
Mismatch Repair Status in Patient-Derived Colorectal Cancer Organoids Does Not Affect Intrinsic Tumor Cell Sensitivity to Systemic Therapy

 , ,
, ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Human Specimens
2.2. In Vitro Organoid Culture
2.3. Immunohistochemistry (IHC)
2.4. DNA Isolation and Whole-Genome-Sequencing (WGS)
2.5. Drug Screens
2.6. Dose–Response Curve (DRC) Fitting and Statistical Analysis
3. Results
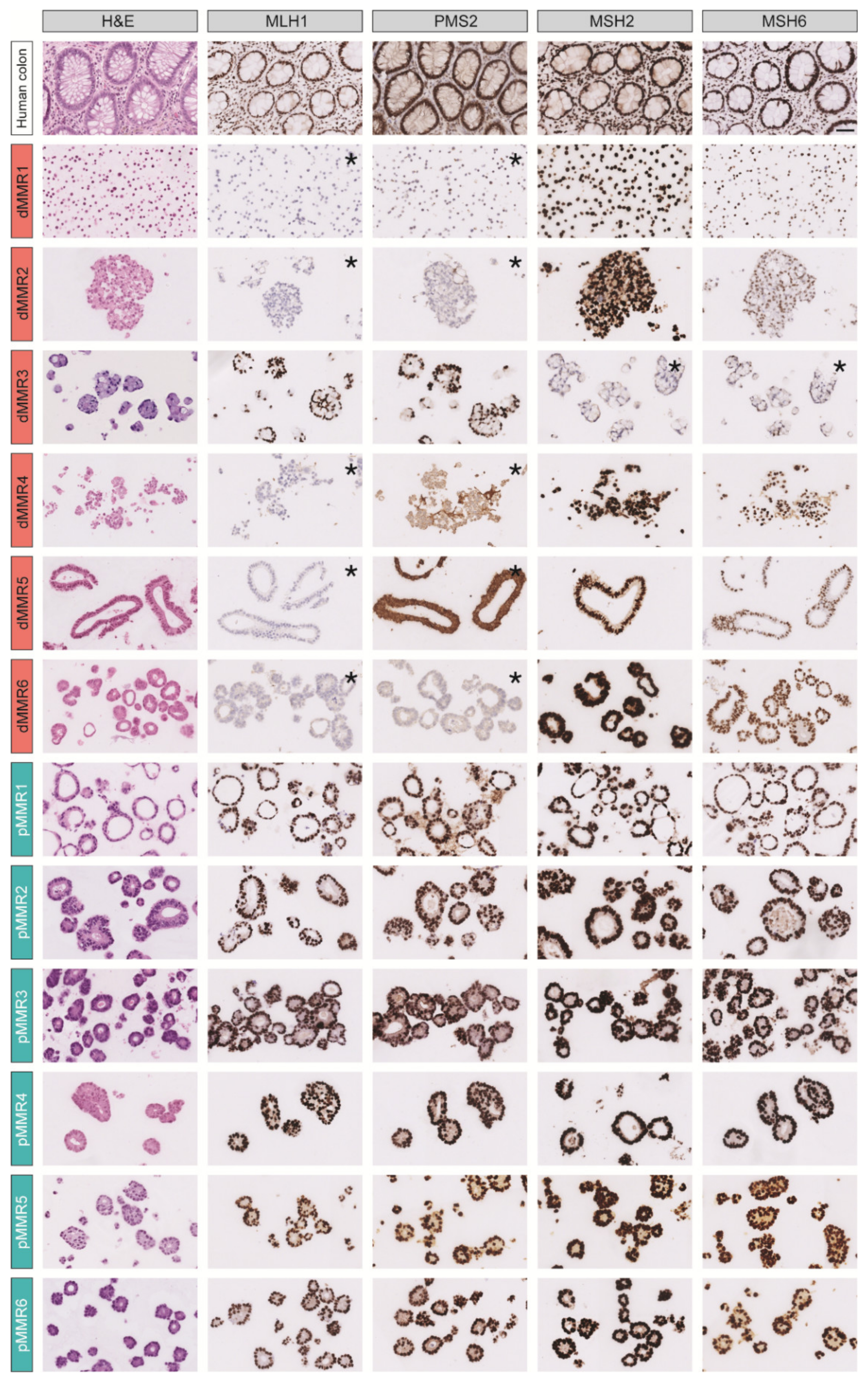
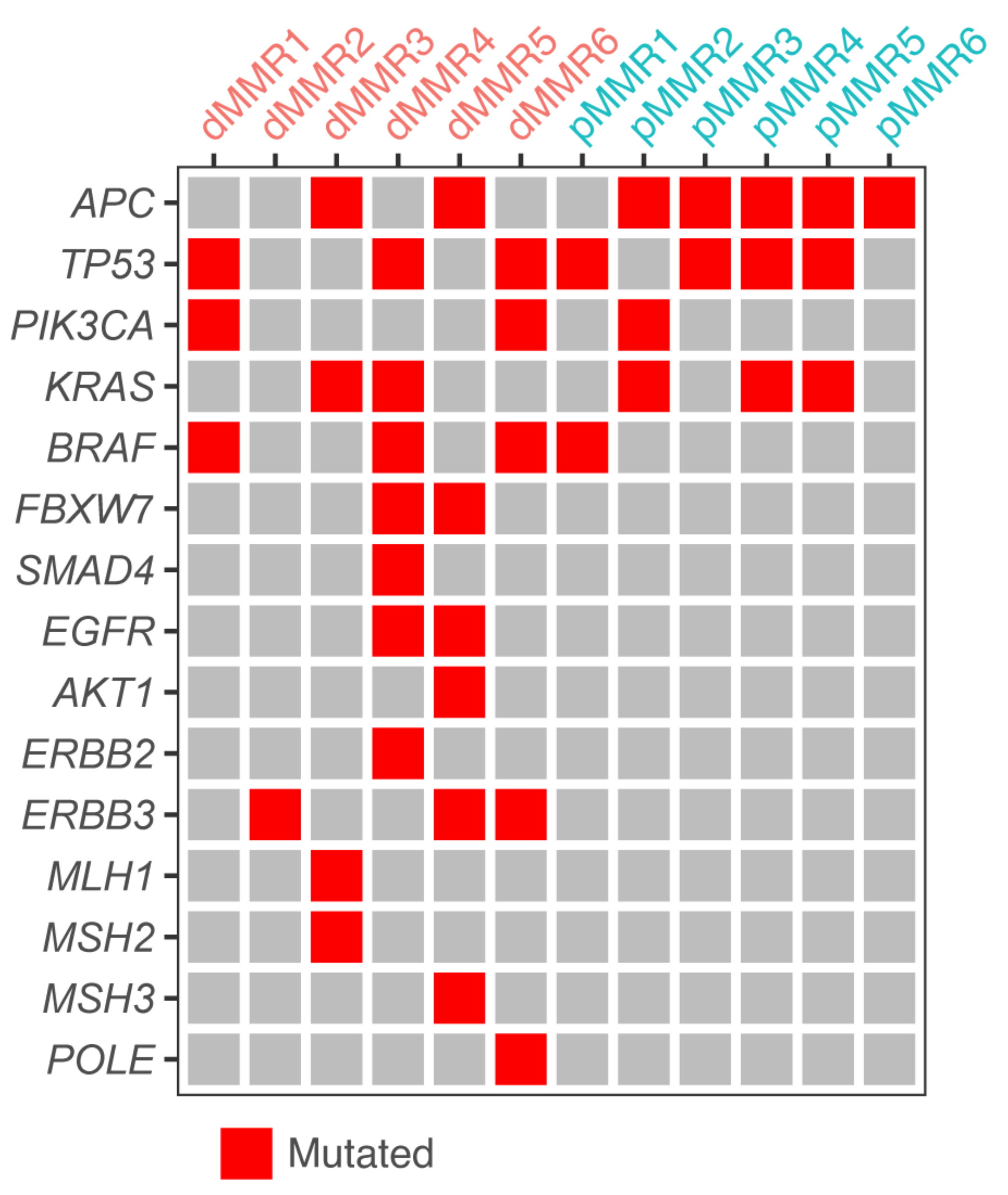
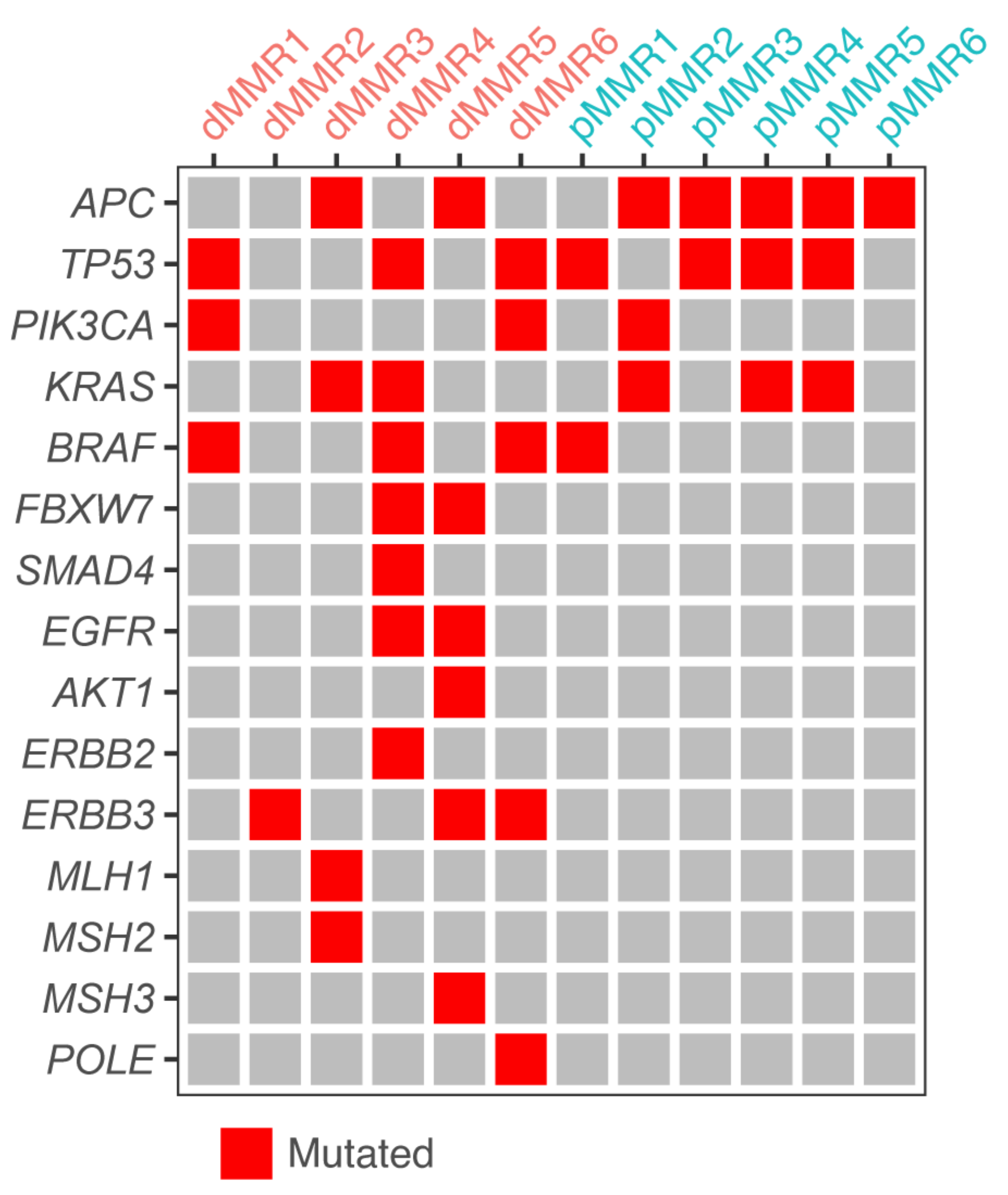
3.1. A Collection of dMMR and pMMR CRC Patient-Derived Organoids
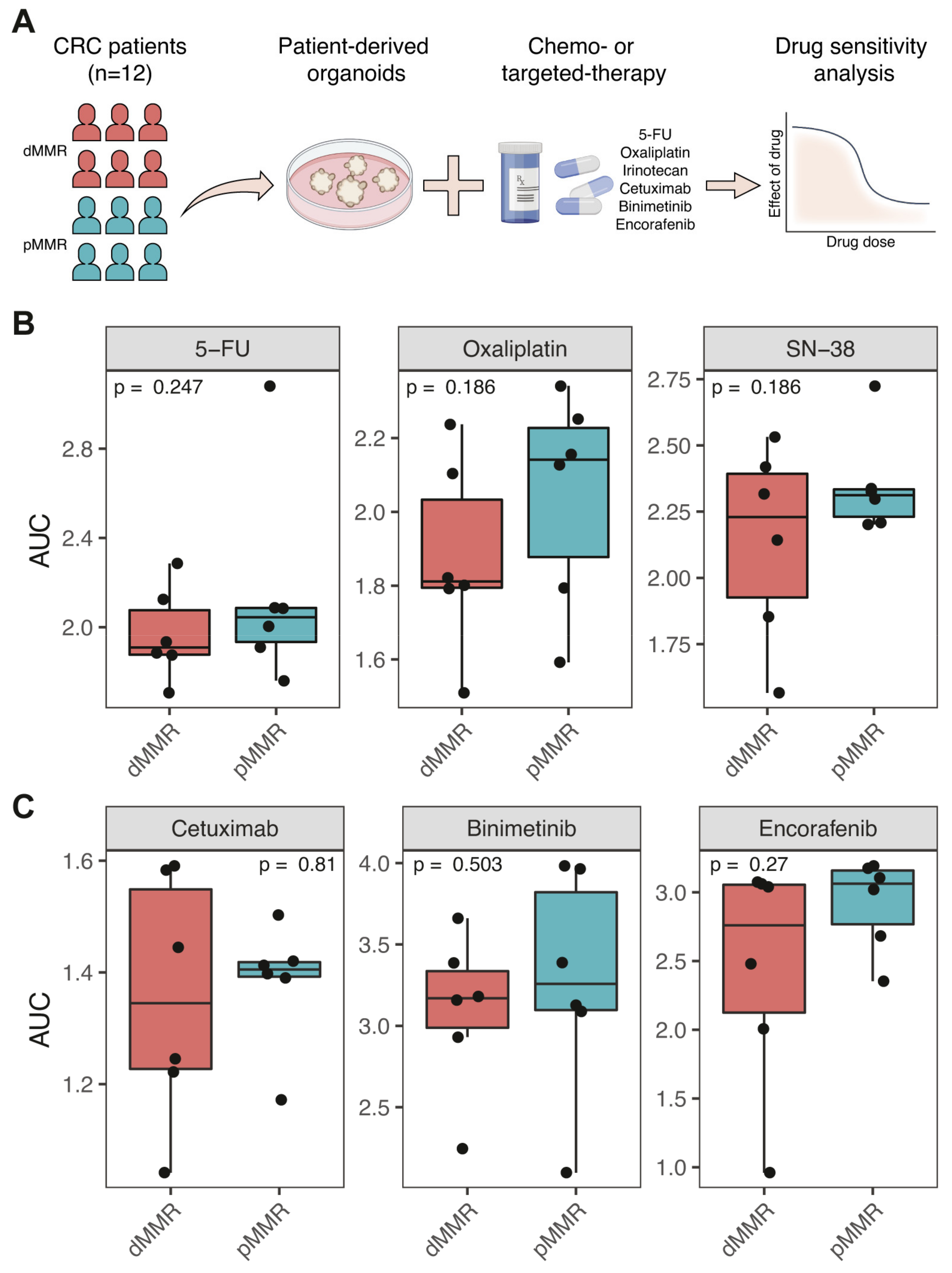
3.2. Comparable Sensitivity of dMMR and pMMR PDOs to Chemotherapy and Targeted Therapy
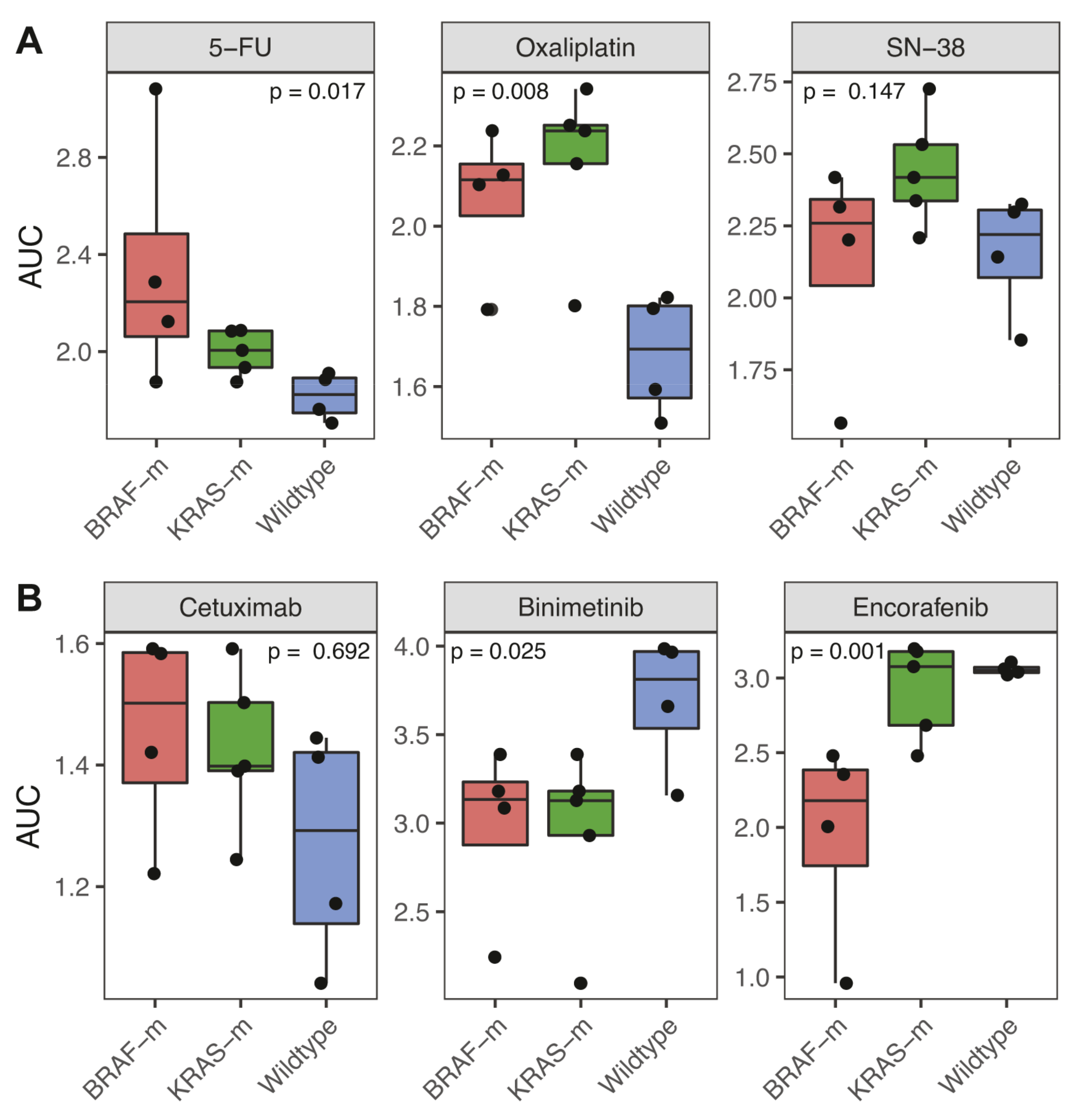
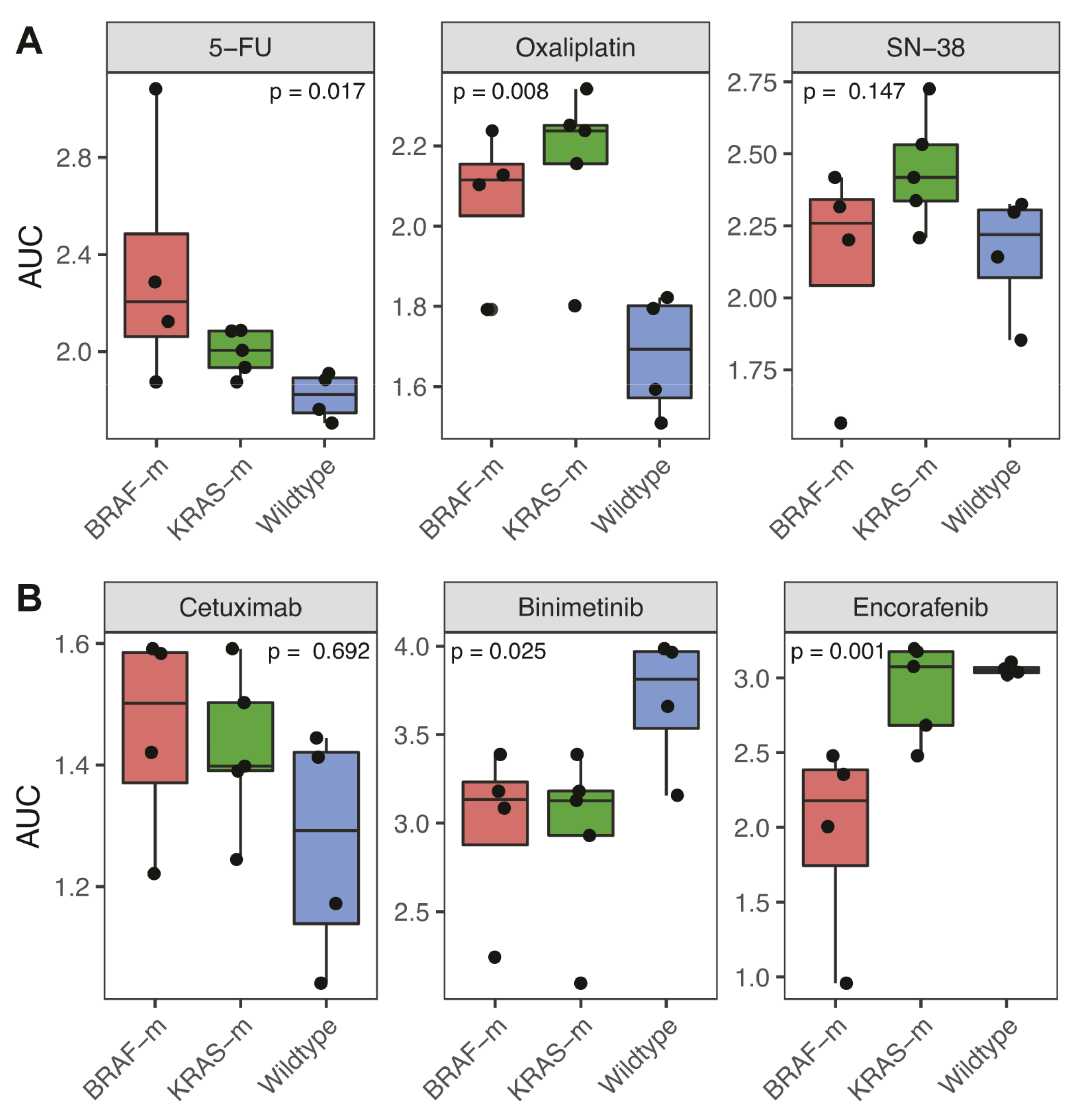
3.3. Association of BRAF and KRAS Status with Response to Chemotherapy and Targeted Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Popat, S.; Hubner, R.; Houlston, R. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, F.; Zhou, X.; Ma, Y.; Fu, W. Is microsatellite instability-high really a favorable prognostic factor for advanced colorectal cancer? A meta-analysis. World J. Surg. Oncol. 2019, 17, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venderbosch, S.; Nagtegaal, I.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.S.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensink, E.; Bond, M.; Kucukkose, E.; May, A.; Vink, G.; Koopman, M.; Kranenburg, O.; Roodhart, J. A review of the sensitivity of metastatic colorectal cancer patients with deficient mismatch repair to standard-of-care chemotherapy and monoclonal antibodies, with recommendations for future research. Cancer Treat. Rev. 2021, 95, 102174. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, K.; Dragomir, A.; Sundström, M.; Mezheyeuski, A.; Edqvist, P.; Eide, G.E.; Ponten, F.; Pfeiffer, P.; Glimelius, B.; Sorbye, H. Consequences of a high incidence of microsatellite instability and BRAF-mutated tumors: A population-based cohort of metastatic colorectal cancer patients. Cancer Med. 2019, 8, 3623–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tougeron, D.; Sueur, B.; Zaanan, A.; de la Fouchardiere, C.; Sefrioui, D.; LeComte, T.; Aparicio, T.; Guetz, G.D.; Artru, P.; Hautefeuille, V.; et al. Prognosis and chemosensitivity of deficient MMR phenotype in patients with metastatic colorectal cancer: An AGEO retrospective multicenter study. Int. J. Cancer 2020, 147, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Taieb, J.; Fiskum, J.; Yothers, G.; Goldberg, R.; Yoshino, T.; Alberts, S.; Allegra, C.; de Gramont, A.; Seitz, J.-F.; et al. Microsatellite Instability in Patients with Stage III Colon Cancer Receiving Fluoropyrimidine with or Without Oxaliplatin: An ACCENT Pooled Analysis of 12 Adjuvant Trials. J. Clin. Oncol. 2021, 39, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective Mismatch Repair as a Predictive Marker for Lack of Efficacy of Fluorouracil-Based Adjuvant Therapy in Colon Cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [Green Version]
- Ribic, C.M.; Sargent, D.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Andre, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J. Clin. Oncol. 2020, 38, LBA4. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.; Ferrante, M.; Vries, R.G.; van Es, J.H.; Brink, S.V.D.; van Houdt, W.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Wensink, G.E.; Elias, S.G.; Mullenders, J.; Koopman, M.; Boj, S.F.; Kranenburg, O.W.; Roodhart, J.M.L. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis. Oncol. 2021, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef] [PubMed]
- Mpindi, J.-P.; Swapnil, P.; Dmitrii, B.; Jani, S.; Saeed, K.; Wennerberg, K.; Aittokallio, T.; Östling, P.; Kallioniemi, O. Impact of normalization methods on high-throughput screening data with high hit rates and drug testing with dose-response data. Bioinformatics 2015, 31, 3815–3821. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Goldlust, I.; Mills, G.; Koh, S.-B.; Bramhall, J.; Richards, F.M.; Jodrell, D.I. An automated fitting procedure and software for dose-response curves with multiphasic features. Sci. Rep. 2015, 5, 14701. [Google Scholar] [CrossRef] [Green Version]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.E.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D Mutation with Outcome in Patients with Chemotherapy-Refractory Metastatic Colorectal Cancer Treated with Cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-Type BRAF Is Required for Response to Panitumumab or Cetuximab in Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Vilaró, A.; Jacobs, B.; Pomella, V.; Asbagh, L.A.; Kirkland, R.; Michel, J.; Singh, S.; Liu, X.; Kim, P.; Weitsman, G.; et al. Feedback activation of HER3 attenuates response to EGFR inhibitors in colon cancer cells. Oncotarget 2016, 8, 4277–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour–stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.S.; Richman, S.D.; Quirke, P.; Daly, C.; Adlard, J.; Elliott, F.; Barrett, J.; Selby, P.; Meade, A.M.; Stephens, R.J.; et al. Predictive Biomarkers of Chemotherapy Efficacy in Colorectal Cancer: Results from the UK MRC FOCUS Trial. J. Clin. Oncol. 2008, 26, 2690–2698. [Google Scholar] [CrossRef]
- Innocenti, F.; Ou, F.-S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients with Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.A.; Van Krieken, J.H.J.M. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.G.; Fisher, D.; Claes, B.; Maughan, T.S.; Idziaszczyk, S.; Peuteman, G.; Harris, R.; James, M.D.; Meade, A.; Jasani, B.; et al. Somatic Profiling of the Epidermal Growth Factor Receptor Pathway in Tumors from Patients with Advanced Colorectal Cancer Treated with Chemotherapy ± Cetuximab. Clin. Cancer Res. 2013, 19, 4104–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punt, C.J.A.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2016, 14, 235–246. [Google Scholar] [CrossRef]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, N.J.; Ng, K.; Hollis, D.; Saltz, L.; Mayer, R.J.; Schaefer, P.; Whittom, R.; et al. Predictive and Prognostic Roles of BRAF Mutation in Stage III Colon Cancer: Results from Intergroup Trial CALGB 89803. Clin. Cancer Res. 2011, 18, 890–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, G.; Southward, K.; Handley, K.; Magill, L.; Beaumont, C.; Stahlschmidt, J.; Richman, S.; Chambers, P.; Seymour, M.; Kerr, D.; et al. Value of Mismatch Repair, KRAS, and BRAF Mutations in Predicting Recurrence and Benefits from Chemotherapy in Colorectal Cancer. J. Clin. Oncol. 2011, 29, 1261–1270. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organoid | MMR Status (IHC) | KRAS Status | BRAF Status | Source of Tissue | Primary Tumor Location (Sidedness) |
|---|---|---|---|---|---|
| dMMR1 | MLH1/PMS2 | WT | V600E | Metastatic | Left |
| (liver) | |||||
| dMMR2 | MLH1/PMS2 | WT | WT | Metastatic | Left |
| (inguinal lymph node) | |||||
| dMMR3 | MSH2/MSH6 | A146T | WT | Metastatic | Right |
| (peritoneum) | |||||
| dMMR4 | MLH1/PMS2 | A146T | V600E | Primary | Right |
| (synchronous metastatic disease) | |||||
| dMMR5 | MLH1/PMS2 | WT | WT | Primary | Right |
| (unknown if synchronous) | |||||
| dMMR6 | MLH1/PMS2 | WT | V600E | Primary | Right |
| (unknown if synchronous) | |||||
| pMMR1 | Normal | WT | V600E | Primary | Right |
| (synchronous metastatic disease) | |||||
| pMMR2 | Normal | G12A | WT | Metastatic | Right |
| (liver) | |||||
| pMMR3 | Normal | WT | WT | Metastatic | Right |
| (liver) | |||||
| pMMR4 | Normal | G12V | WT | Primary | Unknown |
| (unknown if synchronous) | |||||
| pMMR5 | Normal | G12A | WT | Primary | Right |
| (synchronous metastatic disease) | |||||
| pMMR6 | Normal | WT | WT | Primary | Unknown |
| (unknown if synchronous) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Küçükköse, E.; Wensink, G.E.; Roelse, C.M.; van Schelven, S.J.; Raats, D.A.E.; Boj, S.F.; Koopman, M.; Laoukili, J.; Roodhart, J.M.L.; Kranenburg, O. Mismatch Repair Status in Patient-Derived Colorectal Cancer Organoids Does Not Affect Intrinsic Tumor Cell Sensitivity to Systemic Therapy. Cancers 2021, 13, 5434. https://doi.org/10.3390/cancers13215434
Küçükköse E, Wensink GE, Roelse CM, van Schelven SJ, Raats DAE, Boj SF, Koopman M, Laoukili J, Roodhart JML, Kranenburg O. Mismatch Repair Status in Patient-Derived Colorectal Cancer Organoids Does Not Affect Intrinsic Tumor Cell Sensitivity to Systemic Therapy. Cancers. 2021; 13(21):5434. https://doi.org/10.3390/cancers13215434
Chicago/Turabian StyleKüçükköse, Emre, G. Emerens Wensink, Celine M. Roelse, Susanne J. van Schelven, Daniëlle A. E. Raats, Sylvia F. Boj, Miriam Koopman, Jamila Laoukili, Jeanine M. L. Roodhart, and Onno Kranenburg. 2021. "Mismatch Repair Status in Patient-Derived Colorectal Cancer Organoids Does Not Affect Intrinsic Tumor Cell Sensitivity to Systemic Therapy" Cancers 13, no. 21: 5434. https://doi.org/10.3390/cancers13215434
APA StyleKüçükköse, E., Wensink, G. E., Roelse, C. M., van Schelven, S. J., Raats, D. A. E., Boj, S. F., Koopman, M., Laoukili, J., Roodhart, J. M. L., & Kranenburg, O. (2021). Mismatch Repair Status in Patient-Derived Colorectal Cancer Organoids Does Not Affect Intrinsic Tumor Cell Sensitivity to Systemic Therapy. Cancers, 13(21), 5434. https://doi.org/10.3390/cancers13215434