
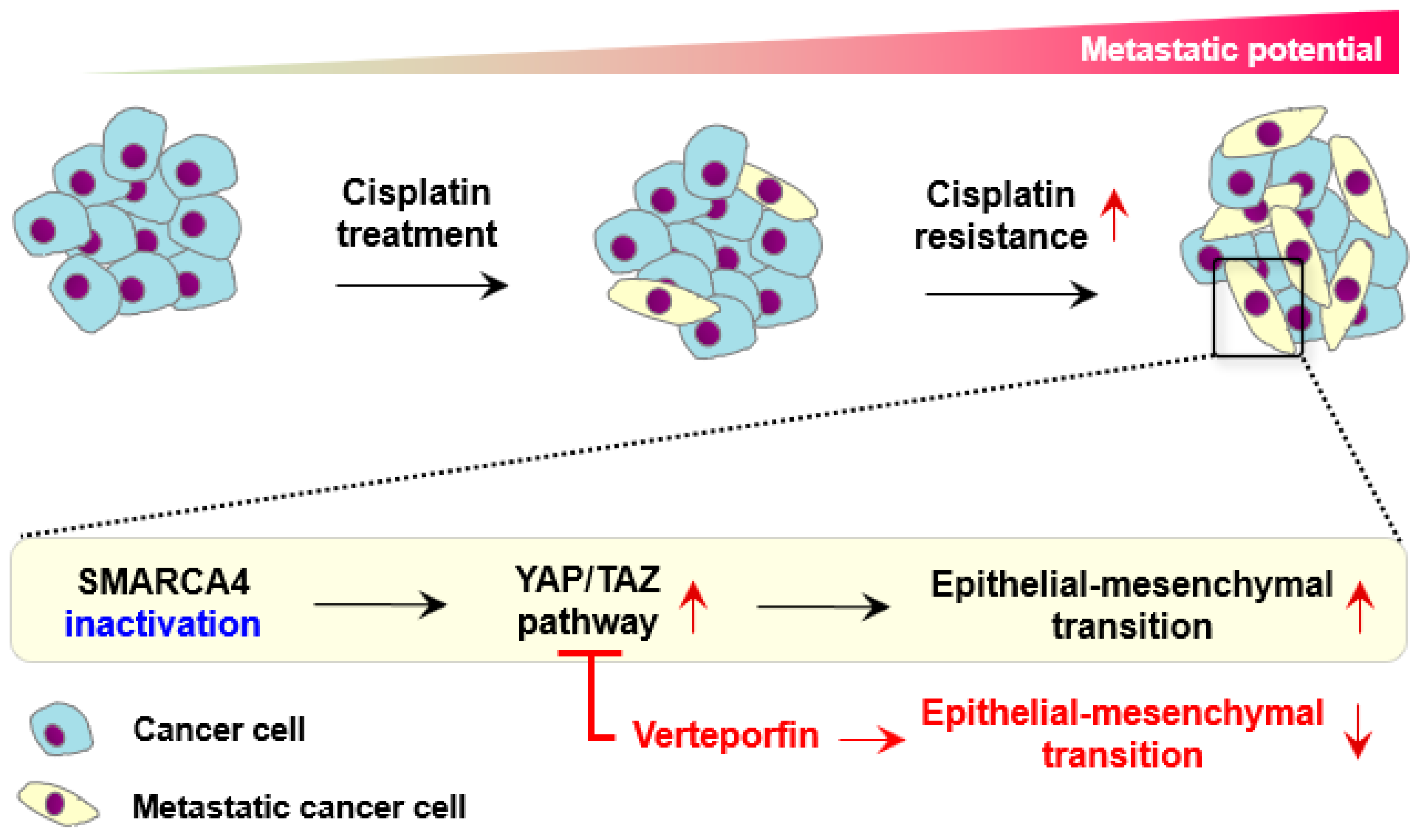
SMARCA4 Depletion Induces Cisplatin Resistance by Activating YAP1-Mediated Epithelial-to-Mesenchymal Transition in Triple-Negative Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. TNBC Patient Cohort and Sample Preprocessing
2.2. Variant Analysis of Cisplatin-Treated TNBC Patients
2.3. Gene Expression Analysis Based on SMARCA4 Activity and BC Subtype
2.4. SMARCA4 Knockout Cells and Cisplatin Response
2.4.1. Cell Culture and Reagents
2.4.2. Generation of Stable SMARCA4 KO Cell Lines Using CRISPR/Cas9
2.4.3. Cell Viability Analysis
2.4.4. Quantitative Reverse Transcription PCR (RT-qPCR) and Immunoblot Analysis
2.4.5. Flow Cytometry
2.4.6. Cell Migration Assay and Wound-Healing Assay
2.4.7. Immunofluorescence Assay
2.5. RNA-Seq Analysis of SMARCA4 KO TNBC Cells
3. Results
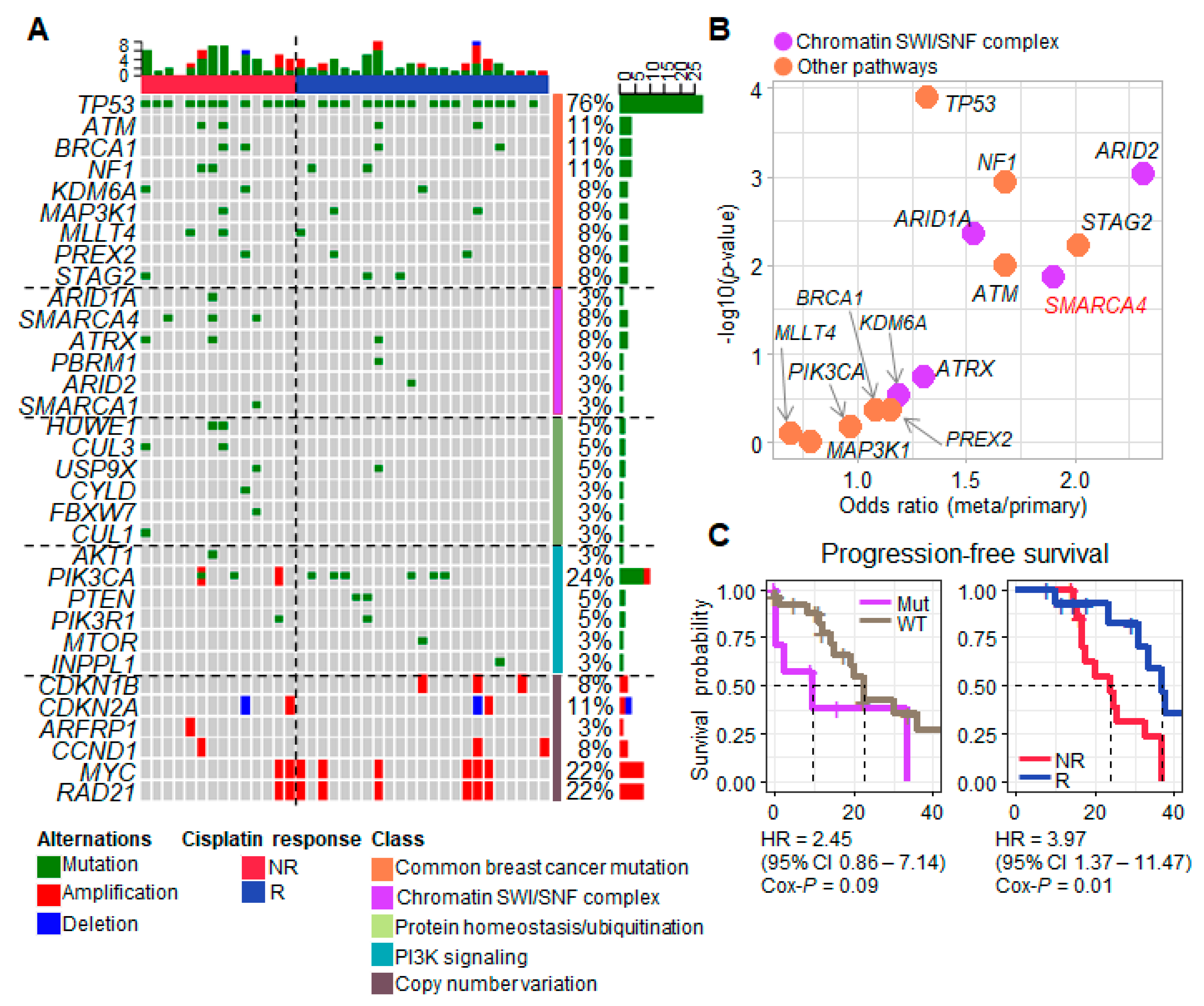
3.1. Progression-Associated Driver Gene Variants Identified in Samples from TNBC Patients Treated with Neoadjuvant Cisplatin
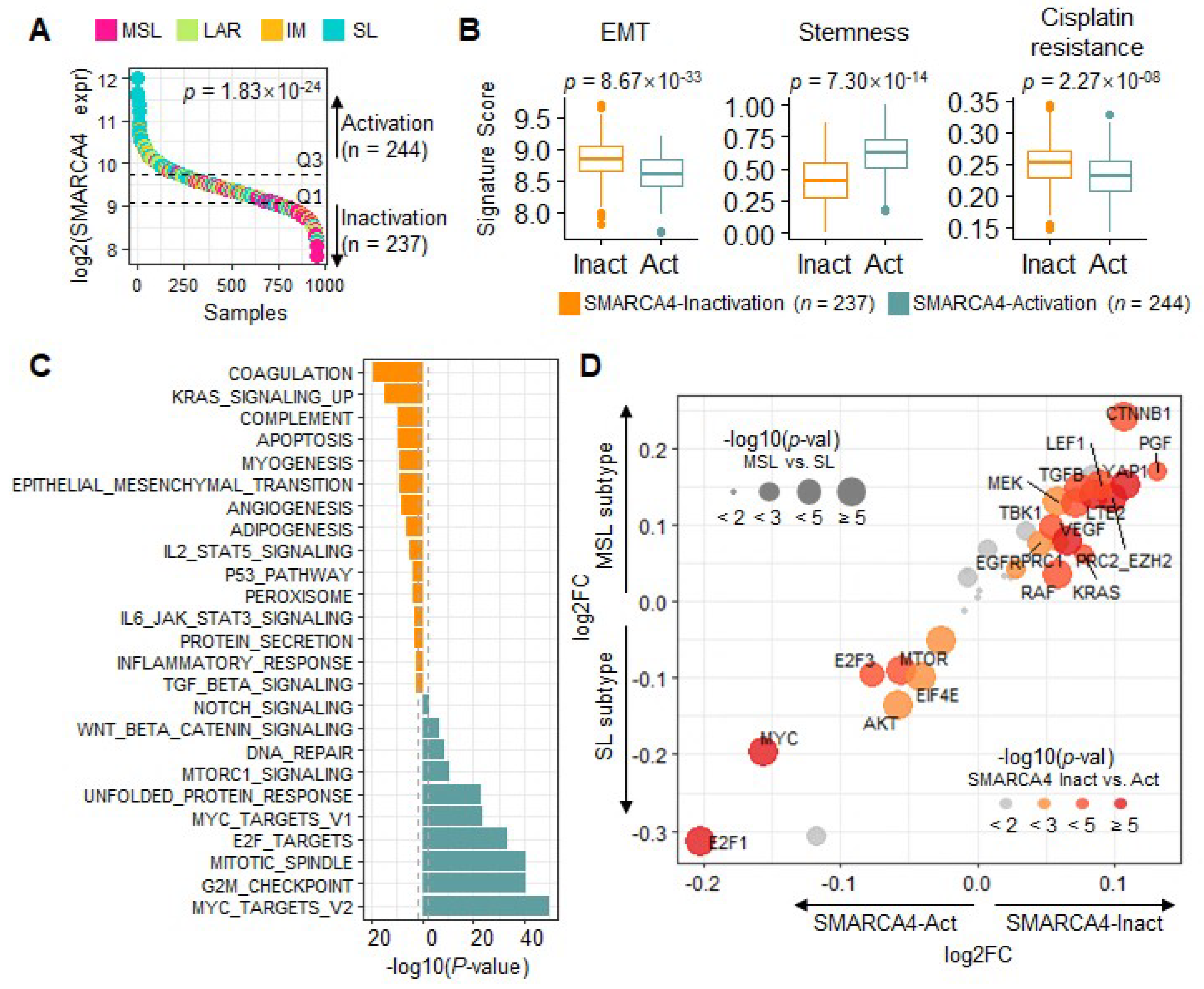
3.2. Transcriptional Characteristics and Subtypes of TNBC Are Regulated by SMARCA4
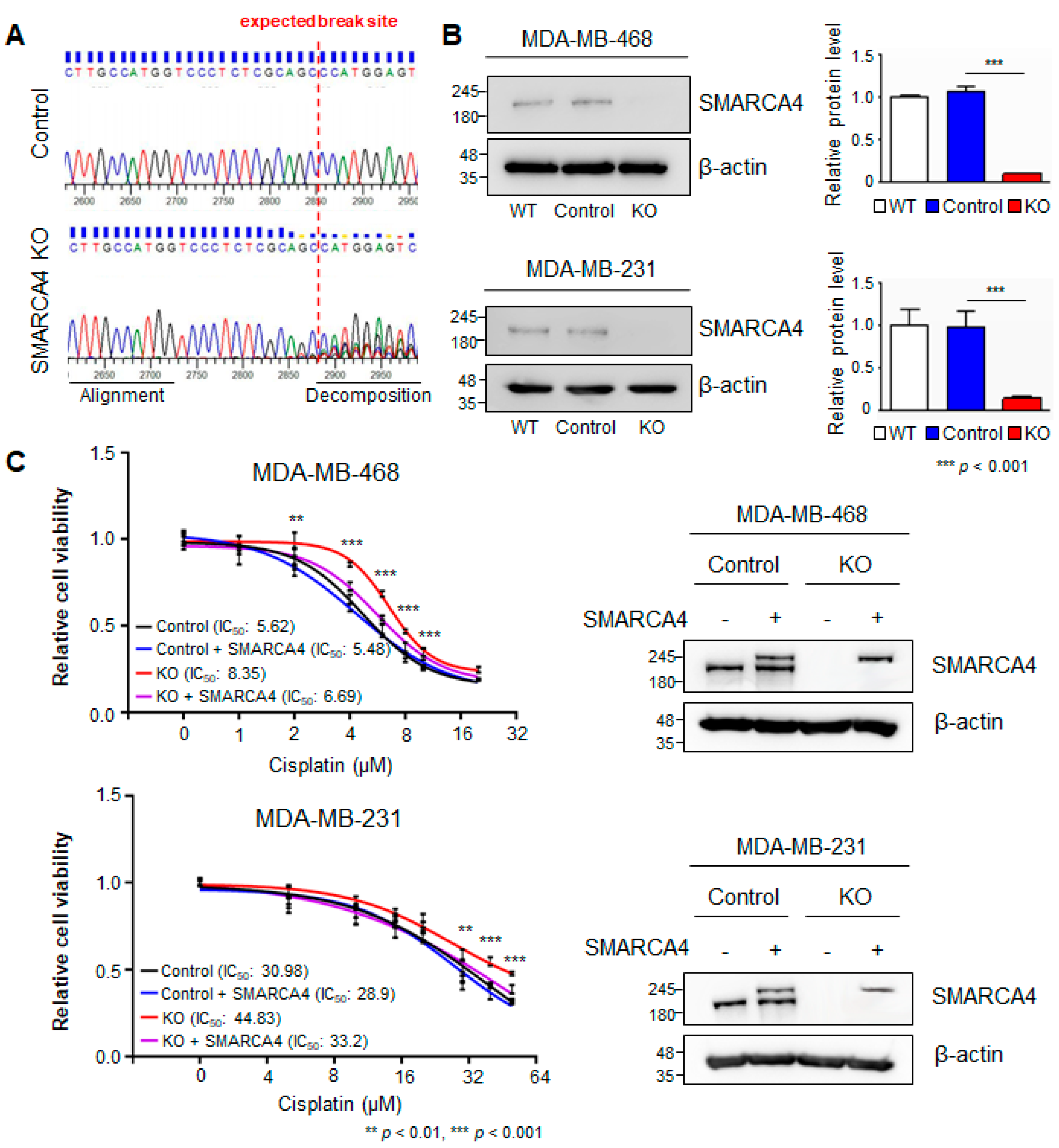
3.3. SMARCA4 Knock-Out to Evaluate Cisplatin Resistance
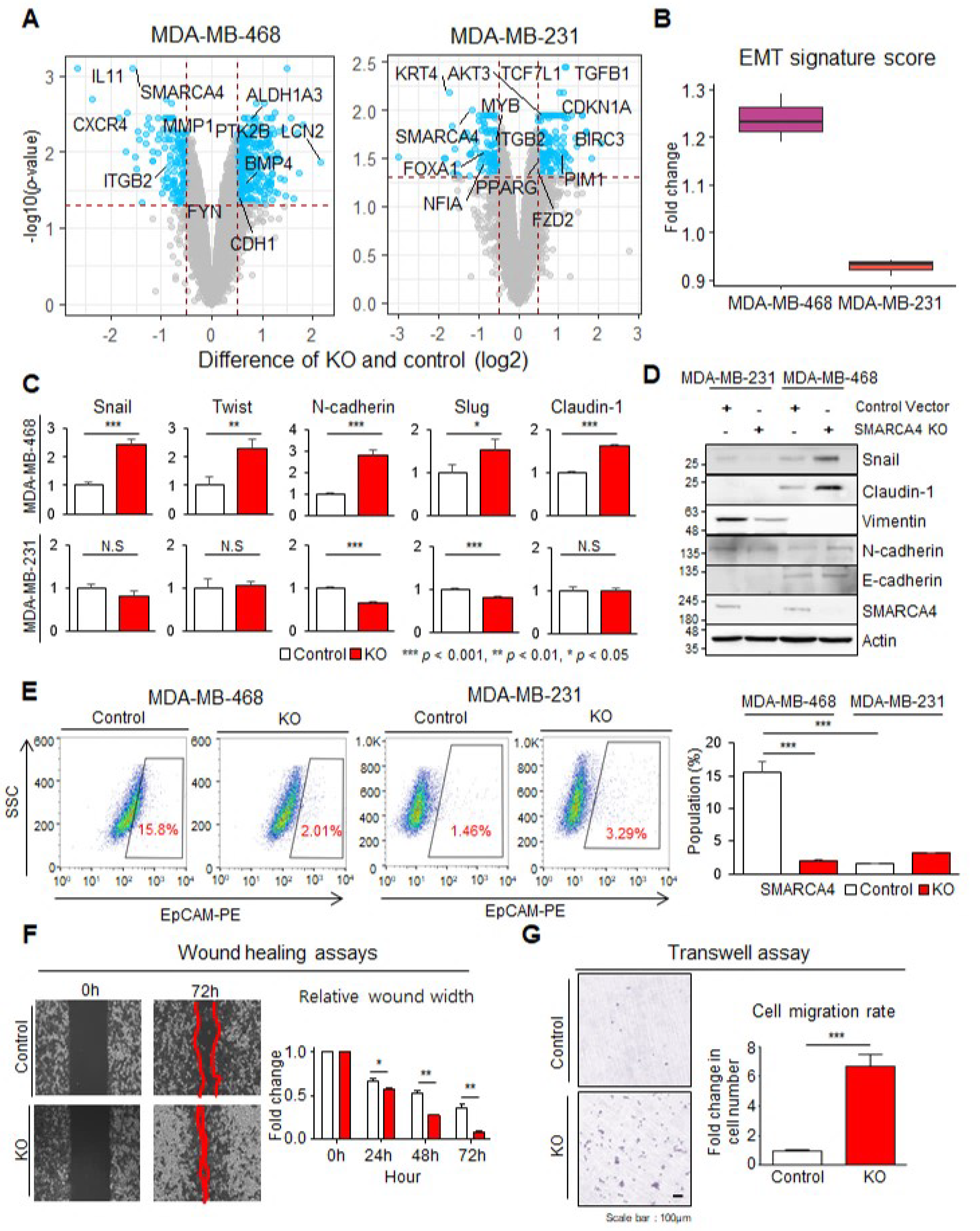
3.4. SMARCA4 KO Transforms Basal-like MDA-MB-468 Cells to an EMT Activated Cell Line
3.5. Hippo Pathway Activation in Response to SMARCA4 KO and the Response to YAP/TAZ Inhibitor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pistelli, M.; Pagliacci, A.; Battelli, N.; Santinelli, A.; Biscotti, T.; Ballatore, Z.; Berardi, R.; Cascinu, S. Prognostic factors in early-stage triple-negative breast cancer: Lessons and limits from clinical practice. Anticancer Res. 2013, 33, 2737–2742. [Google Scholar]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional Relapse and Distant Metastasis in Conservatively Managed Triple Negative Early-Stage Breast Cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Caparica, R.; Lambertini, M.; de Azambuja, E. How I treat metastatic triple-negative breast cancer. ESMO Open 2019, 4, e000504. [Google Scholar] [CrossRef] [Green Version]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yu, D.; Kwon, Y.; Lee, K.S.; Sim, S.H.; Kong, S.Y.; Lee, E.S.; Park, I.H.; Park, C. Genomic characteristics of triple-negative breast cancer nominate molecular subtypes that predict chemotherapy response. Mol. Cancer Res. 2020, 18, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 557. [Google Scholar] [CrossRef]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gonen, M.; Soslow, R.A.; et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Hodges, H.C.; Stanton, B.Z.; Cermakova, K.; Chang, C.Y.; Miller, E.L.; Kirkland, J.G.; Ku, W.L.; Veverka, V.; Zhao, K.; Crabtree, G.R. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 2018, 25, 61–72. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.H.; Chakraborty, A.R.; Mo, X.; Liu, Z.; Shilo, K.; Kirste, S.; Stegmaier, P.; McNulty, M.; Karachaliou, N.; Rosell, R.; et al. SMARCA4/BRG1 is a novel prognostic biomarker predictive of cisplatin-based chemotherapy outcomes in resected non-small cell lung cancer. Clin. Cancer Res. 2016, 22, 2396–2404. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Bandlamudi, C.; Lavery, J.A.; Montecalvo, J.; Namakydoust, A.; Rizvi, H.; Egger, J.; Concepcion, C.P.; Paul, S.; Arcila, M.E.; et al. The genomic landscape of SMARCA4 alterations and associations with outcomes in patients with lung cancer. Clin. Cancer Res. 2021, 26, 5701–5708. [Google Scholar] [CrossRef]
- Guerrero-Martínez, J.A.; Reyes, J.C. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci. Rep. 2018, 8, 2043. [Google Scholar] [CrossRef] [Green Version]
- Barutcu, A.R.; Lajoie, B.R.; Fritz, A.J.; McCord, R.P.; Nickerson, J.A.; Van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Dekker, J.; Stein, G.S.; et al. SMARCA4 regulates gene expression and higherorder chromatin structure in proliferating mammary epithelial cells. Genome Res. 2016, 26, 1188–1201. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, M. Introducing the Metastatic Breast Cancer Project: A novel patient-partnered initiative to accelerate understanding of MBC. ESMO Open 2018, 3, e000452. [Google Scholar] [CrossRef] [Green Version]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.H.; Lichtenstein, L.; Gupta, M.; Lawrence, M.S.; Pugh, T.J.; Saksena, G.; Meyerson, M.; Getz, G. Oncotator: Cancer variant annotation tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef]
- D’Aurizio, R.; Pippucci, T.; Tattini, L.; Giusti, B.; Pellegrini, M.; Magi, A. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 2016, 44, e154. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, 690–705.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database Hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, M.S.; Huang, R.Y.-J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; van Steensel, B. Rapid Quantitative Evaluation of CRISPR Genome Editing by TIDE and TIDER. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NY, USA, 2019; Volume 1961, pp. 29–44. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A cytoscape app for pathway and network-based data analysis. F1000Research 2014, 3, 146. [Google Scholar]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.-S.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, 14732. [Google Scholar] [CrossRef] [Green Version]
- Dantas-Barbosa, C.; Lesluyes, T.; Le Loarer, F.; Chibon, F.; Treilleux, I.; Coindre, J.M.; Meeus, P.; Brahmi, M.; Bally, O.; Ray-Coquard, I.; et al. Expression and role of TYRO3 and AXL as potential therapeutical targets in leiomyosarcoma. Br. J. Cancer 2017, 117, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, P.P.; Ivy, K.S.; Musi, E.; de Stanchina, E.; Schwartz, G.K. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget 2016, 7, 4093–4109. [Google Scholar] [CrossRef] [Green Version]
- Morice, S.; Mullard, M.; Brion, R.; Dupuy, M.; Renault, S.; Tesfaye, R.; Le Royer, B.B.; Ory, B.; Redini, F.; Verrecchia, F. The YAP/tead axis as a new therapeutic target in osteosarcoma: Effect of verteporfin and CA3 on primary tumor growth. Cancers 2020, 12, 3847. [Google Scholar] [CrossRef]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; Reynolds, T.; Baker, E.J.; Bult, C.J.; Chesler, E.J.; et al. Cisplatin-resistant triple-negative breast cancer subtypes: Multiple mechanisms of resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, G.A.; Angus, S.P.; Khella, C.A.; Tong, K.; Khanna, P.; Dixon, S.A.H.; Verzi, M.P.; Johnson, G.L.; Gatza, M.L. SOX4 and SMARCA4 cooperatively regulate PI3k signaling through transcriptional activation of TGFBR2. NPJ Breast Cancer 2021, 7, 40. [Google Scholar] [CrossRef]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Gala, K.; Li, Q.; Sinha, A.; Razavi, P.; Dorso, M.; Sanchez-Vega, F.; Chung, Y.R.; Hendrickson, R.; Hsieh, J.J.; Berger, M.; et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene 2018, 37, 4692–4710. [Google Scholar] [CrossRef] [Green Version]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Milton, C.C.; Humbert, P.O.; Harvey, K.F. Transcriptional output of the Salvador/Warts/Hippo pathway is controlled in distinct fashions in Drosophila melanogaster and mammalian cell lines. Cancer Res. 2009, 69, 6033–6041. [Google Scholar] [CrossRef] [Green Version]
- Natsuizaka, M.; Ohashi, S.; Wong, G.S.; Ahmadi, A.; Kalman, R.A.; Budo, D.; Klein-Szanto, A.J.; Herlyn, M.; Diehl, J.A.; Nakagawa, H. Insulin-like growth factor-binding protein-3 promotes transforming growth factor-β1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis 2010, 31, 1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, L.; Wen, Y.; Miao, X.; Yang, Z. NT5E and FcGBP as key regulators of TGF-1-induced epithelial-mesenchymal transition (EMT) are associated with tumor progression and survival of patients with gallbladder cancer. Cell Tissue Res. 2014, 355, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Bastien, E.; De Jesus, J.P.S.; Dierge, E.; Martherus, R.; Linden, C.V.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.R.; Choi, H.M.; Lee, Y.W.; Joo, H.S.; Park, C.W.; Choi, J.W.; Kim, D.H.; Kang, H.N.; Pyo, K.; Shin, E.J.; et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK-rearranged lung cancer. EMBO Mol. Med. 2019, 11, e10581. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Li, X. Verteporfin inhibits cell proliferation and induces apoptosis in different subtypes of breast cancer cell lines without light activation. BMC Cancer 2020, 20, 1042. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ transcriptional coactivators create therapeutic vulnerability to verteporfin in EGFR-mutant glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
- Glaros, S.; Cirrincione, G.M.; Palanca, A.; Metzger, D.; Reisman, D. Targeted knockout of BRG1 potentiates lung cancer development. Cancer Res. 2008, 68, 3689–3696. [Google Scholar] [CrossRef] [Green Version]
- Schneppenheim, R.; Frühwald, M.C.; Gesk, S.; Hasselblatt, M.; Jeibmann, A.; Kordes, U.; Kreuz, M.; Leuschner, I.; Subero, J.I.M.; Obser, T.; et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am. J. Hum. Genet. 2010, 86, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, X.; Yu, L.; Zhang, C.; Ye, L.; Ren, D.; Li, Y.; Sun, X.; Yu, L.; Ouyang, Y.; et al. Zinc finger protein 367 promotes metastasis by inhibiting the Hippo pathway in breast cancer. Oncogene 2020, 39, 2568–2582. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Jang, G.; Sim, S.H.; Park, I.H.; Kim, K.; Park, C. SMARCA4 Depletion Induces Cisplatin Resistance by Activating YAP1-Mediated Epithelial-to-Mesenchymal Transition in Triple-Negative Breast Cancer. Cancers 2021, 13, 5474. https://doi.org/10.3390/cancers13215474
Kim J, Jang G, Sim SH, Park IH, Kim K, Park C. SMARCA4 Depletion Induces Cisplatin Resistance by Activating YAP1-Mediated Epithelial-to-Mesenchymal Transition in Triple-Negative Breast Cancer. Cancers. 2021; 13(21):5474. https://doi.org/10.3390/cancers13215474
Chicago/Turabian StyleKim, Jihyun, Gyubeom Jang, Sung Hoon Sim, In Hae Park, Kyungtae Kim, and Charny Park. 2021. "SMARCA4 Depletion Induces Cisplatin Resistance by Activating YAP1-Mediated Epithelial-to-Mesenchymal Transition in Triple-Negative Breast Cancer" Cancers 13, no. 21: 5474. https://doi.org/10.3390/cancers13215474
APA StyleKim, J., Jang, G., Sim, S. H., Park, I. H., Kim, K., & Park, C. (2021). SMARCA4 Depletion Induces Cisplatin Resistance by Activating YAP1-Mediated Epithelial-to-Mesenchymal Transition in Triple-Negative Breast Cancer. Cancers, 13(21), 5474. https://doi.org/10.3390/cancers13215474