
Machine Learning Based on Morphological Features Enables Classification of Primary Intestinal T-Cell Lymphomas

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
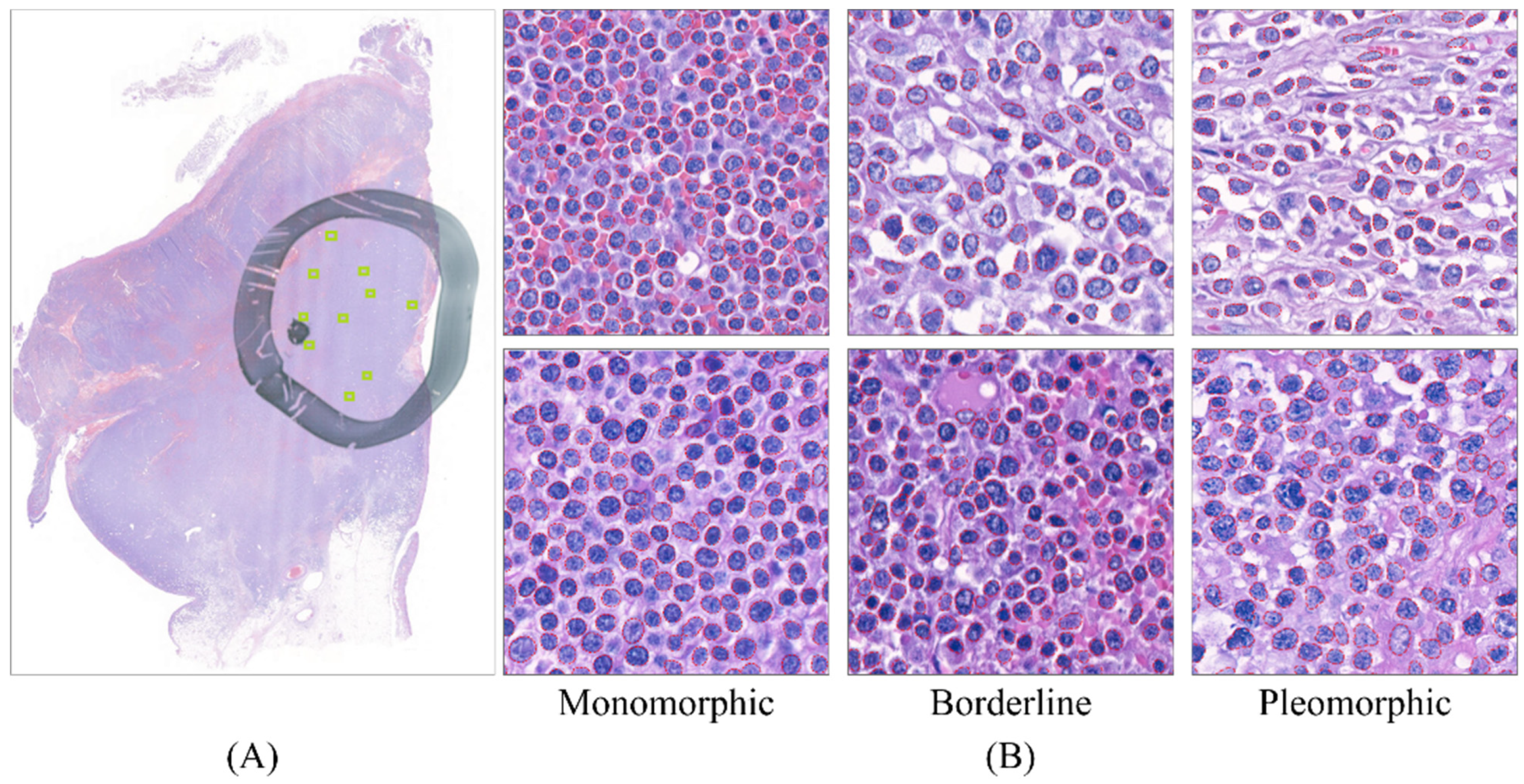
2.2. Dataset Preparation
2.3. Lymphocyte Detection Model
2.4. Computation of Nuclear Morphometrics
2.5. T-cell Lymphoma Classification Model
2.6. Modeling for Diagnostic Prediction from the Feature Profile
3. Results
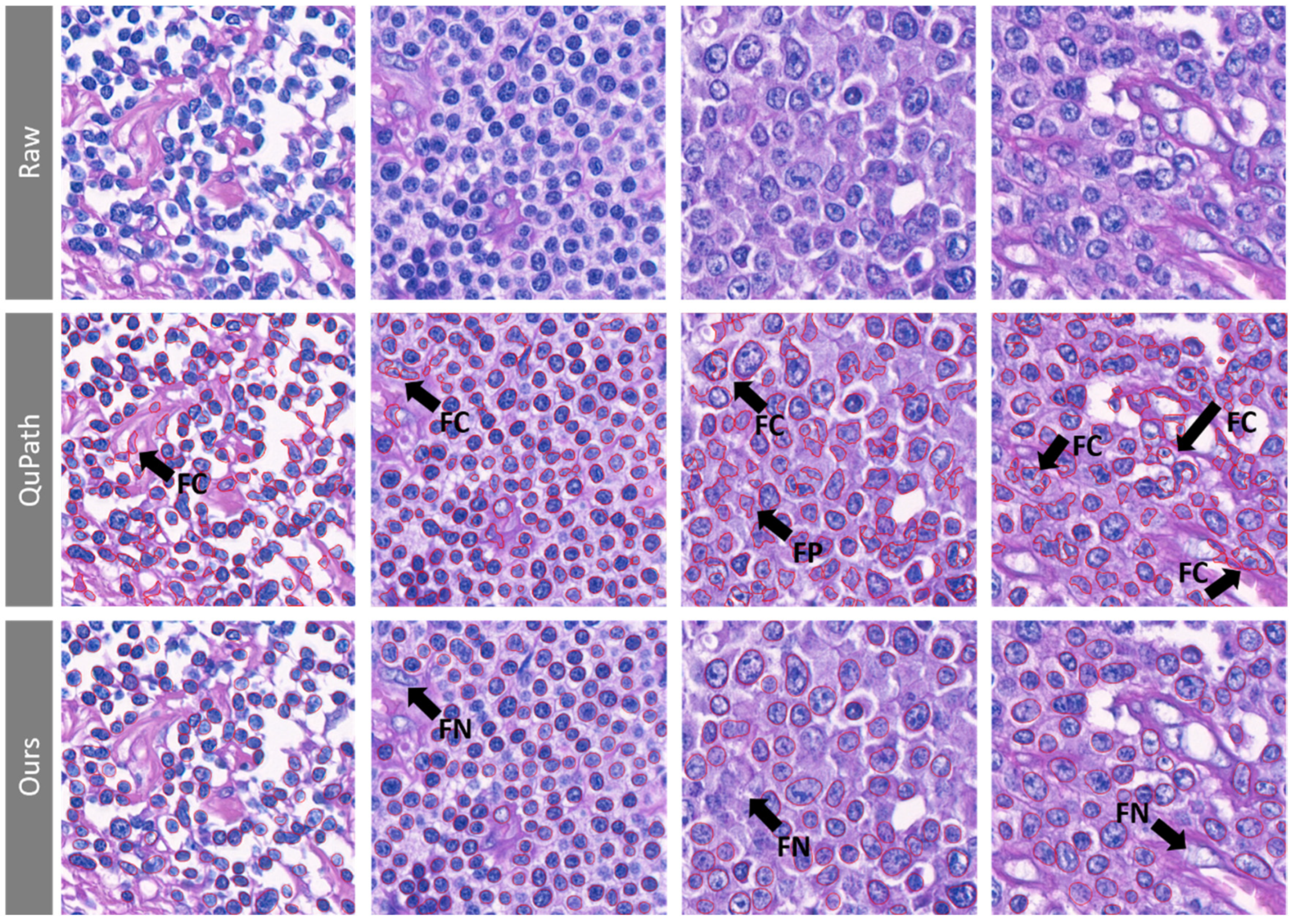
3.1. The Lymphoma Nucleus Detection Model Shows High Sensitivity along with High Positive Predictive Value
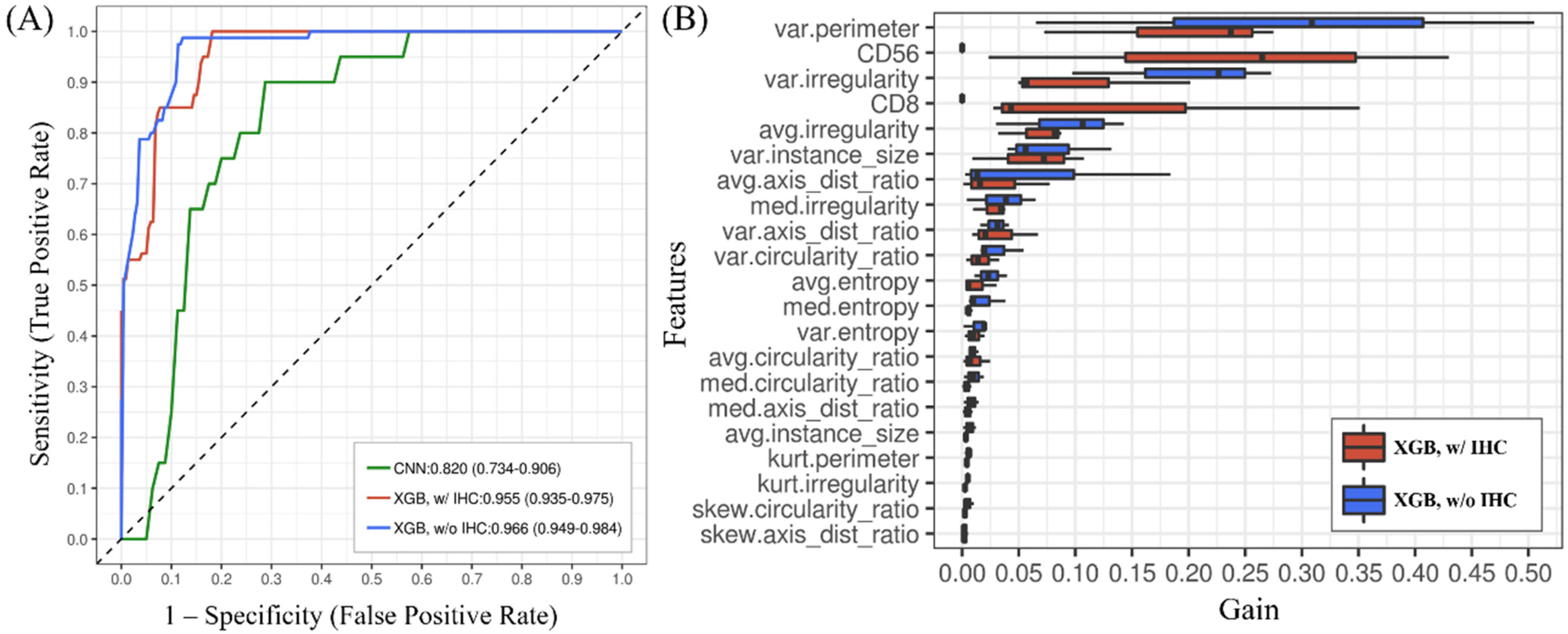
3.2. The T-Cell Lymphoma Classification Model Discriminated MEITL and ITCL-NOS Cases and Showed Higher Accuracy Than the CNN
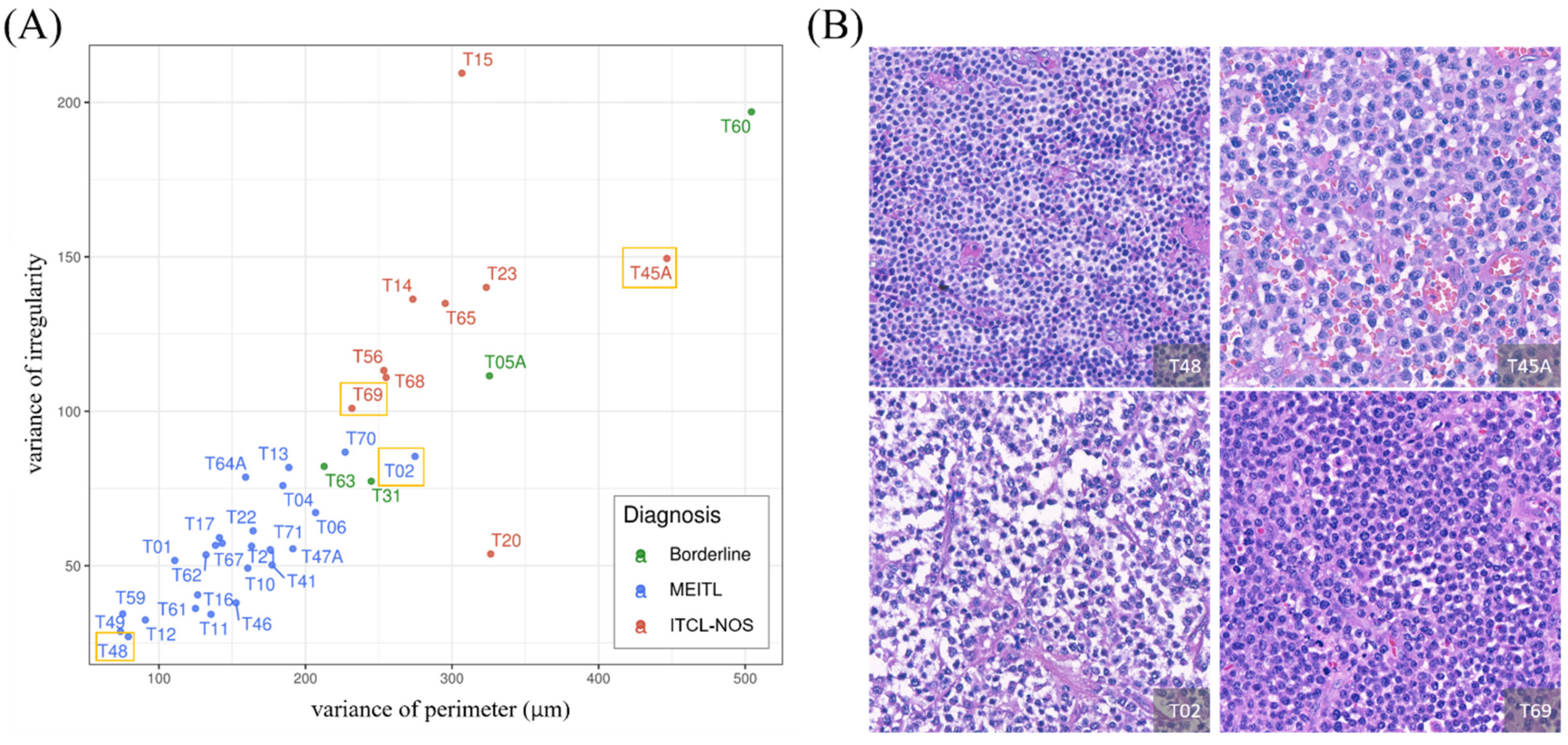
3.3. The Importance of Features Obtained from the XGBoost Model Can Be Ranked
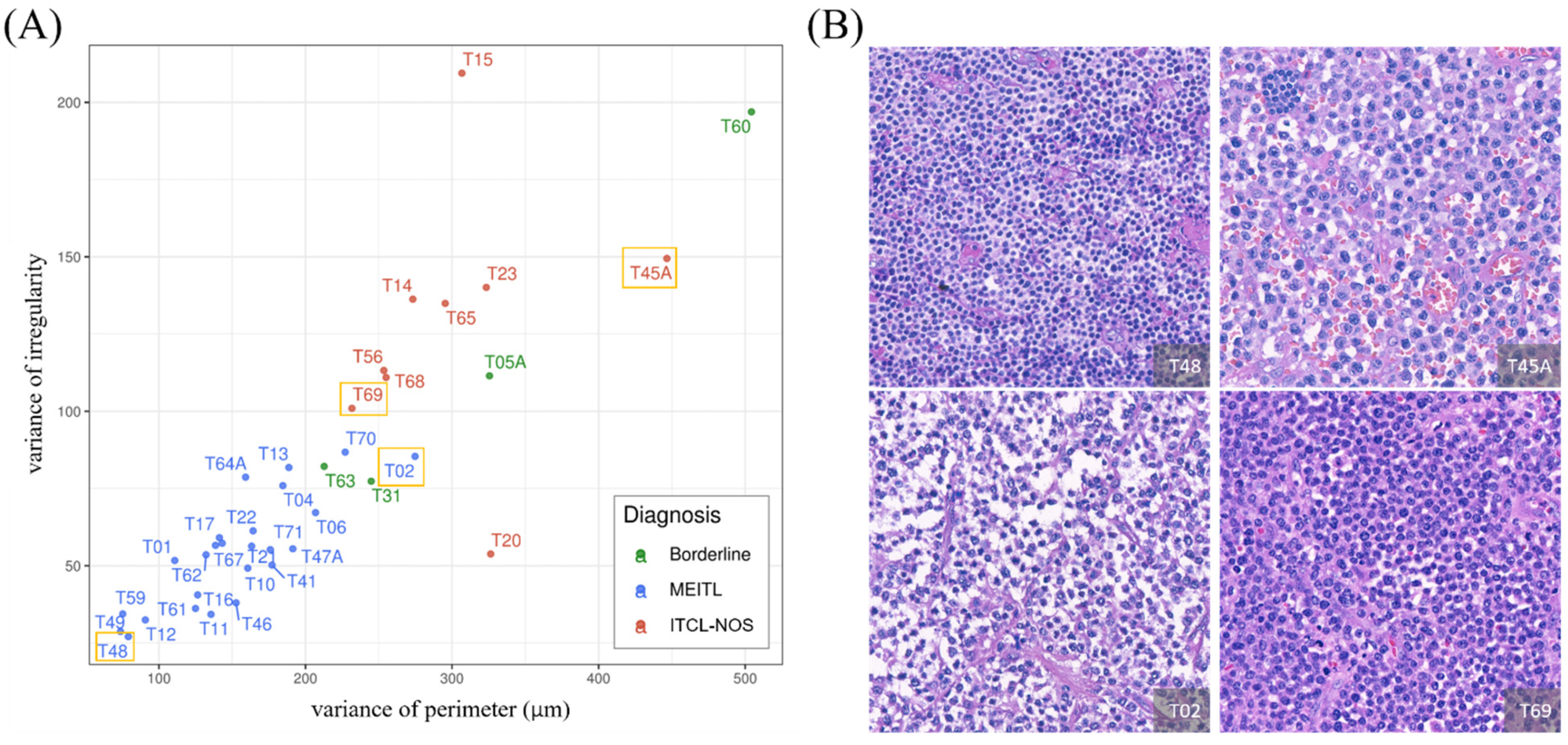
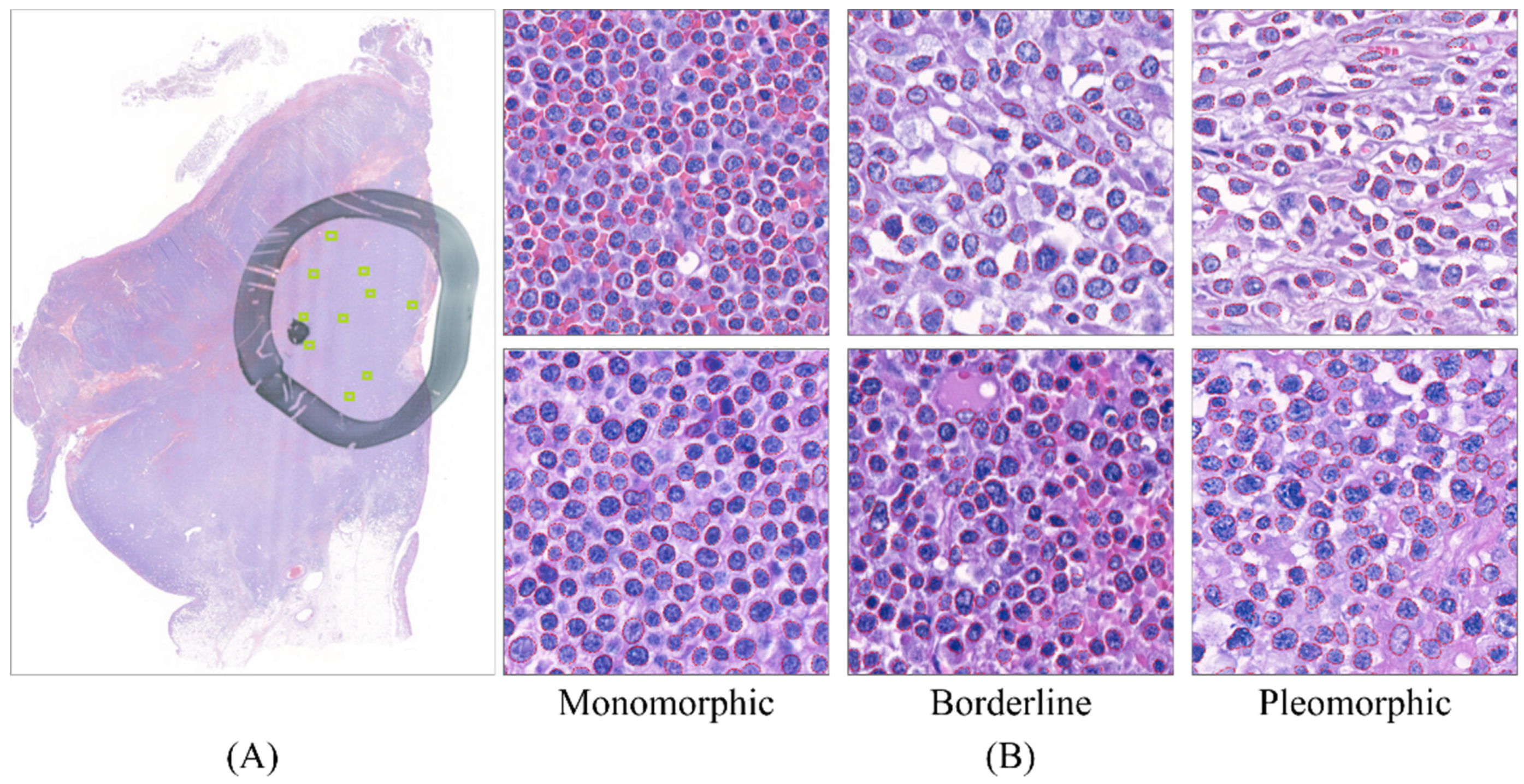
3.4. Feature Analysis Using the GLM Enabled Explicit Interpretation of the Morphological Features
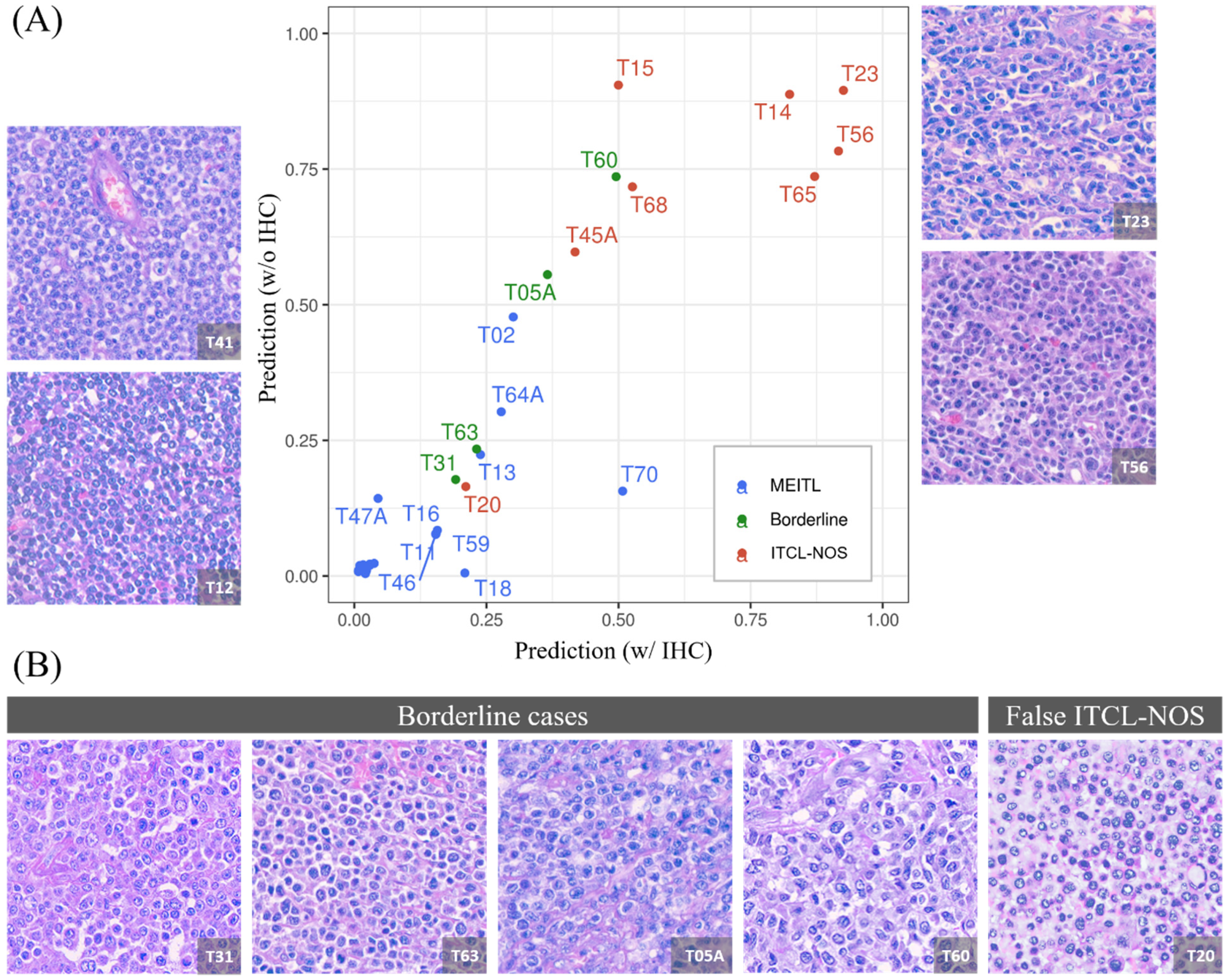
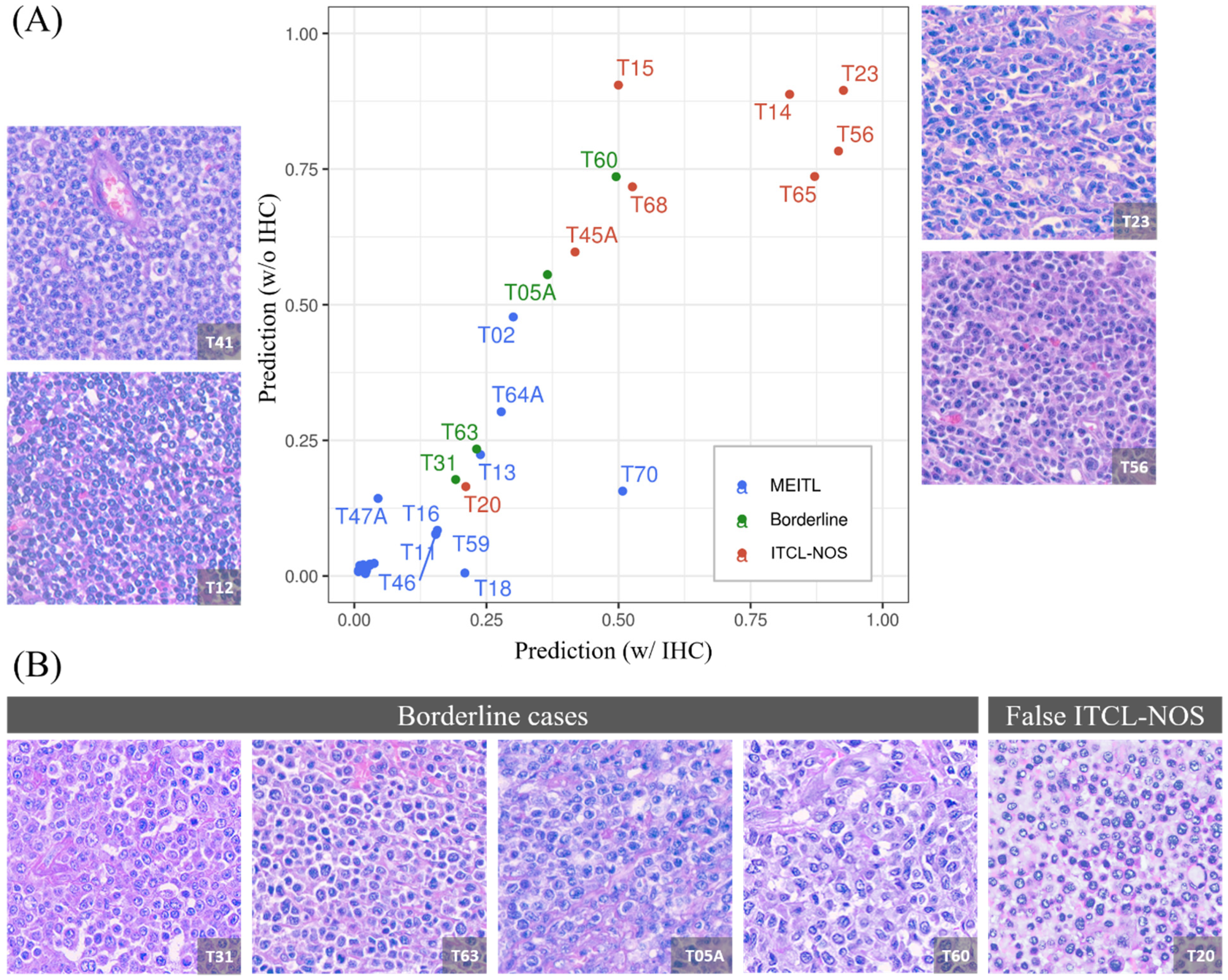
3.5. The Model Produced a 1:1 Ratio Prediction for the Four Borderline Cases
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jaffe, E.S.C.A.; Ott, G.; Chan, J.K.C.; Bhaat, G.; Tan, S.T.; Stein, H.; Isaacson, P.G. Intestinal T-cell lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H.C.E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., Orazi, A., et al., Eds.; IARC: Lyon, Frane, 2017; Volume 2, pp. 372–380. [Google Scholar]
- Skinnider, B.F. Lymphoproliferative disorders of the gastrointestinal tract. Arch. Pathol. Lab. Med. 2018, 142, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.; Chuang, S.S.; Tang, T.; Tan, L.; Ko, Y.H.; Chuah, K.L.; Ng, S.B.; Chng, W.J.; Gatter, K.; Loong, F.; et al. Type II EATL (epitheliotropic intestinal T-cell lymphoma): A neoplasm of intra-epithelial T-cells with predominant CD8alphaalpha phenotype. Leukemia 2013, 27, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Tse, E.; Gill, H.; Loong, F.; Kim, S.J.; Ng, S.B.; Tang, T.; Ko, Y.H.; Chng, W.J.; Lim, S.T.; Kim, W.S.; et al. Type II enteropathy-associated T-cell lymphoma: A multicenter analysis from the Asia Lymphoma Study Group. Am. J. Hematol. 2012, 87, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Chuang, S.S.; Chang, S.T.; Chuang, W.Y.; Huang, W.T.; Hsieh, P.P.; Tsou, M.H.; Liao, Y.L.; Lin, S.H.; Hsieh, Y.C.; Lu, C.L.; et al. NK-cell lineage predicts poor survival in primary intestinal NK-cell and T-cell lymphomas. Am. J. Surg. Pathol. 2009, 33, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Alom, M.Z.; Aspiras, T.; Taha, T.M.; Asari, V.K.; Bowen, T.J.; Billiter, D.; Arkell, S. Advanced deep convolutional neural network approaches for digital pathology image analysis: A comprehensive evaluation with different use cases. arXiv 2019, arXiv:1904.09075. Available online: https://arxiv.org/abs/1904.09075 (accessed on 1 September 2021).
- Banaeeyan, R.; Fauzi, M.F.A.; Chen, W.; Knight, D.; Hampel, H.; Frankel, W.L.; Gurcan, M.N. Tumor budding detection in H E-stained images using deep semantic learning. In Proceedings of the 2020 IEEE Region 10 Conference (TENCON), Osaka, Japan, 16–19 November 2020; pp. 52–56. [Google Scholar]
- Nateghi, R.; Danyali, H.; Helfroush, M.S. A deep learning approach for mitosis detection: Application in tumor proliferation prediction from whole slide images. Artif. Intell. Med. 2021, 114, 102048. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, B.; LaRiviere, M.J.; Cohen, E.A.; Buckingham, T.H.; Yee, S.S.; Black, T.A.; Chien, A.L.; Noël, P.; Hwang, W.-T.; Katz, S.I.; et al. Combining radiomic phenotypes of non-small cell lung cancer with liquid biopsy data may improve prediction of response to EGFR inhibitors. Sci. Rep. 2021, 11, 9984. [Google Scholar] [CrossRef] [PubMed]
- Limkin, E.J.; Sun, R.; Dercle, L.; Zacharaki, E.I.; Robert, C.; Reuzé, S.; Schernberg, A.; Paragios, N.; Deutsch, E.; Ferté, C. Promises and challenges for the implementation of computational medical imaging (radiomics) in oncology. Ann. Oncol. 2017, 28, 1191–1206. [Google Scholar] [CrossRef]
- Popovich, M.J. Ultrasound-guided vascular access in critical care: Can a choice of real-time imaging axis view overcome the “curse of dimensionality”?*. Crit. Care Med. 2015, 43, 920–921. [Google Scholar] [CrossRef]
- Das, A.; Nair, M.S.; Peter, S.D. Computer-aided histopathological image analysis techniques for automated nuclear atypia scoring of breast cancer: A review. J. Digit. Imaging 2020, 33, 1091–1121. [Google Scholar] [CrossRef]
- Faridi, P.; Danyali, H.; Helfroush, M.S.; Jahromi, M.A. An automatic system for cell nuclei pleomorphism segmentation in histopathological images of breast cancer. In Proceedings of the 2016 IEEE Signal Processing in Medicine and Biology Symposium (SPMB), Philadelphia, PA, USA, 3 December 2016; pp. 1–5. [Google Scholar]
- Maqlin, P.; Thamburaj, R.; Mammen, J.J.; Manipadam, M.T. Automated nuclear pleomorphism scoring in breast cancer histopathology images using deep neural networks. In International Conference on Mining Intelligence and Knowledge Exploration; Springer: Cham, Germany, 2015; pp. 269–276. [Google Scholar]
- Moran, J.M.T.; Biecek, P.; Donizy, P.; Wu, C.-L.; Kopczynski, J.; Pieniazek, M.; Ryś, J.; Hoang, M.P. Large nuclear size correlated with better overall survival, Merkel cell polyomavirus positivity, and terminal deoxynucleotidyl transferase expression in Merkel cell carcinoma. J. Am. Acad. Dermatol. 2021, 84, 550–552. [Google Scholar] [CrossRef]
- Wang, L.-W.; Chen, Y.-W.; Ho, C.-Y.; Hsueh Liu, Y.-W.; Chou, F.-I.; Liu, Y.-H.; Liu, H.-M.; Peir, J.-J.; Jiang, S.-H.; Chang, C.-W.; et al. Fractionated boron neutron capture therapy in locally recurrent head and neck cancer: A prospective phase I/II trial. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 396–403. [Google Scholar] [CrossRef]
- Chen, K.; Pang, J.; Wang, J.; Xiong, Y.; Li, X.; Sun, S.; Feng, W.; Liu, Z.; Shi, J.; Ouyang, W.; et al. Hybrid task cascade for instance segmentation. arXiv 2019, arXiv:1901.07518. Available online: https://ieeexplore.ieee.org/document/8954166 (accessed on 1 September 2021).
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. arXiv 2015, arXiv:1512.03385. Available online: https://ieeexplore.ieee.org/document/7780459 (accessed on 1 September 2021).
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Khoshdeli, M.; Cong, R.; Parvin, B. Detection of nuclei in h&e stained sections using convolutional neural networks. IEEE EMBS Int. Conf. Biomed. Health Inf. 2017, 2017, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Verma, R.; Anand, D.; Zhou, Y.; Onder, O.F.; Tsougenis, E.; Chen, H.; Heng, P.-A.; Li, J.; Hu, Z.; et al. A multi-organ nucleus segmentation challenge. IEEE Trans. Med. Imaging 2020, 39, 1380–1391. [Google Scholar] [CrossRef]
- Verma, R.; Kumar, N.; Patil, A.; Kurian, N.C.; Rane, S.; Graham, S.; Vu, Q.D.; Zwager, M.; Raza, S.E.A.; Rajpoot, N.; et al. MoNuSAC2020: A multi-organ nuclei segmentation and classification challenge. IEEE Trans. Med. Imaging 2021. [Google Scholar] [CrossRef]
- Gann, P.H.; Deaton, R.; Amatya, A.; Mohnani, M.; Rueter, E.E.; Yang, Y.; Ananthanarayanan, V. Development of a nuclear morphometric signature for prostate cancer risk in negative biopsies. PLoS ONE 2013, 8, e69457. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Damodaran, K.; Soylemezoglu, A.C.; Uhler, C.; Shivashankar, G.V. Machine learning for nuclear mechano-morphometric biomarkers in cancer diagnosis. Sci. Rep. 2017, 7, 17946. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, P.; Murphy, J.; McGinley, J.; Zhu, Z.; Jiang, W.; Gottschall, E.B.; Thompson, H.J. Using nuclear morphometry to discriminate the tumorigenic potential of cells: A comparison of statistical methods. Cancer Epidemiol. Prev. Biomark. 2004, 13, 976–988. [Google Scholar]
- Kervarrec, T.; Tallet, A.; Miquelestorena-Standley, E.; Houben, R.; Schrama, D.; Gambichler, T.; Berthon, P.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Aubin, F.; et al. Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and negative Merkel cell carcinoma. Mod. Pathol. 2019, 32, 1605–1616. [Google Scholar] [CrossRef]
- Weisenburger, D.D.; Savage, K.J.; Harris, N.L.; Gascoyne, R.D.; Jaffe, E.S.; MacLennan, K.A.; Rudiger, T.; Pileri, S.; Nakamura, S.; Nathwani, B.; et al. Peripheral T-cell lymphoma, not otherwise specified: A report of 340 cases from the International Peripheral T-cell Lymphoma Project. Blood 2011, 117, 3402–3408. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Bledsoe, J.R.; Zeng, Y.; Liu, W.; Hu, Y.; Bi, K.; Liang, A.; Li, S. A deep learning diagnostic platform for diffuse large B-cell lymphoma with high accuracy across multiple hospitals. Nat. Commun. 2020, 11, 6004. [Google Scholar] [CrossRef]
- Syrykh, C.; Abreu, A.; Amara, N.; Siegfried, A.; Maisongrosse, V.; Frenois, F.X.; Martin, L.; Rossi, C.; Laurent, C.; Brousset, P. Accurate diagnosis of lymphoma on whole-slide histopathology images using deep learning. NPJ Digit. Med. 2020, 3, 1–8. [Google Scholar] [CrossRef]
- Li, H.N.; Wang, R.C.; Chen, J.P.; Chang, S.T.; Chuang, S.S. Density and size of lymphoid follicles are useful clues in differentiating primary intestinal follicular lymphoma from intestinal reactive lymphoid hyperplasia. Diagn. Pathol. 2020, 15, 82. [Google Scholar] [CrossRef]
- Pham, B.; Gaonkar, B.; Whitehead, W.; Moran, S.; Dai, Q.; Macyszyn, L.; Edgerton, V.R. Cell counting and segmentation of immunohistochemical images in the spinal cord: Comparing deep learning and traditional approaches. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2018, 2018, 842–845. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
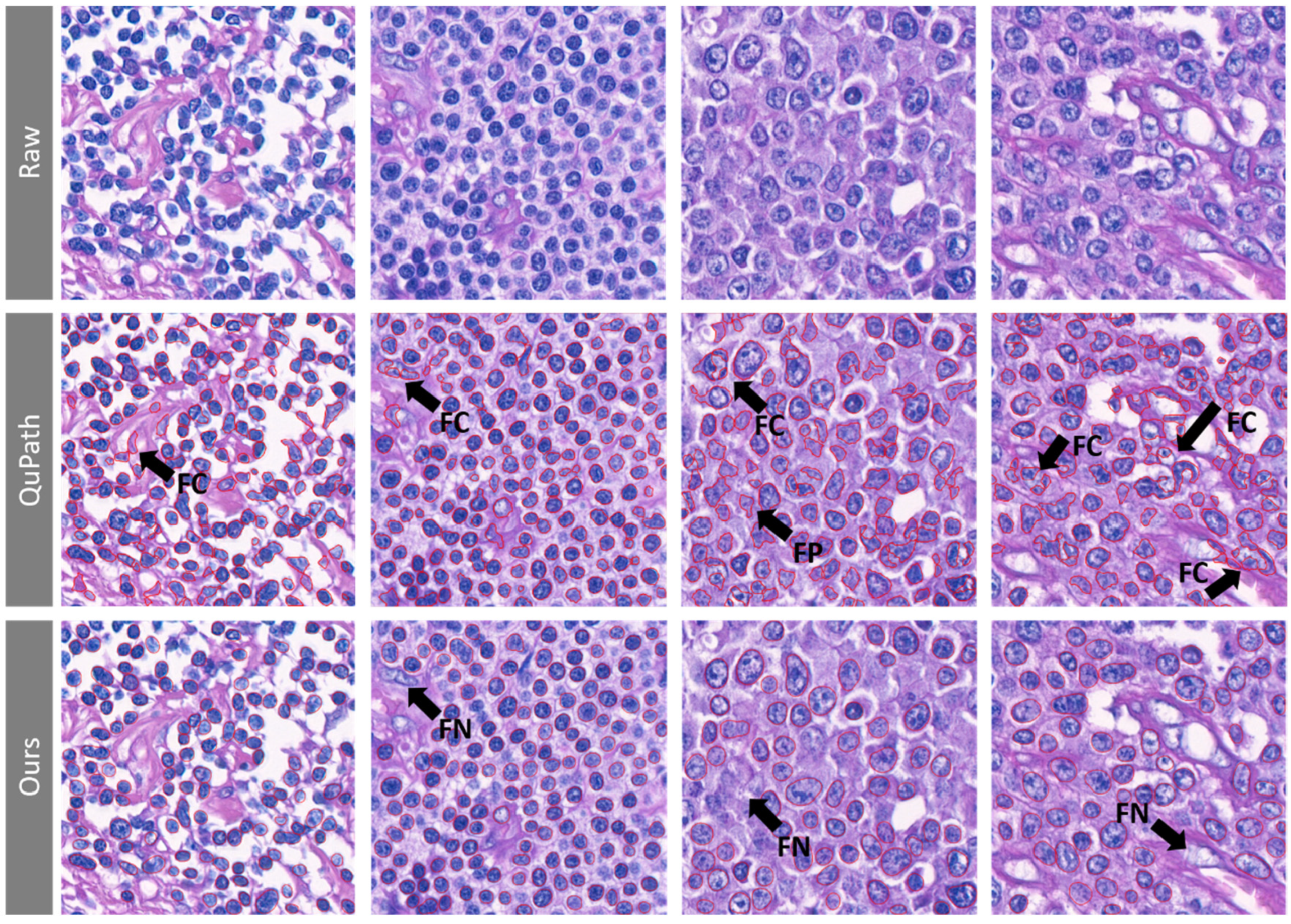{kind=link}
| Case no./Sex/Age | T05A/Male/49 | T31/Female/49 | T60/Male/64 | T63/Male/56 |
|---|---|---|---|---|
| Tumor site | Ileum | Ileum | Jejunum and ileum | Ileum |
| Perforation | Present | Present | Present | Present |
| Cell size | Medium to large | Medium to large | Large | Medium to large |
| Morphology (mono vs. pleomorphic) | Pleomorphic | Pleomorphic | Mildly pleomorphic | Pleomorphic |
| CD8 | + | + | + | + |
| CD30 | - | - | - | - |
| CD56 | + | + | + | + |
| TIA-1 | - | + | + | + |
| Granzyme B | + | + | - | + |
| Lugano stage * | Stage IE | Stage IE | Stage IE | Stage IV |
| Follow-up (month) | DOD (34) | DOD (2) | DOD (6.5) | DOD (5.8) |
| Lymphocyte Detection Model | Feature Extraction | T Cell Lymphoma Classification Model (XGBoost and CNN) | |
|---|---|---|---|
| No. of cases (WSIs) | 18 | 40 | 40 |
| MEITL | 14 | 26 | 26 |
| ITCL-NOS | 3 | 10 | 10 |
| Borderline | 1 | 4 | 4 |
| No. of ROIs | 33 | 400 | 400 |
| ROI size | 768 × 768 pixels | 2304 × 2304 pixels (HPF) | 2304 × 2304 pixels (HPF) |
| Data level | ROI-level | HPF-level | HPF-level |
| Data set | 33 ROIs | 400 HPFs (28 features per HPF) | 36 cases (360 HPFs) 4 borderline cases were excluded |
| Training set | 27 ROIs | - | 3-fold cross-validation |
| Validation set | 3 ROIs | ||
| Testing set | 3 ROIs |
| Attributes | Definition |
|---|---|
| Ratio of axis length | The ratio of the longest axis and the second-longest axis |
| Circularity | Ratio of overlapping pixels between the concentric circle and size of the cell |
| Entropy | Measure of the randomness of pixels in the cell |
| Area | Total numbers of pixels within the boundary of the cell. |
| Irregularity | Variance of length from the center of cell to each vertex of the boundary |
| Perimeter | Estimated total numbers of pixels along the cell boundary |
| Orientation | Cell orientation of the longest axis |
| Model | Model Comparison | XGB-1 (Morphology-only) | XGB-2 (Morphology + IHC) | CNN |
|---|---|---|---|---|
| AUC | 0.966 | 0.955 | 0.820 | |
| 95% CI | 0.949–0.984 | 0.935–0.975 | 0.734–0.906 | |
| p-value (Delong’s Test) | XGB-1 VS. XGB-2 | 0.166 | ||
| XGB-1 VS. CNN | 0.003 * | |||
| XGB-2 VS. CNN | 0.001 * | |||
| Disease Type | F-Test | ||||||
|---|---|---|---|---|---|---|---|
| Overall | MEITL | ITCL-NOS | F-Statistics | p-Value | |||
| No. | Attributes | Moments (Mean ± SD) | N = 36 | N = 26 | N = 10 | ||
| 1 | Ratio of axis length | Mean | 1.29 ± 0.06 | 1.27 ± 0.03 | 1.34 ± 0.06 | 11.53 | <0.001 * |
| Variance | 0.04 ± 0.02 | 0.04 ± 0.01 | 0.06 ± 0.02 | 13.51 | <0.001 * | ||
| Skewness | 1.52 ± 0.35 | 1.59 ± 0.22 | 1.48 ± 0.34 | 1.01 | 0.374 | ||
| Kurtosis | 6.79 ± 2.80 | 7.18 ± 1.51 | 6.78 ± 2.50 | 0.6 | 0.557 | ||
| 2 | Circularity | Mean | 0.75 ± 0.03 | 0.76 ± 0.02 | 0.72 ± 0.03 | 10.25 | <0.001 * |
| Variance | 0.01 ± 0.00 | 0.008 ± 0.001 | 0.010 ± 0.002 | 14.95 | <0.001 * | ||
| Skewness | −0.75 ± 0.26 | −0.83 ± 0.19 | −0.61 ± 0.27 | 3.64 | 0.037* | ||
| Kurtosis | 3.57 ± 0.70 | 3.68 ± 0.49 | 3.21 ± 0.69 | 2.86 | 0.071 | ||
| 3 | Entropy | Mean | 5.85 ± 0.10 | 5.86 ± 0.09 | 5.84 ± 0.10 | 0.31 | 0.734 |
| Variance | 0.07 ± 0.01 | 0.07 ± 0.01 | 0.08 ± 0.01 | 4.24 | 0.023 | ||
| Skewness | −0.19 ± 0.14 | −0.20 ± 0.09 | −0.21 ± 0.10 | 2.84 | 0.072 | ||
| Kurtosis | 3.39 ± 0.38 | 3.40 ± 0.22 | 3.38 ± 0.17 | 0.02 | 0.976 | ||
| 4 | Nuclear area | Mean | 41.72 ± 9.00 | 38.13 ± 7.25 | 45.84 ± 6.93 | 11.63 | <0.001 * |
| Variance | 234.16 ± 155.71 | 157.74 ± 70.25 | 362.70 ± 118.91 | 24.09 | <0.001 * | ||
| Skewness | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.02 ± 0.01 | 2.27 | 0.119 | ||
| Kurtosis | 0.14 ± 0.07 | 0.16 ± 0.07 | 0.12 ± 0.02 | 4.32 | 0.021 * | ||
| 5 | Irregularity | Mean | 1.15 ± 0.40 | 0.94 ± 0.22 | 1.55 ± 0.30 | 22.18 | <0.001 * |
| Variance | 1.75 ± 1.06 | 1.22 ± 0.40 | 2.87 ± 0.94 | 23.87 | <0.001 * | ||
| Skewness | 0.45 ± 0.12 | 0.50 ± 0.08 | 0.40 ± 0.07 | 8.67 | 0.001 * | ||
| Kurtosis | 2.63 ± 1.57 | 3.16 ± 0.97 | 2.32 ± 1.00 | 7.14 | 0.003 * | ||
| 6 | Perimeter | Mean | 24.32 ± 2.66 | 23.26 ± 2.23 | 25.64 ± 1.90 | 11 | <0.001 * |
| Variance | 20.32 ± 9.91 | 15.33 ± 4.80 | 30.40 ± 6.42 | 25.97 | <0.001 * | ||
| Skewness | 0.09 ± 0.07 | 0.11 ± 0.06 | 0.08 ± 0.03 | 3.35 | 0.047 * | ||
| Kurtosis | 0.66 ± 0.24 | 0.74 ± 0.19 | 0.55 ± 0.07 | 7.03 | 0.003 * | ||
| 7 | Orientation | Mean | 1.55 ± 0.13 | 1.54 ± 0.12 | 1.57 ± 0.14 | 0.52 | 0.599 |
| Variance | 0.71 ± 0.15 | 0.72 ± 0.14 | 0.69 ± 0.16 | 0.14 | 0.870 | ||
| Skewness | 0.001 ± 0.004 | 0.0009 ± 0.07 | −0.0002 ± 0.04 | 0.50 | 0.610 | ||
| Kurtosis | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.62 | 0.543 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.-H.; Li, C.-H.; Wang, R.-C.; Yeh, C.-Y.; Chuang, S.-S. Machine Learning Based on Morphological Features Enables Classification of Primary Intestinal T-Cell Lymphomas. Cancers 2021, 13, 5463. https://doi.org/10.3390/cancers13215463
Yu W-H, Li C-H, Wang R-C, Yeh C-Y, Chuang S-S. Machine Learning Based on Morphological Features Enables Classification of Primary Intestinal T-Cell Lymphomas. Cancers. 2021; 13(21):5463. https://doi.org/10.3390/cancers13215463
Chicago/Turabian StyleYu, Wei-Hsiang, Chih-Hao Li, Ren-Ching Wang, Chao-Yuan Yeh, and Shih-Sung Chuang. 2021. "Machine Learning Based on Morphological Features Enables Classification of Primary Intestinal T-Cell Lymphomas" Cancers 13, no. 21: 5463. https://doi.org/10.3390/cancers13215463
APA StyleYu, W.-H., Li, C.-H., Wang, R.-C., Yeh, C.-Y., & Chuang, S.-S. (2021). Machine Learning Based on Morphological Features Enables Classification of Primary Intestinal T-Cell Lymphomas. Cancers, 13(21), 5463. https://doi.org/10.3390/cancers13215463