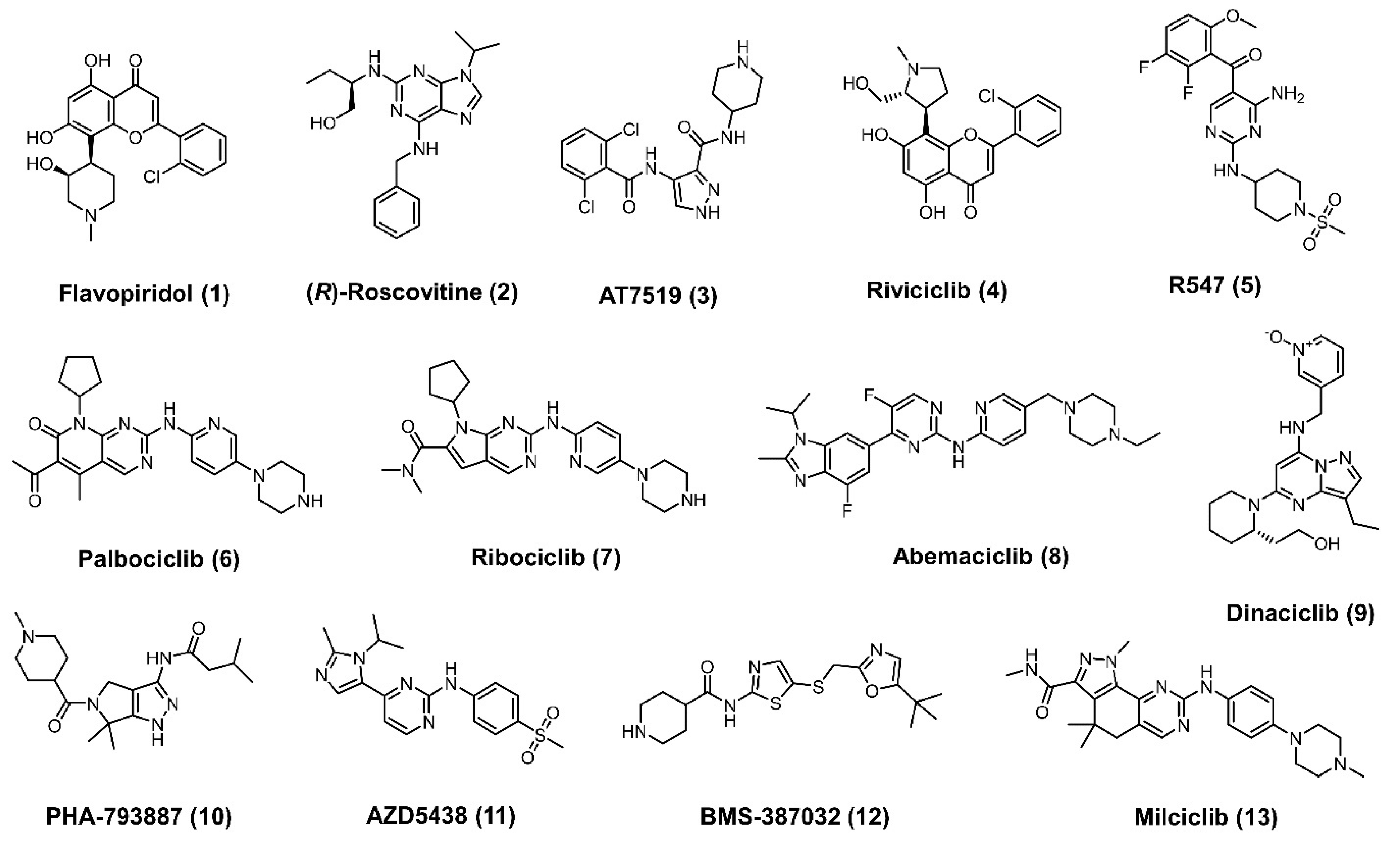
1. Introduction
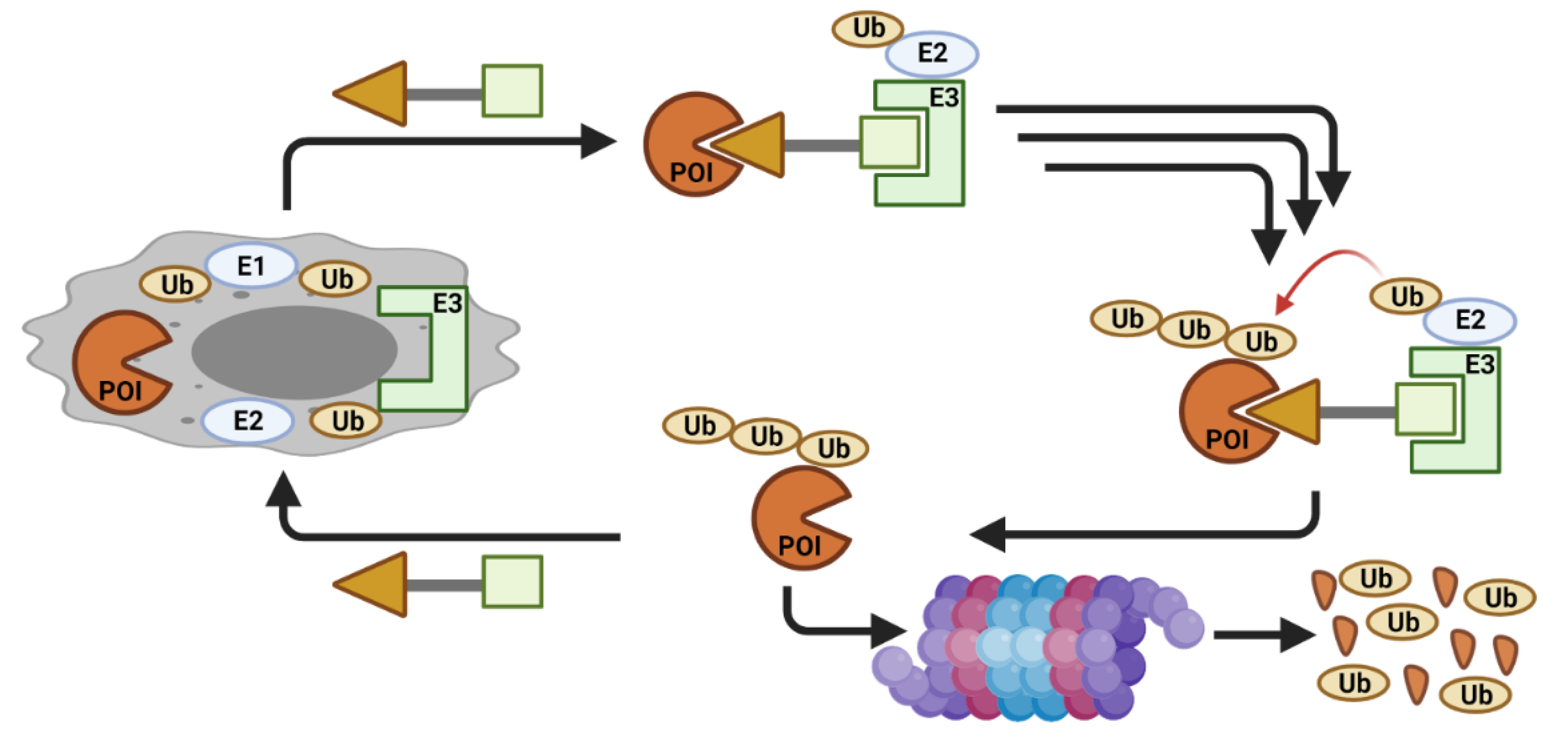
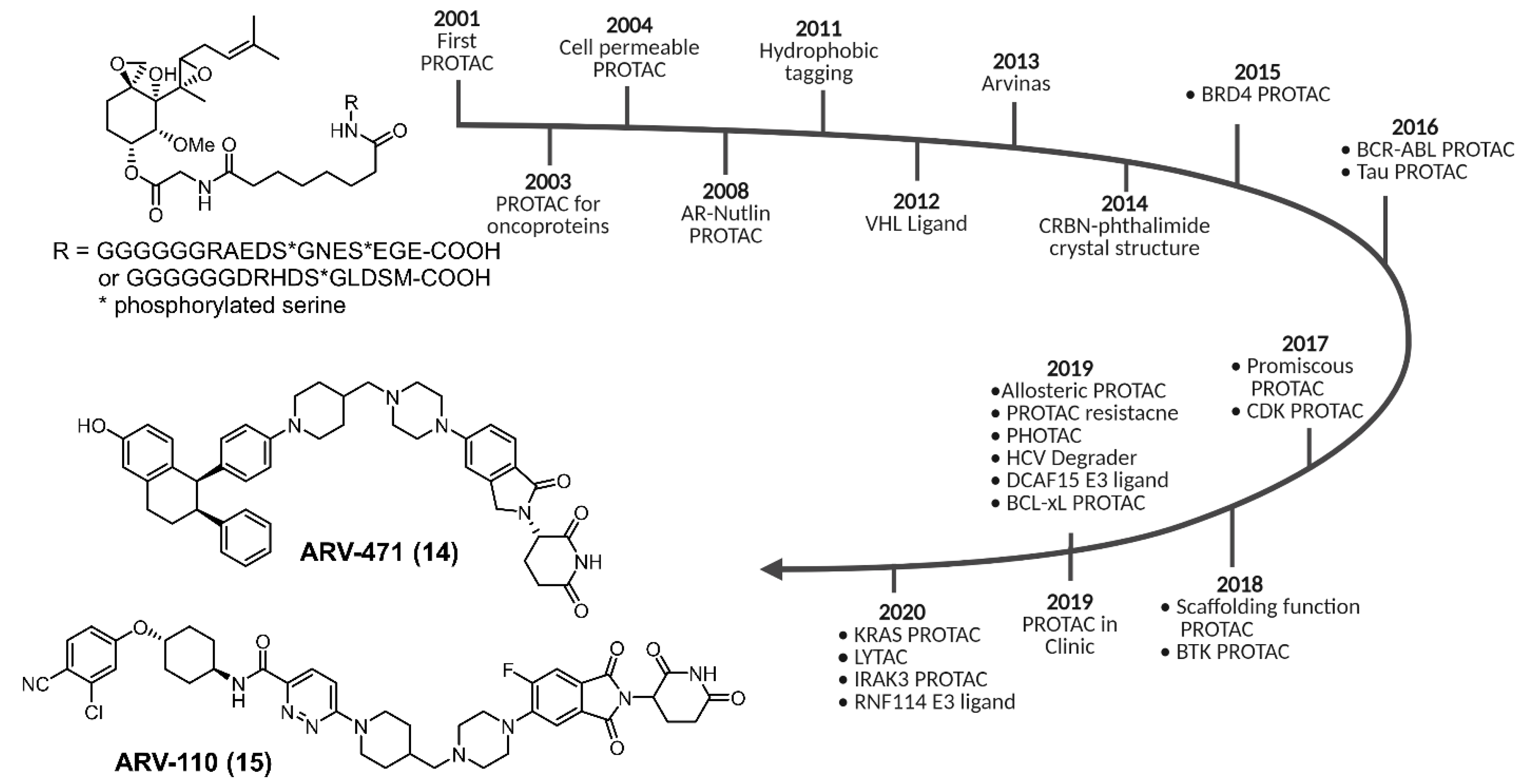
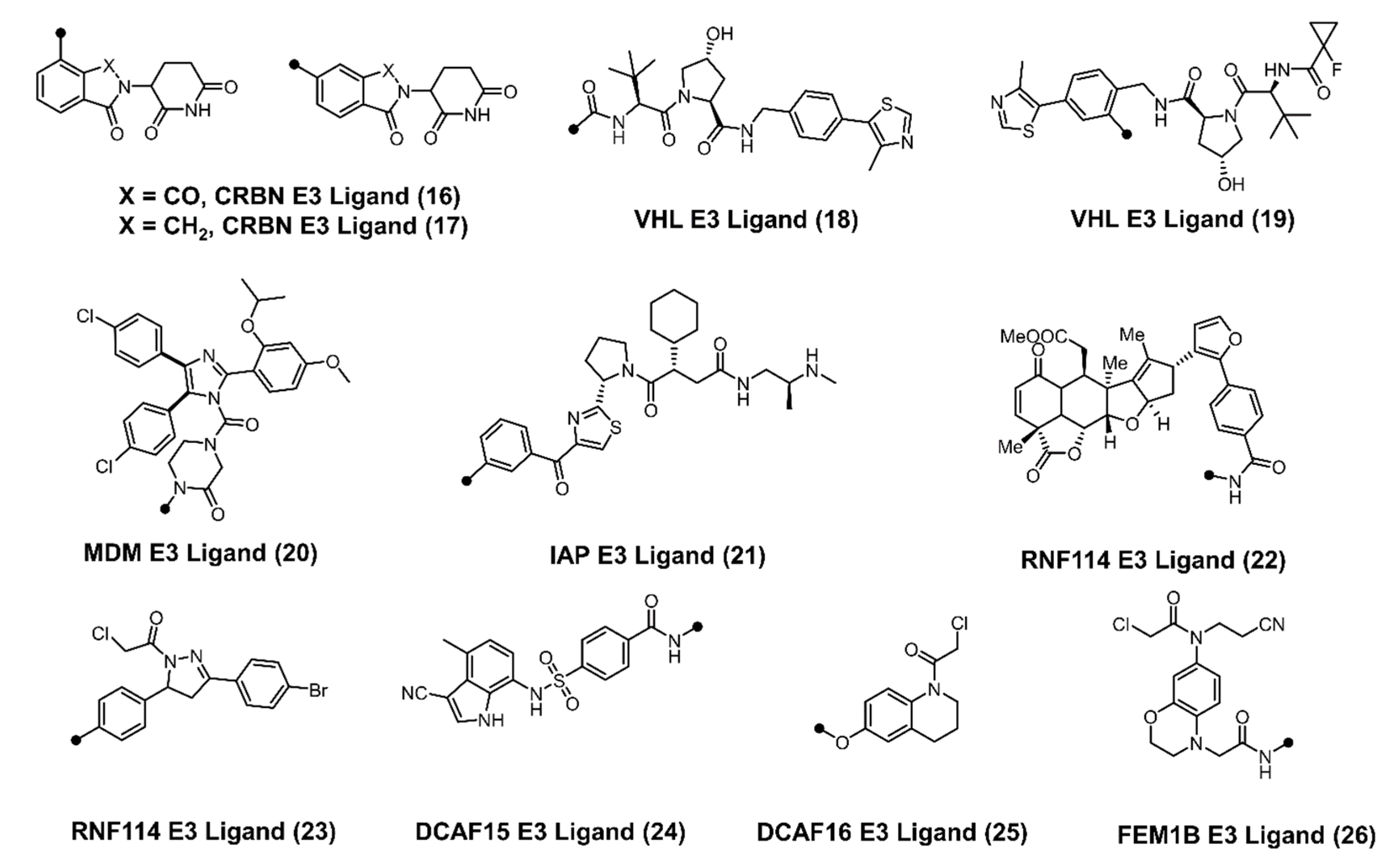
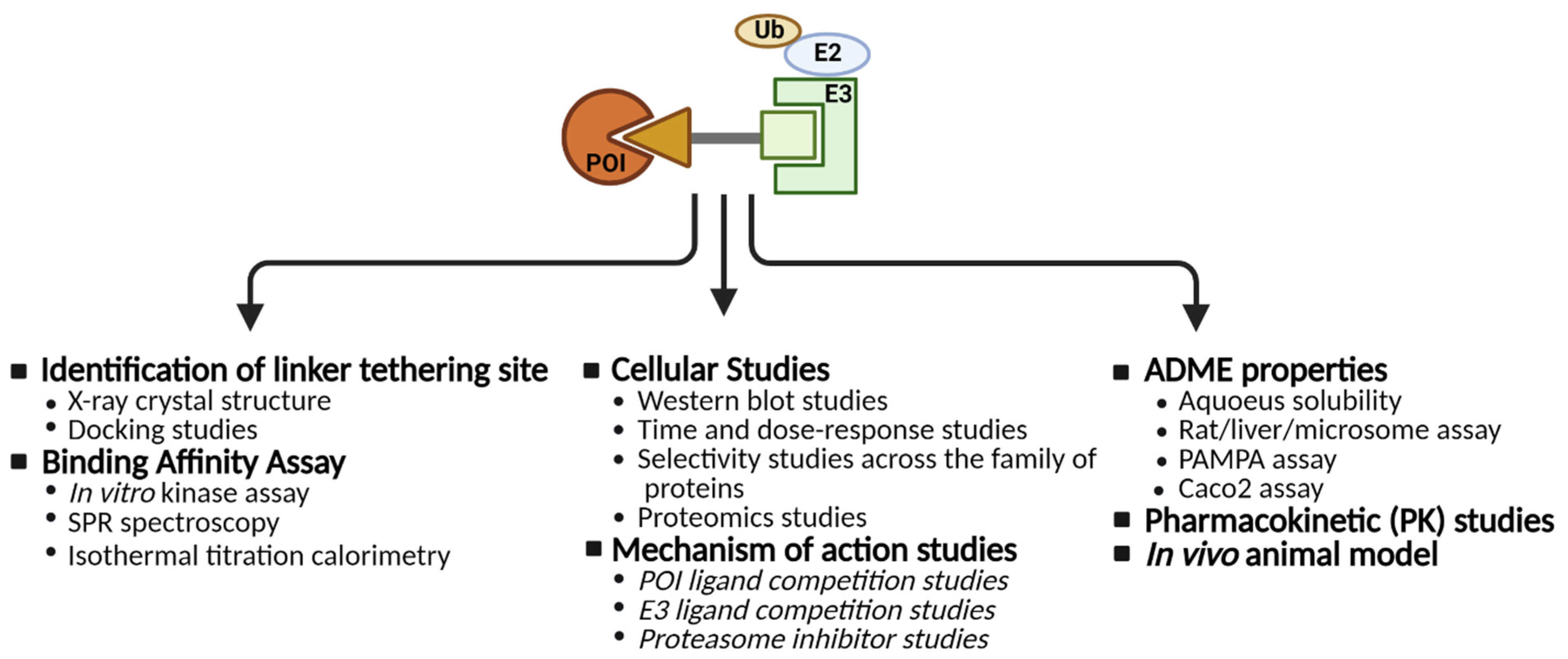
2. CDK Degradation Using PROTAC
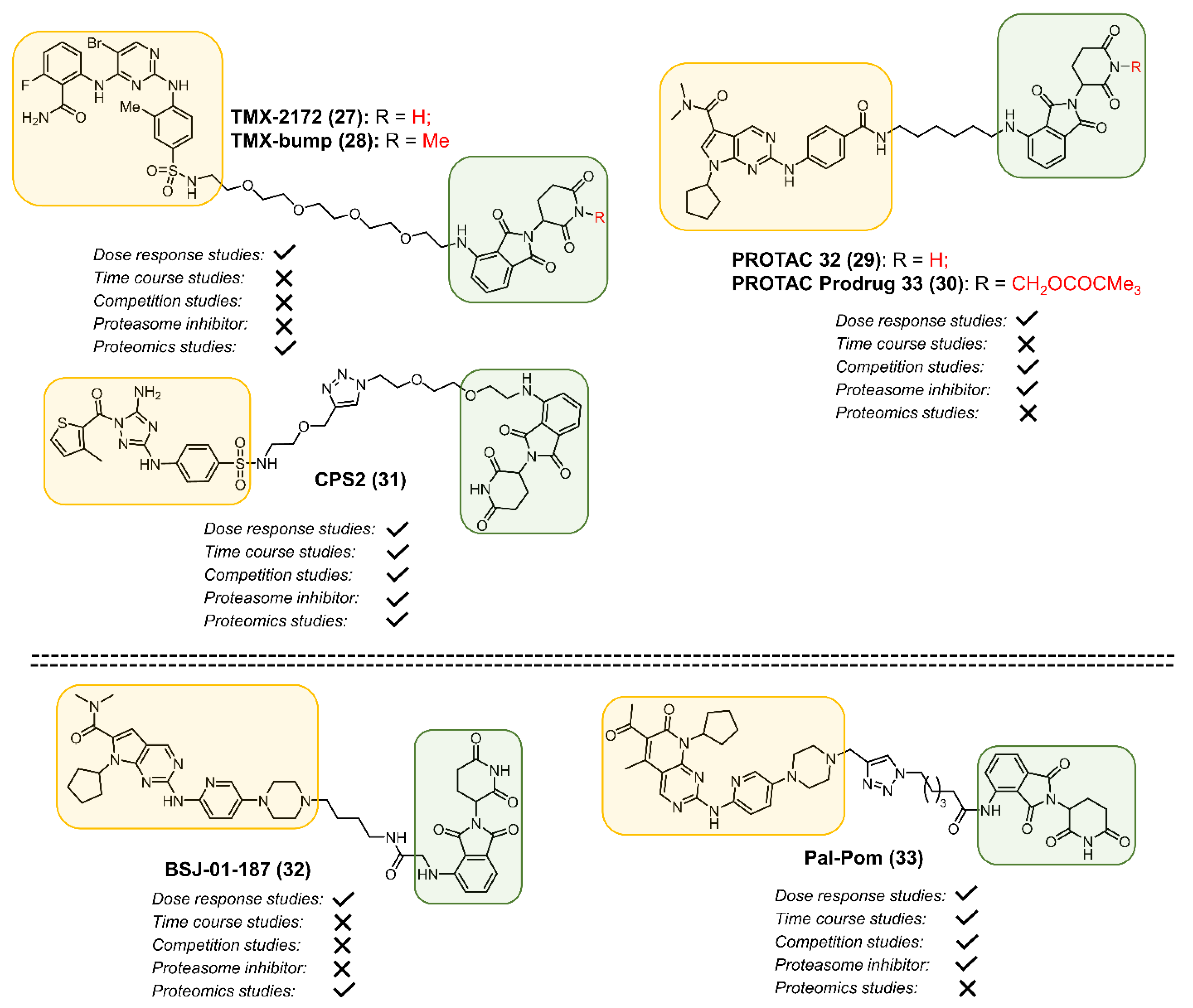
2.1. CDK2
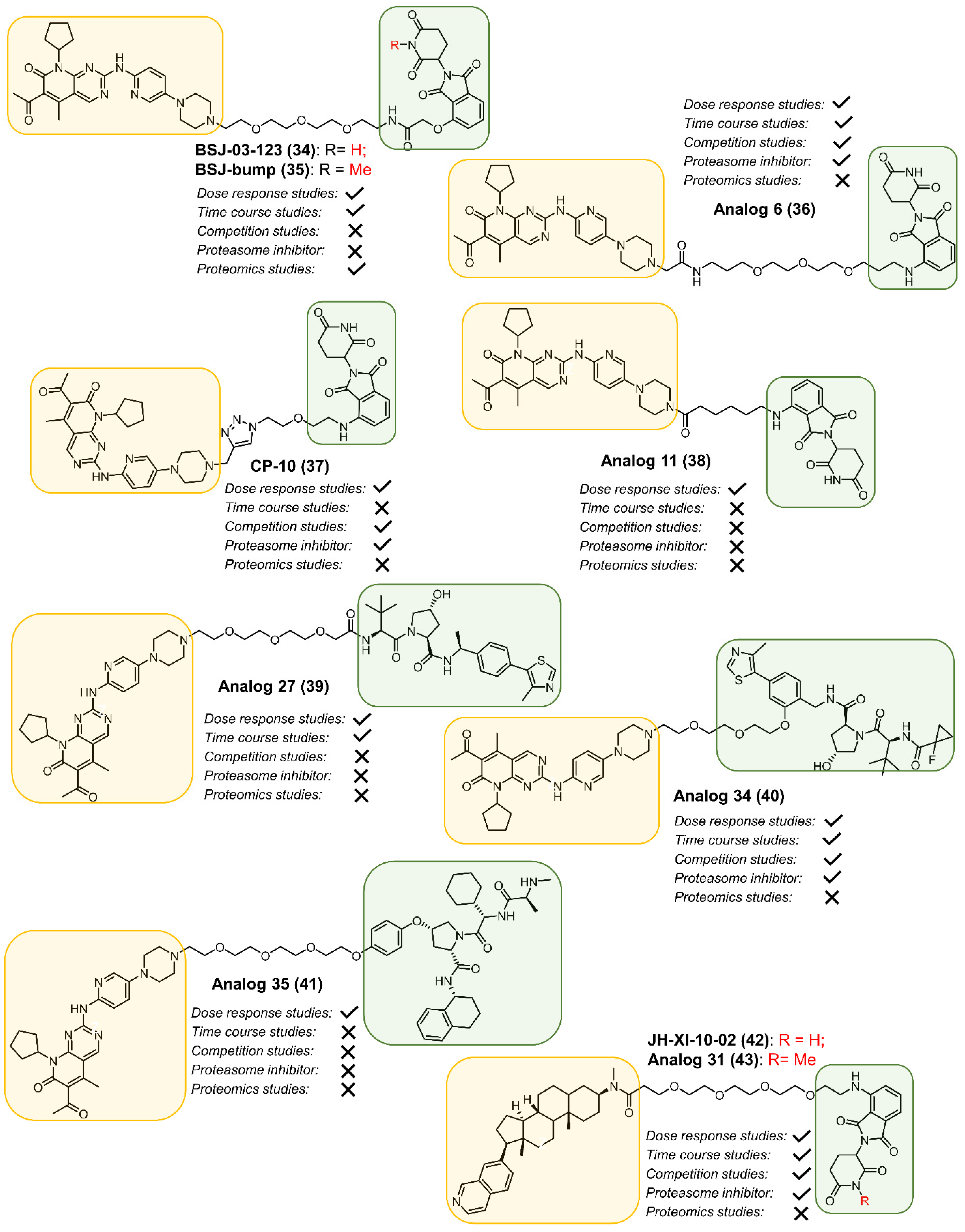
2.2. CDK4
2.3. CDK6
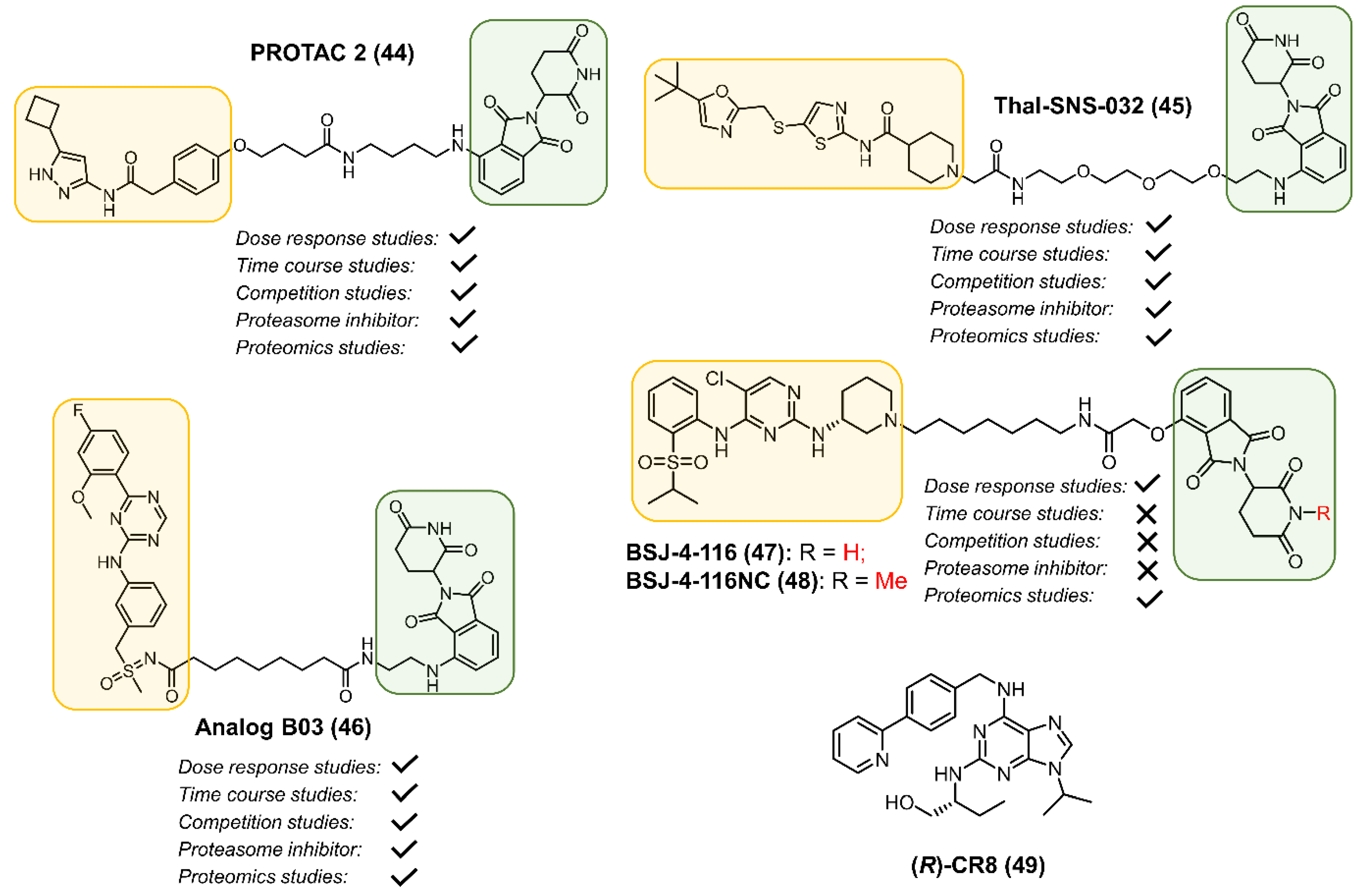
2.4. CDK8
2.5. CDK9
2.6. CDK12
3. Perturbing CDK Function by a Molecular Glue
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. 2013, 60, 2–9. [Google Scholar] [CrossRef]
- Gao, R.; Stock, A.M. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 2009, 63, 133–154. [Google Scholar] [CrossRef]
- Ferris, H.U.; Dunin-Horkawicz, S.; Hornig, N.; Hulko, M.; Martin, J.; Schultz, J.E.; Zeth, K.; Lupas, A.N.; Coles, M. Mechanism of regulation of receptor histidine kinases. Structure 2012, 20, 56–66. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef]
- Benn, C.L.; Dawson, L.A. Clinically Precedented Protein Kinases: Rationale for Their Use in Neurodegenerative Disease. Front. Aging Neurosci. 2020, 12, 242. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Modi, V.; Dunbrack, R.L., Jr. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef]
- Bernetti, M.; Masetti, M.; Rocchia, W.; Cavalli, A. Kinetics of Drug Binding and Residence Time. Annu. Rev. Phys. Chem. 2019, 70, 143–171. [Google Scholar] [CrossRef]
- Potashman, M.H.; Duggan, M.E. Covalent modifiers: An orthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef]
- Abdeldayem, A.; Raouf, Y.S.; Constantinescu, S.N.; Moriggl, R.; Gunning, P.T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Sonawane, Y.A.; Taylor, M.A.; Napoleon, J.V.; Rana, S.; Contreras, J.I.; Natarajan, A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J. Med. Chem. 2016, 59, 8667–8684. [Google Scholar] [CrossRef]
- Robb, C.M.; Kour, S.; Contreras, J.I.; Agarwal, E.; Barger, C.J.; Rana, S.; Sonawane, Y.; Neilsen, B.K.; Taylor, M.; Kizhake, S.; et al. Characterization of CDK(5) inhibitor, 20-223 (aka CP668863) for colorectal cancer therapy. Oncotarget 2018, 9, 5216–5232. [Google Scholar] [CrossRef]
- Chi, Y.; Carter, J.H.; Swanger, J.; Mazin, A.V.; Moritz, R.L.; Clurman, B.E. A novel landscape of nuclear human CDK2 substrates revealed by in situ phosphorylation. Sci. Adv. 2020, 6, eaaz9899. [Google Scholar] [CrossRef] [PubMed]
- Loyer, P.; Trembley, J.H.; Katona, R.; Kidd, V.J.; Lahti, J.M. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell. Signal. 2005, 17, 1033–1051. [Google Scholar] [CrossRef]
- Kour, S.; Rana, S.; Contreras, J.I.; King, H.M.; Robb, C.M.; Sonawane, Y.A.; Bendjennat, M.; Crawford, A.J.; Barger, C.J.; Kizhake, S.; et al. CDK5 Inhibitor Downregulates Mcl-1 and Sensitizes Pancreatic Cancer Cell Lines to Navitoclax. Mol. Pharmacol. 2019, 96, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef]
- Corona, S.P.; Generali, D. Abemaciclib: A CDK4/6 inhibitor for the treatment of HR+/HER2- advanced breast cancer. Drug Des. Dev. Ther. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 31 March 2021).
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef]
- Wang, S.; Han, L.; Han, J.; Li, P.; Ding, Q.; Zhang, Q.-J.; Liu, Z.-P.; Chen, C.; Yu, Y. Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nat. Chem. Biol. 2019, 15, 1223–1231. [Google Scholar] [CrossRef]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.-N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef]
- Hu, J.; Hu, B.; Wang, M.; Xu, F.; Miao, B.; Yang, C.-Y.; Wang, M.; Liu, Z.; Hayes, D.F.; Chinnaswamy, K.; et al. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 2019, 62, 1420–1442. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Fischer, E.S.; Boehm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDBI-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1a. Angew. Chem. Int. Ed. 2012, 51, 11463–11467. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1 alpha Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.L.; Vu, B.T.; Qing, W.G.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.-Y. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef]
- Tong, B.; Spradlin, J.N.; Novaes, L.F.T.; Zhang, E.; Hu, X.; Moeller, M.; Brittain, S.M.; McGregor, L.M.; McKenna, J.M.; Tallarico, J.A.; et al. A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem. Biol. 2020, 15, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Spradlin, J.N.; Boike, L.; Tong, B.; Brittain, S.M.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol. 2021, 28, 559–566. [Google Scholar] [CrossRef]
- Li, L.; Mi, D.; Pei, H.; Duan, Q.; Wang, X.; Zhou, W.; Jin, J.; Li, D.; Liu, M.; Chen, Y. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct. Target. Ther. 2020, 5, 129. [Google Scholar] [CrossRef]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Manford, A.G.; Spradlin, J.N.; Brittain, S.M.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Rape, M.; Nomura, D.K. Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications. bioRxiv 2021. [Google Scholar] [CrossRef]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306. [Google Scholar] [CrossRef]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Stevens, S.L.S.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79, 4744–4753. [Google Scholar] [CrossRef]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the Scaffold to Improve Focal Adhesion Kinase-Targeted Cancer Therapeutics. Sci. Signal. 2013, 6, pe10. [Google Scholar] [CrossRef]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef]
- Demizu, Y.; Okuhira, K.; Motoi, H.; Ohno, A.; Shoda, T.; Fukuhara, K.; Okuda, H.; Naito, M.; Kurihara, M. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett. 2012, 22, 1793–1796. [Google Scholar] [CrossRef]
- Demizu, Y.; Shibata, N.; Hattori, T.; Ohoka, N.; Motoi, H.; Misawa, T.; Shoda, T.; Naito, M.; Kurihara, M. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett. 2016, 26, 4865–4869. [Google Scholar] [CrossRef] [PubMed]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.A.; Trachet, E.; Albassam, M.; Zheng, X.X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1437. [Google Scholar] [PubMed]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; Xu, T.; Wang, M.; Yang, C.-Y.; Chinnaswamy, K.; Stuckey, J.; et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J. Med. Chem. 2019, 62, 11218–11231. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.-R.; Rasmusson, T.; Robinson, J.; Pachl, F.; Read, J.; Kawatkar, S.; O’Donovan, D.H.; Bagal, S.; Code, E.; Rawlins, P.; et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol. 2020, 27, 41–46. [Google Scholar] [CrossRef]
- Humphreys, P.G.; Bamborough, P.; Chung, C.-w.; Crags, P.D.; Gordon, L.; Grandi, P.; Hayhow, T.G.; Hussain, J.; Jones, K.L.; Lindon, M.; et al. Discovery of a Potent, Cell Penetrant, and Selective p300/CBP-Associated Factor (PCAF)/General Control Nonderepressible 5 (GCN5) Bromodomain Chemical Probe. J. Med. Chem. 2017, 60, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. 2019, 58, 6321–6326. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X-L PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.-H.; Ko, E.; Jang, J.; et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem. Biol. 2018, 25, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 2020, 27, 19–31 e16. [Google Scholar] [CrossRef]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Ma, T.; Van Tine, B.A.; Wei, Y.; Garrett, M.D.; Nelson, D.; Adams, P.D.; Wang, J.; Qin, J.; Chow, L.T.; Harper, J.W. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000, 14, 2298–2313. [Google Scholar] [CrossRef]
- Okuda, M.; Horn, H.F.; Tarapore, P.; Tokuyama, Y.; Smulian, A.G.; Chan, P.K.; Knudsen, E.S.; Hofmann, I.A.; Snyder, J.D.; Bove, K.E.; et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 2000, 103, 127–140. [Google Scholar] [CrossRef]
- Scaltriti, M.; Eichhorn, P.J.; Cortes, J.; Prudkin, L.; Aura, C.; Jimenez, J.; Chandarlapaty, S.; Serra, V.; Prat, A.; Ibrahim, Y.H.; et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl. Acad. Sci. USA 2011, 108, 3761–3766. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Australian Ovarian Cancer Study Group; Davis, S.; D’Andrea, A.D.; Simpson, K.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Yamakawa, M.; Takeda, H.; Kimura, S.; Takahashi, T. Expression of cell cycle markers in colorectal carcinoma: Superiority of cyclin A as an indicator of poor prognosis. Int. J. Cancer 1999, 84, 225–233. [Google Scholar] [CrossRef]
- Michalides, R.; van Tinteren, H.; Balkenende, A.; Vermorken, J.B.; Benraadt, J.; Huldij, J.; van Diest, P. Cyclin A is a prognostic indicator in early stage breast cancer with and without tamoxifen treatment. Br. J. Cancer 2002, 86, 402–408. [Google Scholar] [CrossRef]
- Han, Y.; Wei, Y.; Yao, J.; Chu, Y.Y.; Li, C.W.; Hsu, J.L.; Nie, L.; Hung, M.C. Inhibition of CDK2 reduces EZH2 phosphorylation and reactivates ERα expression in high-grade serous ovarian carcinoma. Am. J. Cancer Res. 2020, 10, 1194–1206. [Google Scholar] [PubMed]
- Ying, M.; Shao, X.; Jing, H.; Liu, Y.; Qi, X.; Cao, J.; Chen, Y.; Xiang, S.; Song, H.; Hu, R.; et al. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood 2018, 131, 2698–2711. [Google Scholar] [CrossRef]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 knockout mice are viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2019, 62, 4233–4251. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Jiang, J.; He, Z.; Kwiatkowski, N.P.; Donovan, K.A.; Mills, C.E.; Victor, C.; Hatcher, J.M.; Fischer, E.S.; Sorger, P.K.; et al. Development of CDK2 and CDK5 Dual Degrader TMX-2172. Angew. Chem. Int. Ed. Engl. 2020, 59, 13865–13870. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zhao, R.; Cao, Y.; Wei, Y.; Li, M.; Dong, Z.; Liu, Y.; Ruan, H.; Li, Y.; Cao, S.; et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur. J. Med. Chem. 2021, 209, 112903. [Google Scholar] [CrossRef]
- Wang, L.; Shao, X.; Zhong, T.; Wu, Y.; Xu, A.; Sun, X.; Gao, H.; Liu, Y.; Lan, T.; Tong, Y.; et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat. Chem. Biol. 2021, 17, 567–575. [Google Scholar] [CrossRef]
- Corces, M.R.; Chang, H.Y.; Majeti, R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 263. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Schmidt, E.E.; Ichimura, K.; Reifenberger, G.; Collins, V.P. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994, 54, 6321–6324. [Google Scholar]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Zhao, B.; Burgess, K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. 2019, 55, 2704–2707. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Bendjennat, M.; Kour, S.; King, H.M.; Kizhake, S.; Zahid, M.; Natarajan, A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379. [Google Scholar] [CrossRef]
- Lu, H.; Schulze-Gahmen, U. Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J. Med. Chem. 2006, 49, 3826–3831. [Google Scholar] [CrossRef]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.A.; Cryan, J.; Ahmed, A.; Dai, H.; McGonagle, G.A.; Rozier, C.; Benowitz, A.B. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg. Med. Chem. Lett. 2020, 30, 127106. [Google Scholar] [CrossRef]
- Steinebach, C.; Ng, Y.L.D.; Sosic, I.; Lee, C.S.; Chen, S.; Lindner, S.; Vu, L.P.; Bricelj, A.; Haschemi, R.; Monschke, M.; et al. Systematic exploration of different E3 ubiquitin ligases: An approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11, 3474–3486. [Google Scholar] [CrossRef]
- Firestein, R.; Shima, K.; Nosho, K.; Irahara, N.; Baba, Y.; Bojarski, E.; Giovannucci, E.L.; Hahn, W.C.; Fuchs, C.S.; Ogino, S. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 2010, 126, 2863–2873. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Donner, A.J.; Szostek, S.; Hoover, J.M.; Espinosa, J.M. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 2007, 27, 121–133. [Google Scholar] [CrossRef]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 2004, 16, 509–520. [Google Scholar] [CrossRef]
- Alarcón, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Philip, S.; Kumarasiri, M.; Teo, T.; Yu, M.; Wang, S. Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? J. Med. Chem. 2018, 61, 5073–5092. [Google Scholar] [CrossRef]
- Menzl, I.; Witalisz-Siepracka, A.; Sexl, V. CDK8-Novel Therapeutic Opportunities. Pharmaceuticals 2019, 12, 92. [Google Scholar] [CrossRef]
- Dannappel, M.V.; Sooraj, D.; Loh, J.J.; Firestein, R. Molecular and in vivo Functions of the CDK8 and CDK19 Kinase Modules. Front. Cell Dev. Biol. 2018, 6, 171. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wang, E.S.; Johannessen, L.; Kwiatkowski, N.; Sim, T.; Gray, N.S. Development of Highly Potent and Selective Steroidal Inhibitors and Degraders of CDK8. ACS Med. Chem. Lett. 2018, 9, 540–545. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Sun, Y.P.; Peng, X.S.; Polet, D.; Chen, D.Y. Total synthesis of (+)-cortistatin A. Angew. Chem. Int. Ed. Engl. 2008, 47, 7310–7313. [Google Scholar] [CrossRef]
- Shenvi, R.A.; Guerrero, C.A.; Shi, J.; Li, C.C.; Baran, P.S. Synthesis of (+)-cortistatin A. J. Am. Chem. Soc. 2008, 130, 7241–7243. [Google Scholar] [CrossRef][Green Version]
- Simmons, E.M.; Hardin-Narayan, A.R.; Guo, X.; Sarpong, R. Formal total synthesis of (+/−)-cortistatin A. Tetrahedron 2010, 66, 4696–4700. [Google Scholar] [CrossRef][Green Version]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Albert, T.K.; Rigault, C.; Eickhoff, J.; Baumgart, K.; Antrecht, C.; Klebl, B.; Mittler, G.; Meisterernst, M. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. Br. J. Pharmacol. 2014, 171, 55–68. [Google Scholar] [CrossRef]
- Yin, T.; Lallena, M.J.; Kreklau, E.L.; Fales, K.R.; Carballares, S.; Torrres, R.; Wishart, G.N.; Ajamie, R.T.; Cronier, D.M.; Iversen, P.W.; et al. A novel CDK9 inhibitor shows potent antitumor efficacy in preclinical hematologic tumor models. Mol. Cancer Ther. 2014, 13, 1442–1456. [Google Scholar] [CrossRef]
- Contreras, J.I.; Robb, C.M.; King, H.M.; Baxter, J.; Crawford, A.J.; Kour, S.; Kizhake, S.; Sonawane, Y.A.; Rana, S.; Hollingsworth, M.A.; et al. Chemical Genetic Screens Identify Kinase Inhibitor Combinations that Target Anti-Apoptotic Proteins for Cancer Therapy. ACS Chem. Biol. 2018, 13, 1148–1152. [Google Scholar] [CrossRef]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef]
- Franco, L.C.; Morales, F.; Boffo, S.; Giordano, A. CDK9: A key player in cancer and other diseases. J. Cell Biochem. 2018, 119, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Lam, F.; Zhong, L.; Teo, T.; Adams, J.; Yu, M.; Milne, R.W.; Pepper, C.; Lokman, N.A.; Ricciardelli, C.; et al. Targeting CDK9 for treatment of colorectal cancer. Mol. Oncol. 2019, 13, 2178–2193. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019, 33, 5990–6000. [Google Scholar] [CrossRef]
- Krystof, V.; Uldrijan, S. Cyclin-dependent kinase inhibitors as anticancer drugs. Curr. Drug Targets 2010, 11, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Sonawane, Y.A.; Taylor, M.A.; Kizhake, S.; Zahid, M.; Natarajan, A. Synthesis of aminopyrazole analogs and their evaluation as CDK inhibitors for cancer therapy. Bioorg. Med. Chem. Lett. 2018, 28, 3736–3740. [Google Scholar] [CrossRef]
- King, H.M.; Rana, S.; Kubica, S.P.; Mallareddy, J.R.; Kizhake, S.; Ezell, E.L.; Zahid, M.; Naldrett, M.J.; Alvarez, S.; Law, H.C.; et al. Aminopyrazole based CDK9 PROTAC sensitizes pancreatic cancer cells to venetoclax. Bioorg. Med. Chem. Lett. 2021, 43, 128061. [Google Scholar] [CrossRef]
- Lopez, H.; Zhang, L.; George, N.M.; Liu, X.; Pang, X.; Evans, J.J.D.; Targy, N.M.; Luo, X. Perturbation of the Bcl-2 network and an induced Noxa/Bcl-xL interaction trigger mitochondrial dysfunction after DNA damage. J. Biol. Chem. 2010, 285, 15016–15026. [Google Scholar] [CrossRef]
- Rana, S.; Kour, S.; Sonawane, Y.A.; Robb, C.M.; Contreras, J.I.; Kizhake, S.; Zahid, M.; Karpf, A.R.; Natarajan, A. Symbiotic prodrugs (SymProDs) dual targeting of NFkappaB and CDK. Chem. Biol. Drug Des. 2020, 96, 773–784. [Google Scholar] [CrossRef]
- MacCallum, D.E.; Melville, J.; Frame, S.; Watt, K.; Anderson, S.; Gianella-Borradori, A.; Lane, D.P.; Green, S.R. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [Google Scholar] [CrossRef]
- Abid, M.; Sonawane, Y.A.; Contreras, J.I.; Rana, S.; Natarajan, A. Recent Advances in Cancer Drug Development: Targeting Induced Myeloid Cell Leukemia-1 (Mcl-1) Differentiation Protein. Curr. Med. Chem. 2017, 24, 4488–4514. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, L.; Cao, C.; Yu, J.; Luo, X.; Zhou, P.; Zhao, L.; Du, W.; Cheng, J.; Xie, Y.; et al. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur. J. Med. Chem. 2020, 187, 111952. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.N.; Xiao, H.Y.; Kim, K.S.; Lu, S.; Han, W.C.; Barbosa, S.A.; Hunt, J.T.; Rawlins, D.B.; Shan, W.; Ahmed, S.Z.; et al. N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J. Med. Chem. 2004, 47, 1719–1728. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Barsanti, P.A.; Hu, C.; Jeff, J.; Keyes, R.; Kucejko, R.; Xiaodong, L.; Yue, P.; Pfister Kb, S.M.; Sutton, J.; Lifeng, W. Pyridine and Pyrzaine Derivatives as Protein Kinase Modulators. International Patent PCT/JP2008/073864 (WO/2011/012661), 3 February 2011. [Google Scholar]
- Polier, G.; Ding, J.; Konkimalla, B.V.; Eick, D.; Ribeiro, N.; Kohler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011, 2, e182. [Google Scholar] [CrossRef]
- Bian, J.; Ren, J.; Li, Y.; Wang, J.; Xu, X.; Feng, Y.; Tang, H.; Wang, Y.; Li, Z. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg. Chem. 2018, 81, 373–381. [Google Scholar] [CrossRef]
- Chen, R.; Keating, M.J.; Gandhi, V.; Plunkett, W. Transcription inhibition by flavopiridol: Mechanism of chronic lymphocytic leukemia cell death. Blood 2005, 106, 2513–2519. [Google Scholar] [CrossRef]
- Chen, R.; Guo, L.; Chen, Y.; Jiang, Y.; Wierda, W.G.; Plunkett, W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood 2011, 117, 156–164. [Google Scholar] [CrossRef]
- Qiu, X.; Li, Y.; Yu, B.; Ren, J.; Huang, H.; Wang, M.; Ding, H.; Li, Z.; Wang, J.; Bian, J. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur. J. Med. Chem. 2021, 211, 113091. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Hu, L.; Wu, Z.; Chen, Z.; Liu, S.; Xu, X.; Qian, A. CDK12: A Potent Target and Biomarker for Human Cancer Therapy. Cells 2020, 9, 1483. [Google Scholar] [CrossRef]
- Chen, H.H.; Wang, Y.C.; Fann, M.J. Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol. Cell. Biol. 2006, 26, 2736–2745. [Google Scholar] [CrossRef]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell. Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef]
- Jiang, B.; Gao, Y.; Che, J.; Lu, W.; Kaltheuner, I.H.; Dries, R.; Kalocsay, M.; Berberich, M.J.; Jiang, J.; You, I.; et al. Discovery and resistance mechanism of a selective CDK12 degrader. Nat. Chem. Biol. 2021, 17, 1057–1064. [Google Scholar] [CrossRef]
- den Besten, W.; Lipford, J.R. Prospecting for molecular glues. Nat. Chem. Biol. 2020, 16, 1157–1158. [Google Scholar] [CrossRef]
- Schreiber, S.L. The Rise of Molecular Glues. Cell 2021, 184, 3–9. [Google Scholar] [CrossRef]
- Munn, J.D. Thalidomide and Congenital Malformations. Can. Med. Assoc. J. 1962, 86, 665. [Google Scholar] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Asatsuma-Okumura, T.; Ando, H.; De Simone, M.; Yamamoto, J.; Sato, T.; Shimizu, N.; Asakawa, K.; Yamaguchi, Y.; Ito, T.; Guerrini, L.; et al. p63 is a cereblon substrate involved in thalidomide teratogenicity. Nat. Chem. Biol. 2019, 15, 1077–1084. [Google Scholar] [CrossRef]
- Donovan, K.A.; An, J.; Nowak, R.P.; Yuan, J.C.; Fink, E.C.; Berry, B.C.; Ebert, B.L.; Fischer, E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 2018, 7, e38430. [Google Scholar] [CrossRef] [PubMed]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.-C.; Miller, K.; Fang, W.; Wang, N.-Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Slabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thoma, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. [Google Scholar] [CrossRef] [PubMed]
- Zangari, M.; Elice, F.; Tricot, G. Immunomodulatory drugs in multiple myeloma. Expert Opin. Investig. Drugs 2005, 14, 1411–1418. [Google Scholar] [CrossRef]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef]
- Bussiere, D.E.; Xie, L.; Srinivas, H.; Shu, W.; Burke, A.; Be, C.; Zhao, J.; Godbole, A.; King, D.; Karki, R.G.; et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat. Chem. Biol. 2020, 16, 15–23. [Google Scholar] [CrossRef]
- Du, X.; Volkov, O.A.; Czerwinski, R.M.; Tan, H.; Huerta, C.; Morton, E.R.; Rizzi, J.P.; Wehn, P.M.; Xu, R.; Nijhawan, D.; et al. Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 2019, 27, 1625–1633. [Google Scholar] [CrossRef]
- Faust, T.B.; Yoon, H.; Nowak, R.P.; Donovan, K.A.; Li, Z.; Cai, Q.; Eleuteri, N.A.; Zhang, T.; Gray, N.S.; Fischer, E.S. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat. Chem. Biol. 2020, 16, 7–14. [Google Scholar] [CrossRef]
- Slabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [PubMed]
- Slabicki, M.; Yoon, H.; Koeppel, J.; Nitsch, L.; Roy Burman, S.S.; Di Genua, C.; Donovan, K.A.; Sperling, A.S.; Hunkeler, M.; Tsai, J.M.; et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature 2020, 588, 164–168. [Google Scholar] [CrossRef]
- Rana, S.; Natarajan, A. Small molecule induced polymerization of BCL6 facilitates SIAH1 mediated degradation. Signal Transduct. Target. Ther. 2021, 6, 142. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
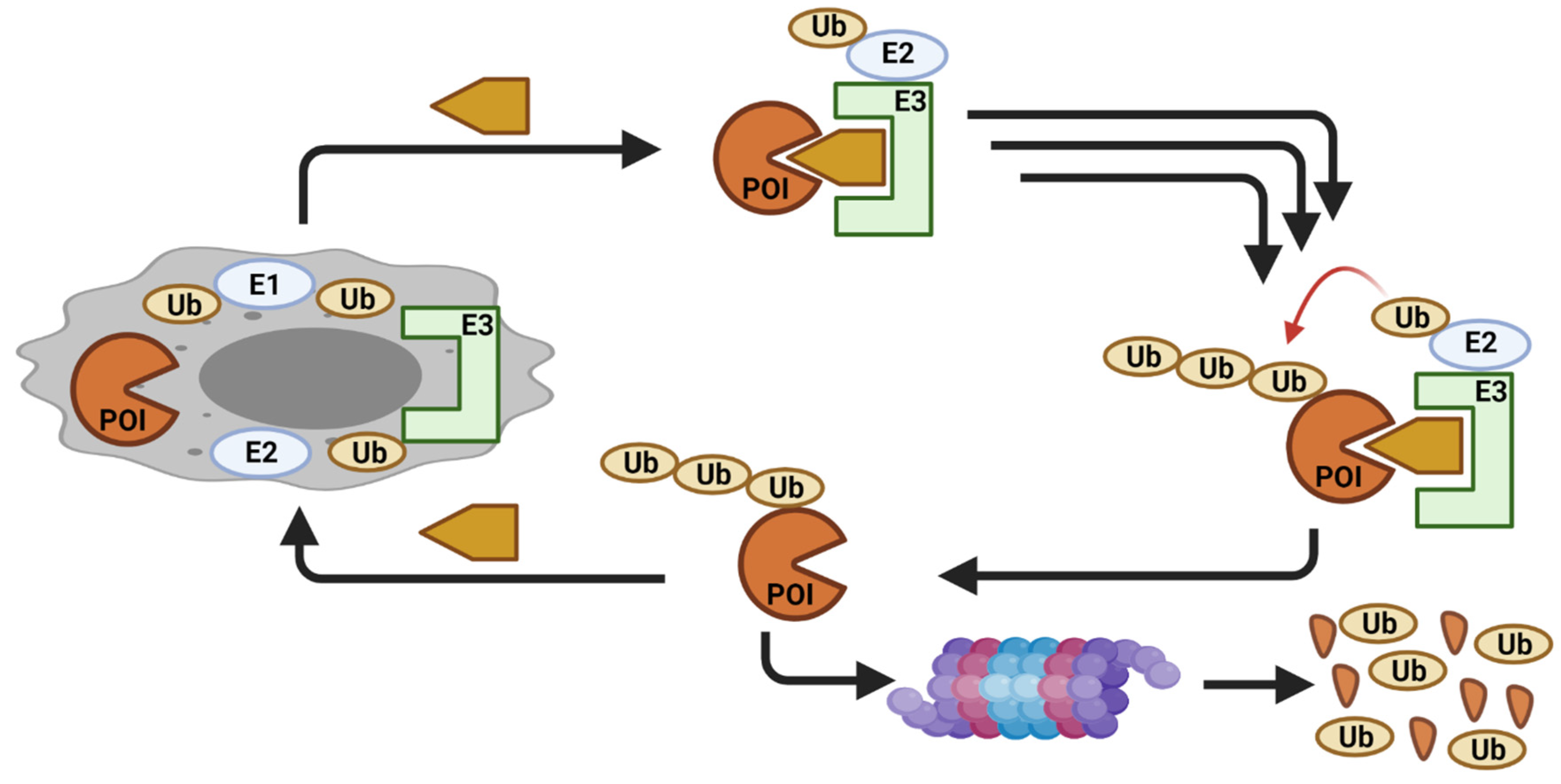{kind=link}
| NCT No. | Conditions | Interventions | Phase |
|---|---|---|---|
| 04802759 a | BC | GDC-9545, Abemaciclib, Ipatasertib, GDC-0077, Ribociclib, Everolimus | I, II |
| 04607668 a | MCC | Trilaciclib | III |
| 04585724 a | BC | Abemaciclib, Palbociclib, Ribociclib | I |
| 04553133 a | SCLC, NSCLC, TNBC, EGFR2, OC | PF-07104091 monotherapy, PF-07104091 + Palbociclib, PF-07104091 + Palbociclib + Letrozole | II |
| 04469764 a | EC, OC | Abemaciclib, Anastrozole, Letrozole | II |
| 04282031 a | ST, BC | BPI-1178 c, Fulvestrant, Letrozole | I, II |
| 04247126 a | ST, BC, SCLC | SY-5609 d, Fulvestrant | I |
| 04116541 a | ST | HDM201, Ribociclib, Cabozantinib, Alectinib | II |
| 04049227 a | OC | Abemaciclib, Letrozole | I |
| 04040205 a | STS | Abemaciclib | II |
| 04010357 a | SCLC | Abemaciclib | II |
| 03959891 a | BC | Ipatasertib, Fulvestrant, Palbociclib | I |
| 03740334 a | ALL | Ribociclib, Dexamethasone, Everolimus | I |
| 03675893 a | EC | Letrozole, Abemaciclib | II |
| 03593915 b | MS | Alvocidib + Decitabine, or Azacitidine | I, II |
| 03455270 b | BC | G1T48, Palbociclib | I |
| 03439735 a | BC | AI and Palbociclib | II |
| 03386929 b | NSCLC | Avelumab e, Axitinib, Palbociclib | I, II |
| 03363893 a | ST | CT7001, CT7001 + Fulvestrant | I, II |
| 03310879 a | Cancer | Abemaciclib | II |
| 03285412 a | BC | Ribociclib, ET | II |
| 03280563 a | BC | Atezolizumab f, Bevacizumab g, Entinostat, Exemestane, Fulvestrant Ipatasertib, Tamoxifen, Abemaciclib | I, II |
| 03227328 a | BC | CDK4/6 inhibitor + ET, chemotherapy + ET | II |
| 03220178 a | BC | Palbociclib, Fulvestrant, Anastrozole, Letrozole, Exemestane | IV |
| 03170206 a | LC | Binimetinib, Palbociclib | I, II |
| 03155997 a | BC | Abemaciclib, ET | III |
| 03110744 a | BC | Palbociclib | II |
| 02712723 a | BC | Letrozole, Ribociclib | II |
| 02644460 a | BT, ST, ES, BC | Abemaciclib | I |
| 02632045 a | BC | LEE-011 (Ribociclib), Fulvestrant | II |
| 02626507 a | BC | Gedatolisib, Faslodex, Palbociclib, Zoladex | I |
| 02603679 a | BC | Paclitaxel, Tamoxifen + Palbociclib, AI + Palbociclib, Goserelin + AI + Palbociclib, | II |
| 02599714 b | BC | AZD2014, Palbociclib, Fulvestrant | I |
| 02592083 b | BC | Tamoxifen, Goserelin, Palbociclib | II |
| 02586675 b | BC | Tamoxifen, Ribociclib, Goserelin | I |
| 02503709 b | ST | AT7519, Onalespib | I |
| 02499146 b | BC | Palbociclib, Letrozole | I |
| 02095132 b | BT | Adavosertib, Irinotecan Hydrochloride | I, II |
| 01864746 b | HR | Palbociclib | III |
| 01723774 a | BC | PD0332991, Anastrozole, Goserelin | II |
| 01676753 b | BC, TNBC | Dinaciclib, Pembrolizumab h | I |
| 01522989 b | ST | PD-0332991, 5-FU, Oxaliplatin | I |
| 01434316 a | ST | Dinaciclib, Veliparib | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, S.; Mallareddy, J.R.; Singh, S.; Boghean, L.; Natarajan, A. Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase. Cancers 2021, 13, 5506. https://doi.org/10.3390/cancers13215506
Rana S, Mallareddy JR, Singh S, Boghean L, Natarajan A. Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase. Cancers. 2021; 13(21):5506. https://doi.org/10.3390/cancers13215506
Chicago/Turabian StyleRana, Sandeep, Jayapal Reddy Mallareddy, Sarbjit Singh, Lidia Boghean, and Amarnath Natarajan. 2021. "Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase" Cancers 13, no. 21: 5506. https://doi.org/10.3390/cancers13215506
APA StyleRana, S., Mallareddy, J. R., Singh, S., Boghean, L., & Natarajan, A. (2021). Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase. Cancers, 13(21), 5506. https://doi.org/10.3390/cancers13215506