
Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy


Abstract
:Simple Summary
Abstract
1. Oncolytic Virus: Multi-Mechanistic Cancer Therapeutics
1.1. Oncolytic Virus: Brief Background and History
1.2. Types of Viruses in Use/Development as Oncolytic Therapeutics
2. Mechanisms of Cancer Cell Tropism of OVs
2.1. Overexpression of Receptor Molecules on the Cancer Cell Surface
2.2. Alteration in Intracellular Signaling Pathways
2.3. Altered Metabolism of Cancer Cells
3. Mechanisms of Antitumor Effects Mediated by OVs
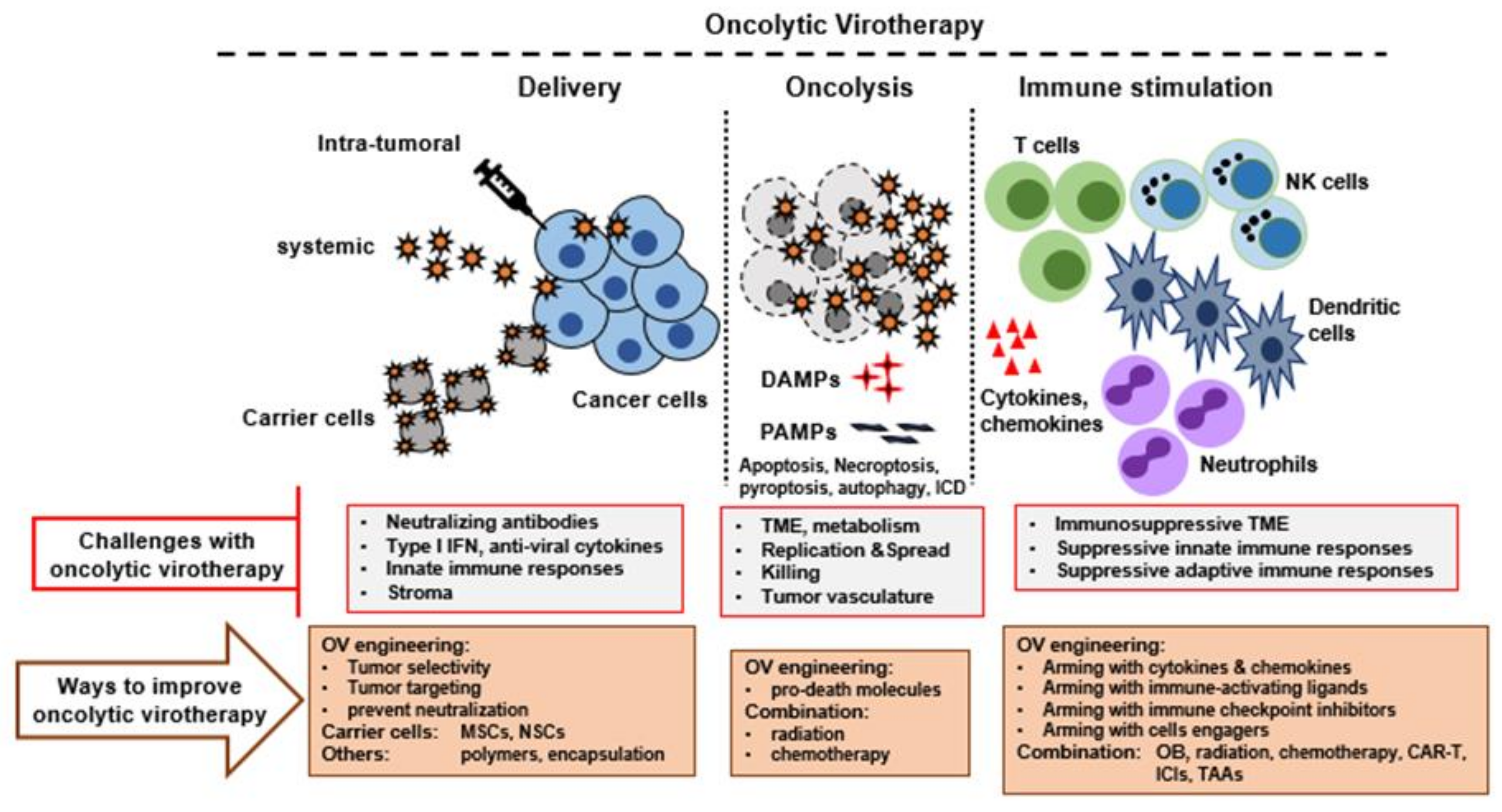
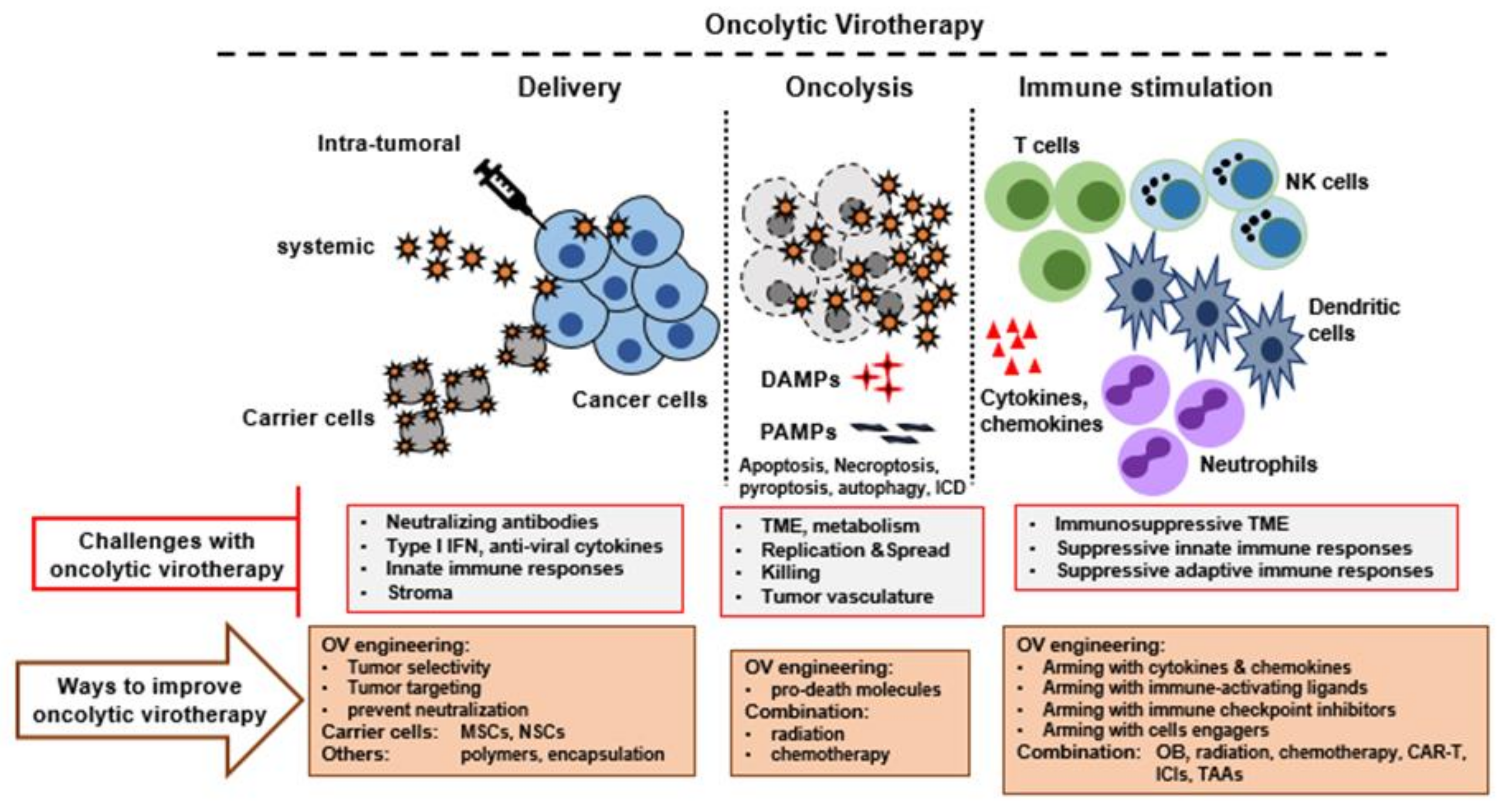
4. Challenges/Limitations with OV to Become Successful as Monotherapy
- Selection of optimal OV candidate: Until now, multiple DNA and RNA viruses have been explored as OV candidates. To be an ideal candidate, there are various properties that the selected virus should have, such as the ability to incorporate transgenes stably, little or no toxicity to normal cells and tissues, immunogenicity, large scale clinical grade amplification, production optimization and appropriate therapeutic targets of the chosen OVs.
- Virus entry, infection, and spread: OVs that use selected cell surface receptors for binding and entry are often not useful for tumors with reduced or no expression of those receptors. Although this barrier has been overcome for some viruses by engineering, a few viruses (e.g., poxviruses) can circumvent this issue by binding to nonspecific determinants like ubiquitously expressed cell surface glycosaminiglycans. At the intracellular level, there are additional complex signaling pathways that are directly or indirectly linked with the antiviral pathways and commonly restrict virus replication (if operative) and spread to the new cells. For example, AKT activation levels regulate MYXV replication in human cancer cells [62,63]. In the tumor bed, the presence of excessive extracellular matrix (ECM) can prevent viral spread. For example, fibrillar collagen in the ECM limits oncolytic HSV spread within tumors [64].
- Delivery of OV: Delivery of OV to the sites of primary and metastatic sites is vital for optimal therapeutic outcomes. In this regard, since only a minority of human cancers are amenable to direct intratumoral (IT) injection, systemic delivery is the preferred route compared to IT injection of the virus. However, there are several barriers to the successful delivery of any OV. The presence of neutralizing antiviral antibodies, complement activation, expression of antiviral cytokines, and natural clearance site of OVs by the liver and spleen are all major obstacles in the systemic OV delivery. Although IT delivery of virus can circumvent some of these barriers, tumor beds can limit virus spread, and the tumor vasculature is also a limiting factor to IT and metastatic sites. One strategy to overcome these issues is to exploit migratory leukocytes as carrier cells to ferry the virus into tumor beds that allow cellular ingress.
- Neutralizing antibodies and antiviral cytokines: Preexisting neutralizing antiviral antibodies are the main obstacle in the context of systemic delivery of free virus to reach the tumor bed [65]. Additionally, the host immune system activates antiviral immunity and limits the oncolytic activity of OVs. The virus-sensing cellular receptor molecules that detect virus particles and virus-infected cells activate type I IFN signaling pathways, which activates antiviral defense pathways in the uninfected cells and limits OV infection and spread. Moreover, the immune clearance of infected cells, including cancerous cells, prevents virus spread although it can be an important feature of activating antitumor immune responses.
- Immunosuppressive TME: Another barrier to OV therapy is the frequent presence of highly immunosuppressive TME. In the tumor bed, various cellular subsets like cancer cells, stromal cells, inhibitory cytokines (e.g., TGF-beta) and infiltrating immune cells (e.g., regulatory T cells and myeloid derived suppressor cells) all contribute to the immunosuppressive TME. Although this is critical for the tumor to evade the host’s innate and adaptive immunological defenses, OVs must function within this immunosuppressive TME. Additionally, some OV infections can further promote the tumor bed’s immunosuppressive environment by activating the immune system. For example, Maraba virus upregulated the PD-1/PD-L1 axis on tumor cells and tumor-infiltrating immune cells [66,67]. Similarly, oncolytic NDV also promoted PD-L1 production in the tumor bed in response to the virus stimulated type I IFN signaling, resulting in an immunosuppressive TME even in distant tumors [68,69].
5. Engineered Oncolytic Viruses
6. Combination Therapy with Oncolytic Virus
6.1. Combination with Traditional Radiotherapy and Chemotherapy
6.2. Systemic Delivery of OV via Carrier Cells
6.3. Combination with Immune Checkpoint Inhibitors (ICIs)
6.4. Combination with Cell Therapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, S.; Rabkin, S.D. The discovery and development of oncolytic viruses: Are they the future of cancer immunotherapy? Expert Opin. Drug Discov. 2021, 16, 391–410. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Bell, J.; McFadden, G. Viruses for tumor therapy. Cell Host Microbe 2014, 15, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef]
- Martinez-Quintanilla, J.; Seah, I.; Chua, M.; Shah, K. Oncolytic viruses: Overcoming translational challenges. J. Clin. Investig. 2019, 129, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Xu, J.; Liu, X.Y.; Chen, Z.N.; Bian, H. Fighting Cancer with Viruses: Oncolytic Virus Therapy in China. Hum. Gene Ther. 2018, 29, 151–159. [Google Scholar] [CrossRef]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene laherparepvec: Review of its mechanism of action and clinical efficacy and safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, K.; Iwai, M.; Ito, H.; Tanaka, M.; Seto, Y.; Todo, T. Oncolytic herpes virus G47Δ works synergistically with CTLA-4 inhibition via dynamic intratumoral immune modulation. Mol. Ther. Oncolytics 2021, 22, 129–142. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Zainutdinov, S.S.; Kochneva, G.V.; Netesov, S.V.; Chumakov, P.M.; Matveeva, O.V. Directed evolution as a tool for the selection of oncolytic RNA viruses with desired phenotypes. Oncolytic Virotherapy 2019, 8, 9–26. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Mueller, S.; Chipman, P.R.; Bator, C.M.; Peng, X.; Bowman, V.D.; Mukhopadhyay, S.; Wimmer, E.; Kuhn, R.J.; Rossmann, M.G. Complexes of poliovirus serotypes with their common cellular receptor, CD155. J. Virol. 2003, 77, 4827–4835. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [Green Version]
- Bergelson, J.M.; Shepley, M.P.; Chan, B.M.; Hemler, M.E.; Finberg, R.W. Identification of the integrin VLA-2 as a receptor for echovirus 1. Science 1992, 255, 1718–1720. [Google Scholar] [CrossRef]
- Ong, H.T.; Timm, M.M.; Greipp, P.R.; Witzig, T.E.; Dispenzieri, A.; Russell, S.J.; Peng, K.W. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp. Hematol. 2006, 34, 713–720. [Google Scholar] [CrossRef]
- Lin, L.T.; Richardson, C.D. The Host Cell Receptors for Measles Virus and Their Interaction with the Viral Hemagglutinin (H) Protein. Viruses 2016, 8, 250. [Google Scholar] [CrossRef]
- Yu, Z.; Adusumilli, P.S.; Eisenberg, D.P.; Darr, E.; Ghossein, R.A.; Li, S.; Liu, S.; Singh, B.; Shah, J.P.; Fong, Y.; et al. Nectin-1 expression by squamous cell carcinoma is a predictor of herpes oncolytic sensitivity. Mol. Ther. 2007, 15, 103–113. [Google Scholar] [CrossRef]
- Chan, W.M.; McFadden, G. Oncolytic Poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; McFadden, G. Oncolytic Virotherapy with Myxoma Virus. J. Clin. Med. 2020, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Engeland, C.E.; Ungerechts, G. Measles Virus as an Oncolytic Immunotherapy. Cancers 2021, 13, 544. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Myxoma Virus-Encoded Host Range Protein M029: A Multifunctional Antagonist Targeting Multiple Host Antiviral and Innate Immune Pathways. Vaccines 2020, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Diaz, E.; Mathew, R.; Hawkinson, D.; Esfandyari, T.; Liu, Z.; Lee, P.W.; Farassati, F. Pro-oncogenic cell signaling machinery as a target for oncolytic viruses. Curr. Pharm. Biotechnol. 2012, 13, 1742–1749. [Google Scholar] [CrossRef] [PubMed]
- Pikor, L.A.; Bell, J.C.; Diallo, J.S. Oncolytic Viruses: Exploiting Cancer’s Deal with the Devil. Trends Cancer 2015, 1, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, O.V.; Chumakov, P.M. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev. Med. Virol. 2018, 28, e2008. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Bénéteau, M.; Zunino, B.; Jacquin, M.A.; Meynet, O.; Chiche, J.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.E. Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc. Natl. Acad. Sci. USA 2012, 109, 20071–20076. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the Keys to the Kitchen: Viral Manipulation of the Host Cell Metabolic Network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef]
- Cai, J.; Zhu, W.; Lin, Y.; Hu, J.; Liu, X.; Xu, W.; Liu, Y.; Hu, C.; He, S.; Gong, S.; et al. Lonidamine potentiates the oncolytic efficiency of M1 virus independent of hexokinase 2 but via inhibition of antiviral immunity. Cancer Cell Int. 2020, 20, 532. [Google Scholar] [CrossRef]
- Al-Ziaydi, A.G.; Al-Shammari, A.M.; Hamzah, M.I.; Kadhim, H.S.; Jabir, M.S. Hexokinase inhibition using D-Mannoheptulose enhances oncolytic newcastle disease virus-mediated killing of breast cancer cells. Cancer Cell Int. 2020, 20, 420. [Google Scholar] [CrossRef]
- Al-Ziaydi, A.G.; Al-Shammari, A.M.; Hamzah, M.I.; Kadhim, H.S.; Jabir, M.S. Newcastle disease virus suppress glycolysis pathway and induce breast cancer cells death. Virusdisease 2020, 31, 341–348. [Google Scholar] [CrossRef]
- Li, C.; Meng, G.; Su, L.; Chen, A.; Xia, M.; Xu, C.; Yu, D.; Jiang, A.; Wei, J. Dichloroacetate blocks aerobic glycolytic adaptation to attenuated measles virus and promotes viral replication leading to enhanced oncolysis in glioblastoma. Oncotarget 2015, 6, 1544–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seegers, S.L.; Frasier, C.; Greene, S.; Nesmelova, I.V.; Grdzelishvili, V.Z. Experimental Evolution Generates Novel Oncolytic Vesicular Stomatitis Viruses with Improved Replication in Virus-Resistant Pancreatic Cancer Cells. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Altomonte, J.; Marozin, S.; Schmid, R.M.; Ebert, O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol. Ther. 2010, 18, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Rogulin, V.; Simon, I.; Rose, J.K.; van den Pol, A.N. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 2010, 84, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.C.; Hwang, T.; Park, B.H.; Bell, J.; Kirn, D.H. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol. Ther. 2008, 16, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Byrne, T.; Inoue, M.; Tait, M.E.; Wall, P.; Wang, A.; Dermyer, M.R.; Laklai, H.; Binder, J.J.; Lees, C.; et al. Oncolytic Vaccinia Virus Gene Modification and Cytokine Expression Effects on Tumor Infection, Immune Response, and Killing. Mol. Cancer Ther. 2021, 20, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Tian, L.; Chen, J.; Wang, J.; Ma, R.; Dong, W.; Li, A.; Zhang, J.; Antonio Chiocca, E.; Kaur, B.; et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat. Commun. 2021, 12, 5908. [Google Scholar] [CrossRef] [PubMed]
- Uche, I.K.; Kousoulas, K.G.; Rider, P.J.F. The Effect of Herpes Simplex Virus-Type-1 (HSV-1) Oncolytic Immunotherapy on the Tumor Microenvironment. Viruses 2021, 13, 1200. [Google Scholar] [CrossRef] [PubMed]
- Boagni, D.A.; Ravirala, D.; Zhang, S.X. Current strategies in engaging oncolytic viruses with antitumor immunity. Mol. Ther. Oncolytics 2021, 22, 98–113. [Google Scholar] [CrossRef]
- Nichols, D.B.; De Martini, W.; Cottrell, J. Poxviruses Utilize Multiple Strategies to Inhibit Apoptosis. Viruses 2017, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Imre, G. Cell death signalling in virus infection. Cell Signal. 2020, 76, 109772. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef]
- De Munck, J.; Binks, A.; McNeish, I.A.; Aerts, J.L. Oncolytic virus-induced cell death and immunity: A match made in heaven? J. Leukoc. Biol. 2017, 102, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 2019, 8, 1591875. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, C.; Zhou, Z.; Jiang, K.; Yu, S.; Jia, L.; Wu, Y.; Liu, Y.; Meng, S.; Ding, C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch. Virol. 2012, 157, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.T.; Tao, X.H.; Fan, Y.B.; Wang, S.B. Crosstalk between oncolytic viruses and autophagy in cancer therapy. Biomed. Pharmacother. 2021, 134, 110932. [Google Scholar] [CrossRef]
- Furukawa, Y.; Takasu, A.; Yura, Y. Role of autophagy in oncolytic herpes simplex virus type 1-induced cell death in squamous cell carcinoma cells. Cancer Gene Ther. 2017, 24, 393–400. [Google Scholar] [CrossRef]
- Zhang, J.; Lai, W.; Li, Q.; Yu, Y.; Jin, J.; Guo, W.; Zhou, X.; Liu, X.; Wang, Y. A novel oncolytic adenovirus targeting Wnt signaling effectively inhibits cancer-stem like cell growth via metastasis, apoptosis and autophagy in HCC models. Biochem. Biophys. Res. Commun. 2017, 491, 469–477. [Google Scholar] [CrossRef]
- Zahedi-Amiri, A.; Malone, K.; Beug, S.T.; Alain, T.; Yeganeh, B. Autophagy in Tumor Immunity and Viral-Based Immunotherapeutic Approaches in Cancer. Cells 2021, 10, 2672. [Google Scholar] [CrossRef]
- Correa, R.J.; Komar, M.; Tong, J.G.; Sivapragasam, M.; Rahman, M.M.; McFadden, G.; Dimattia, G.E.; Shepherd, T.G. Myxoma virus-mediated oncolysis of ascites-derived human ovarian cancer cells and spheroids is impacted by differential AKT activity. Gynecol. Oncol. 2012, 125, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Barrett, J.W.; Stanford, M.; Werden, S.J.; Johnston, J.B.; Gao, X.; Sun, M.; Cheng, J.Q.; McFadden, G. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. USA 2006, 103, 4640–4645. [Google Scholar] [CrossRef] [Green Version]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Q.; Xin, H.Y.; Lyu, Y.N.; Ma, Z.W.; Peng, X.C.; Xiang, Y.; Wang, Y.Y.; Wu, Z.J.; Cheng, J.T.; Ji, J.F.; et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018, 25, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- McGray, A.J.R.; Huang, R.Y.; Battaglia, S.; Eppolito, C.; Miliotto, A.; Stephenson, K.B.; Lugade, A.A.; Webster, G.; Lichty, B.D.; Seshadri, M.; et al. Oncolytic Maraba virus armed with tumor antigen boosts vaccine priming and reveals diverse therapeutic response patterns when combined with checkpoint blockade in ovarian cancer. J. Immunother. Cancer 2019, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamarin, D.; Ricca, J.M.; Sadekova, S.; Oseledchyk, A.; Yu, Y.; Blumenschein, W.M.; Wong, J.; Gigoux, M.; Merghoub, T.; Wolchok, J.D. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J. Clin. Investig. 2018, 128, 1413–1428. [Google Scholar] [CrossRef] [Green Version]
- Chulpanova, D.S.; Solovyeva, V.V.; Kitaeva, K.V.; Dunham, S.P.; Khaiboullina, S.F.; Rizvanov, A.A. Recombinant Viruses for Cancer Therapy. Biomedicines 2018, 6, 94. [Google Scholar] [CrossRef] [Green Version]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers 2020, 12, 1699. [Google Scholar] [CrossRef]
- de Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; van den Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.D.; Janssen, A.B.; van Beusechem, V.W. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin. Biol. Ther. 2015, 15, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geiβ, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, C.; Oronsky, B.; Scicinski, J.; Fanger, G.R.; Stirn, M.; Oronsky, A.; Reid, T.R. Going viral: A review of replication-selective oncolytic adenoviruses. Oncotarget 2015, 6, 19976–19989. [Google Scholar] [CrossRef] [Green Version]
- Pelin, A.; Boulton, S.; Tamming, L.A.; Bell, J.C.; Singaravelu, R. Engineering vaccinia virus as an immunotherapeutic battleship to overcome tumor heterogeneity. Expert Opin. Biol. Ther. 2020, 20, 1083–1097. [Google Scholar] [CrossRef]
- Pol, J.G.; Workenhe, S.T.; Konda, P.; Gujar, S.; Kroemer, G. Cytokines in oncolytic virotherapy. Cytokine Growth Factor Rev. 2020, 56, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Guz-Montgomery, K.; Saha, D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells 2020, 9, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, X.; Wang, J.; Gao, D.; Li, Y.; Li, H.; Chu, Y.; Zhang, Z.; Liu, H.; Jiang, G.; et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017, 8, 1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sheng, Y.; Hou, W.; Sampath, P.; Byrd, D.; Thorne, S.; Zhang, Y. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J. Immunother. Cancer 2020, 8, e000131. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.S.; Kalinski, P.; Thorne, S.H.; Bartlett, D.L. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011, 19, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Ståhle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenthe, J.; Naseri, S.; Hellström, A.C.; Wiklund, H.J.; Eriksson, E.; Loskog, A. Immunostimulatory oncolytic virotherapy for multiple myeloma targeting 4-1BB and/or CD40. Cancer Gene Ther. 2020, 27, 948–959. [Google Scholar] [CrossRef]
- Vijayakumar, G.; McCroskery, S.; Palese, P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Liu, J.; Zhou, H.; Li, J.; Sun, C.; Zhu, W.; Yin, Y.; Li, X. Enhanced anti-tumor response elicited by a novel oncolytic HSV-1 engineered with an anti-PD-1 antibody. Cancer Lett. 2021, 518, 49–58. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef]
- Scott, E.M.; Jacobus, E.J.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R.; et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 320. [Google Scholar] [CrossRef]
- Tong, Y.; You, L.; Liu, H.; Li, L.; Meng, H.; Qian, Q.; Qian, W. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget 2013, 4, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Wang, S.; Xu, N.; Chen, Y.; Wu, G.; Zhang, A.; Chen, X.; Tong, Y.; Qian, W. Enhancing therapeutic efficacy of oncolytic vaccinia virus armed with Beclin-1, an autophagic Gene in leukemia and myeloma. Biomed. Pharmacother. 2020, 125, 110030. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Di Somma, S.; Iannuzzi, C.A.; Pentimalli, F.; Portella, G. Virotherapy: From single agents to combinatorial treatments. Biochem. Pharmacol. 2020, 177, 113986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Cheng, P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol. Cancer 2020, 19, 158. [Google Scholar] [CrossRef]
- Oh, C.M.; Chon, H.J.; Kim, C. Combination Immunotherapy Using Oncolytic Virus for the Treatment of Advanced Solid Tumors. Int. J. Mol. Sci. 2020, 21, 7742. [Google Scholar] [CrossRef]
- Chicas-Sett, R.; Zafra-Martin, J.; Morales-Orue, I.; Castilla-Martinez, J.; Berenguer-Frances, M.A.; Gonzalez-Rodriguez, E.; Rodriguez-Abreu, D.; Couñago, F. Immunoradiotherapy as An Effective Therapeutic Strategy in Lung Cancer: From Palliative Care to Curative Intent. Cancers 2020, 12, 2178. [Google Scholar] [CrossRef]
- Waters, A.M.; Johnston, J.M.; Reddy, A.T.; Fiveash, J.; Madan-Swain, A.; Kachurak, K.; Bag, A.K.; Gillespie, G.Y.; Markert, J.M.; Friedman, G.K. Rationale and Design of a Phase 1 Clinical Trial to Evaluate HSV G207 Alone or with a Single Radiation Dose in Children with Progressive or Recurrent Malignant Supratentorial Brain Tumors. Hum. Gene Ther. Clin. Dev. 2017, 28, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Cathail, S.M.; Pokrovska, T.D.; Maughan, T.S.; Fisher, K.D.; Seymour, L.W.; Hawkins, M.A. Combining Oncolytic Adenovirus with Radiation-A Paradigm for the Future of Radiosensitization. Front. Oncol. 2017, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Simbawa, E.; Al-Johani, N.; Al-Tuwairqi, S. Modeling the Spatiotemporal Dynamics of Oncolytic Viruses and Radiotherapy as a Treatment for Cancer. Comput. Math. Methods Med. 2020, 2020, 3642654. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Velez, N.; Marigil, M.; García-Moure, M.; Gonzalez-Huarriz, M.; Aristu, J.J.; Ramos-García, L.I.; Tejada, S.; Díez-Valle, R.; Patiño-García, A.; Becher, O.J.; et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol. Commun. 2019, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Udayakumar, T.S.; Betancourt, D.M.; Ahmad, A.; Tao, W.; Totiger, T.M.; Patel, M.; Marples, B.; Barber, G.; Pollack, A. Radiation Attenuates Prostate Tumor Antiviral Responses to Vesicular Stomatitis Virus Containing IFNβ, Resulting in Pronounced Antitumor Systemic Immune Responses. Mol. Cancer Res. 2020, 18, 1232–1243. [Google Scholar] [CrossRef]
- Kellish, P.; Shabashvili, D.; Rahman, M.M.; Nawab, A.; Guijarro, M.V.; Zhang, M.; Cao, C.; Moussatche, N.; Boyle, T.; Antonia, S.; et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J. Clin. Investig. 2019, 129, 2279–2292. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.T.; Liu, J.; Li, S.; Rahman, M.M.; Mona, M.; McFadden, G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol. Ther. 2012, 20, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Hall, R.R.; Lesniak, M.S.; Ahmed, A.U. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses 2015, 7, 6200–6217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, D.G.; Bell, J.C. Cell carriers for oncolytic viruses: Current challenges and future directions. Oncolytic Virotherapy 2013, 2, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, Y.; Wang, N.; Wu, X.; Xu, J.; Zhou, Y.; Zhao, X.; He, Z. Mesenchymal stem cell carriers enhance antitumor efficacy induced by oncolytic reovirus in acute myeloid leukemia. Int. Immunopharmacol. 2021, 94, 107437. [Google Scholar] [CrossRef]
- Hammad, M.; Cornejo, Y.R.; Batalla-Covello, J.; Majid, A.A.; Burke, C.; Liu, Z.; Yuan, Y.C.; Li, M.; Dellinger, T.H.; Lu, J.; et al. Neural Stem Cells Improve the Delivery of Oncolytic Chimeric Orthopoxvirus in a Metastatic Ovarian Cancer Model. Mol. Ther. Oncolytics 2020, 18, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, Y.; Li, M.; Dellinger, T.H.; Mooney, R.; Rahman, M.M.; McFadden, G.; Aboody, K.S.; Hammad, M. NSCs are permissive to oncolytic Myxoma virus and provide a delivery method for targeted ovarian cancer therapy. Oncotarget 2020, 11, 4693–4698. [Google Scholar] [CrossRef]
- Mooney, R.; Majid, A.A.; Batalla-Covello, J.; Machado, D.; Liu, X.; Gonzaga, J.; Tirughana, R.; Hammad, M.; Lesniak, M.S.; Curiel, D.T.; et al. Enhanced Delivery of Oncolytic Adenovirus by Neural Stem Cells for Treatment of Metastatic Ovarian Cancer. Mol. Ther. Oncolytics 2019, 12, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 8627. [Google Scholar] [CrossRef] [PubMed]
- Malogolovkin, A.; Gasanov, N.; Egorov, A.; Weener, M.; Ivanov, R.; Karabelsky, A. Combinatorial Approaches for Cancer Treatment Using Oncolytic Viruses: Projecting the Perspectives through Clinical Trials Outcomes. Viruses 2021, 13, 1271. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Chen, K.; Qian, L.; Wang, P. Oncolytic virotherapy reverses the immunosuppressive tumor microenvironment and its potential in combination with immunotherapy. Cancer Cell Int. 2021, 21, 262. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.I.; Park, A.K.; Chaurasiya, S.; Kang, S.; Lu, J.; Yang, A.; Sivanandam, V.; Zhang, Z.; Woo, Y.; Priceman, S.J.; et al. Recombinant Orthopoxvirus Primes Colon Cancer for Checkpoint Inhibitor and Cross-Primes T Cells for Antitumor and Antiviral Immunity. Mol. Cancer Ther. 2021, 20, 173–182. [Google Scholar] [CrossRef]
- Nair, S.; Mazzoccoli, L.; Jash, A.; Govero, J.; Bais, S.S.; Hu, T.; Fontes-Garfias, C.R.; Shan, C.; Okada, H.; Shresta, S.; et al. Zika virus oncolytic activity requires CD8+ T cells and is boosted by immune checkpoint blockade. JCI Insight 2021, 6, e144619. [Google Scholar] [CrossRef] [PubMed]
- Havunen, R.; Kalliokoski, R.; Siurala, M.; Sorsa, S.; Santos, J.M.; Cervera-Carrascon, V.; Anttila, M.; Hemminki, A. Cytokine-Coding Oncolytic Adenovirus TILT-123 Is Safe, Selective, and Effective as a Single Agent and in Combination with Immune Checkpoint Inhibitor Anti-PD-1. Cells 2021, 10, 246. [Google Scholar] [CrossRef]
- Haines, B.B.; Denslow, A.; Grzesik, P.; Lee, J.S.; Farkaly, T.; Hewett, J.; Wambua, D.; Kong, L.; Behera, P.; Jacques, J.; et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Christie, J.D.; Appel, N.; Canter, H.; Achi, J.G.; Elliott, N.M.; de Matos, A.L.; Franco, L.; Kilbourne, J.; Lowe, K.; Rahman, M.M.; et al. Systemic delivery of TNF-armed myxoma virus plus immune checkpoint inhibitor eliminates lung metastatic mouse osteosarcoma. Mol. Ther. Oncolytics 2021, 22, 539–554. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, M.; Zhang, Z.; Tang, X.; Chen, Y.; Peng, A.; Peng, X.; Tong, A.; Zhou, L. Interleukin-7-loaded oncolytic adenovirus improves CAR-T cell therapy for glioblastoma. Cancer Immunol. Immunother. 2021, 70, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.E.; Rosewell Shaw, A.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Name | Virus Type | Host | Year Approved | Country Approved | Indication | Background |
|---|---|---|---|---|---|---|
| Rigvir (ECHO-7) | Picornavirus | Human | 2004 | Latvia | Melanoma | Unmodified |
| Oncorine (H101) | Adenovirus serotype 5 | Human | 2005 | China | Head and neck cancer | Deleted for viral E1B-55K and with four deletions in viral E3 |
| T-VEC (Imlygic) | HSV-1 | Human | 2015 | United States and Europe | Metastatic melanoma | Deletion of ICP34.5 and ICP47; encoding two copies of human GMCSF |
| DELYTACT (teserpaturev/G47Δ) | HSV-1 | Human | 2021 | Japan | Malignant glioma or any primary brain cancer | Triple mutation (Deletion of ICP34.5, ICP6 and α47 genes) |
| Virus | Genome (Size) | Cell Receptor/Binding Determinants | Replication Site | Vertebrate Host | Examples of OV Candidates |
|---|---|---|---|---|---|
| DNA Virus: | |||||
| Adenovirus | dsDNA (35 kb) | CAR | Nucleus | Human, animals | DNX-2401, ONCOS-102, AD-E6E7 |
| Herpesvirus: HSV-1, HSV-2 | dsDNA (154 kb) | HVEM, Nectin 1, Nectin 2 | Nucleus | Human (HSV-1) | T-VEC, OH2, HSVG207, M032 |
| Parvovirus: B19PV, H1PV | ssDNA (5 kb) | Sialic acid residues, P antigens | Nucleus | Human, animals | ParvOryx01 |
| Poxvirus: VACV, MYXV | dsDNA (160–190 kb) | Heparan, laminin, chondroitin, integrin β1, CD98 | Cytoplasm | VACV (unknown), MYXV (rabbit) | Pexa-Vec, JX-594 |
| RNA Virus: | |||||
| Alphavirus: Semiliki Forest virus (SFV), Sindbis virus (SINV), M1 | SS (+) RNA (11–12 kb) | Prohibitin, phosphatidyl serine, GAGs, ATP synthetase β subunit | Cytoplasm | SFV: rodents/human; SINV: birds | SFV-IL12; SINV AR339 |
| Flavivirus: Zika virus | SS (+) RNA (10.8 kb) | GAGS, Heparan sulfate, C-type lectin | Cytoplasm | Monkey | ZIKV-LAV |
| Paramyxo virus: | |||||
| Measles virus | SS (−) RNA (16 kb) | SLAMF1 (CD150), CD46, Nectin 4 | Nucleus | Human | MV-NIS |
| Newcastle disease virus (NDV) | SS (−) RNA (15 kb) | Sialic acid | Nucleus | Birds | MEDI5395 |
| Picornavirus: | |||||
| Coxsackievirus A21 | SS (+) RNA (28 kb) | CAR, ICAM-1, DAF | Cytoplasm | Human | CVA21, CV-B3 |
| Polio virus | SS (+) RNA (7.5 kb) | CD155 | Cytoplasm | Human | PVSRIPO |
| Seneca valley virus (SVV) | SS (+) RNA (7 kb) | Anthrax toxin receptor 1 | Cytoplasm | Pig, cow | SVV-001 |
| Reovirus | dsRNA (23 kb) | Sialic acid, JAM1 | Cytoplasm | Human | Reolysin |
| Rhabdovirus: | |||||
| VSV | SS (−) RNA (11 kb) | LDLR | Cytoplasm | Cattle, horse, pigs | VSV-IFNβ-NIS |
| Maraba virus: MG1 | SS (−) RNA (11 kb) | LDLR | Cytoplasm | Amazonian phlebotomine sand flies | MG1MA3 |
| Virus | Biological Agent | Genetic Modifications/Transgenes | Combination Therapy | Indication (Delivery Route) | Clinical Phase | Clinical Trial No |
|---|---|---|---|---|---|---|
| Adenovirus | LOAd703 | TMZ-CD40L 4-1 BBL | Gemcitabine, nab-paclitaxel, +/− anti-PD-L1 | Pancreatic cancer (IT) | I/II | NCT02705196 |
| DNX-2440 | Recurrent glioblastoma (stereo tactically) | I | NCT03714334 | |||
| TILT-123 | TNF, IL-2 | Solid tumor (IT) | I | NCT04695327 | ||
| TILT-123 | TNF, IL-2 | Adoptive cell therapy with TILs | Metastatic melanoma (IT/IV) | I | NCT04217473 | |
| Enadenotucirev (Ad3) | Capecitabine, radiotherapy | Locally advanced rectal cancer (IT) | I | NCT03916510 | ||
| NG-641 | FAP-TAc antibody + CXCL9/CXCL10/IFNa | Metastatic cancer, epithelial tumors (IT/IV) | I | NCT04053283 | ||
| AdAPT-001 | Solid tumor (IT) | I | NCT04673942 | |||
| Ad5 OBP-301 (Telomelysin) | hTERT | Melanoma stage III/IV (IT) | II | NCT03190824 | ||
| HSV | OH2 (HSV2) | GMCSF | Advanced or metastatic pancreatic cancer | I/II | NCT04637698 | |
| OH2 | GMCSF | HX008 (anti-PD-1) | Advanced solid tumors | I/II | NCT03866525 | |
| OH2 | GMCSF | HX008 (anti-PD-1) | melanoma | I/II | NCT04616443 | |
| OH2 | GMCSF | Keytruda (anti-PD-1) | Advanced solid tumors, melanoma | I/II | NCT04386967 | |
| HSVG207 (HSV-1) | Tumor selective mutation | Low dose of radiation (5 Gy) | Pediatric brain tumor, recurrent or refractory cerebellar brain tumor | I | NCT03911388 | |
| HSVG207 | Tumor selective mutation | Low dose of radiation (5 Gy) | Recurrent high-grade glioma in children | II | NCT04482933 | |
| M032 (NSC733972) | IL-12 | Low dose of radiation (5 Gy) | Recurrent malignant glioma | I | NCT02062827 | |
| Parvovirus (H-1 PV) | ParvOryx | Pancreatic ductal carcinoma | I/II | NCT02653313 | ||
| VACV | Pexa-Vec (JX-594) | GM-CSF (TK inactivated) | Tremelimumab (anti-CTLA4), Durvalumab (anti-PD-L1) | Colorectal cancer | I/II | NCT03206073 |
| TBio-6517/RIVAL-01 | Pembrolizumab (anti-PD-1) | Advanced solid tumors (IT) | I/II | NCT04301011 | ||
| T601 | TK-RR deletion, expression of FCU1 gene | 5-FC | Advanced malignant solid tumors | I/II | NCT04226066 | |
| ASP9801 | IL-7, IL-12 | Metastatic advanced solid tumors (IT) | I | NCT03954067 | ||
| NDV | MEDI5395 | GMCSF | Durvalumab (anti-PD-L1) | Advanced solid tumors (IT) | I | NCT03889275 |
| Reovirus | Reolysin | Anti-PD-1 | Metastatic TNBC | II | NCT04445844 | |
| Reolysin | Chemotherapy (Dexamethasone, carfilzomib), ICIs (Nivolumab) | Relapsed multiple myeloma | I | NCT03605719 | ||
| Reolysin | Chemotherapy (paclitaxel), ICI (anti-PD-L1) | Metastatic breast cancer | II | NCT04215146 | ||
| Reolysin | Chemotherapy (letrozole), ICIs (atzolizumab, trastuzumab) | Breast cancer | I | NCT04102618 | ||
| VSV | VSV-IFNβ/TYRP1 | IFNβ, tyrosinase related protein 1 | Stage III-IV melanoma (IT/IV) | I | NCT03865212 | |
| VSV-IFNβ-NIS | IFNβ, NIS | Avelumab (anti-PD-L1) | Malignant solid tumor (IT) | I | NCT02923466 | |
| VSV-IFNβ-NIS | IFNβ, NIS | Cyclophosphamide, ruxolitinib phosphate | MM, AML, T cell lymphoma (IV) | I | NCT03017820 | |
| VSV-IFNβ-NIS | IFNβ, NIS | Pembrolizumab (anti-PD-1) | Refractory NSCLC, HNSCC, solid tumor (IV) | I | NCT03647163 | |
| Maraba | MG1MA3 (MG1 Maraba/MAGE-A3) | MAGE-A3 | Advanced/metastatic solid tumors | I/II | NCT02285816 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; McFadden, G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers 2021, 13, 5452. https://doi.org/10.3390/cancers13215452
Rahman MM, McFadden G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers. 2021; 13(21):5452. https://doi.org/10.3390/cancers13215452
Chicago/Turabian StyleRahman, Masmudur M., and Grant McFadden. 2021. "Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy" Cancers 13, no. 21: 5452. https://doi.org/10.3390/cancers13215452
APA StyleRahman, M. M., & McFadden, G. (2021). Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers, 13(21), 5452. https://doi.org/10.3390/cancers13215452