
The Urokinase Receptor (uPAR) as a “Trojan Horse” in Targeted Cancer Therapy: Challenges and Opportunities



Simple Summary
Abstract
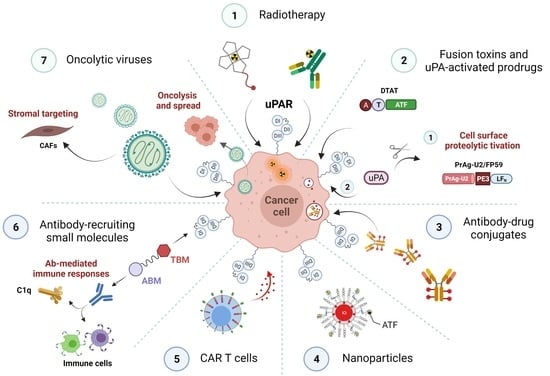
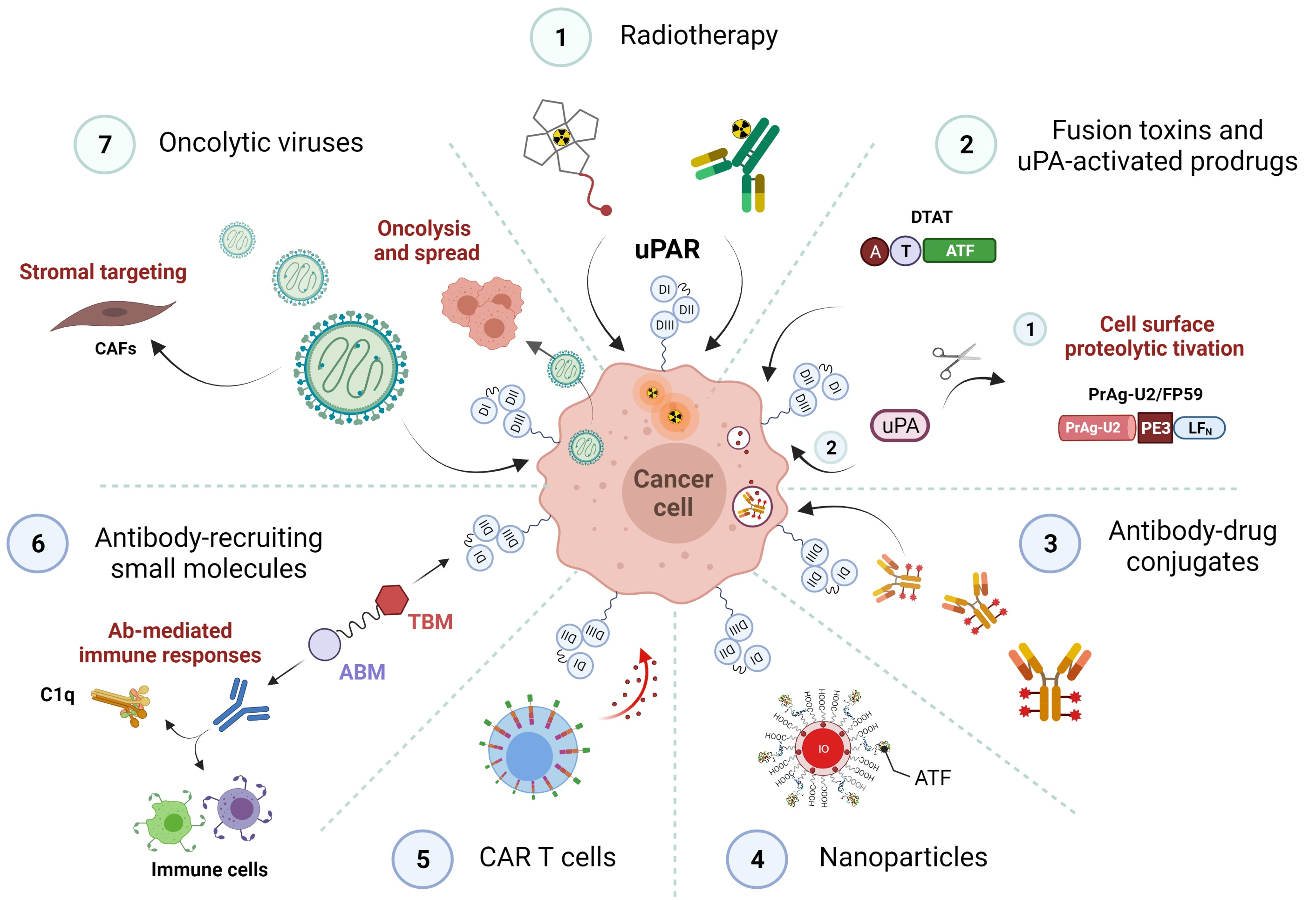
1. Introduction
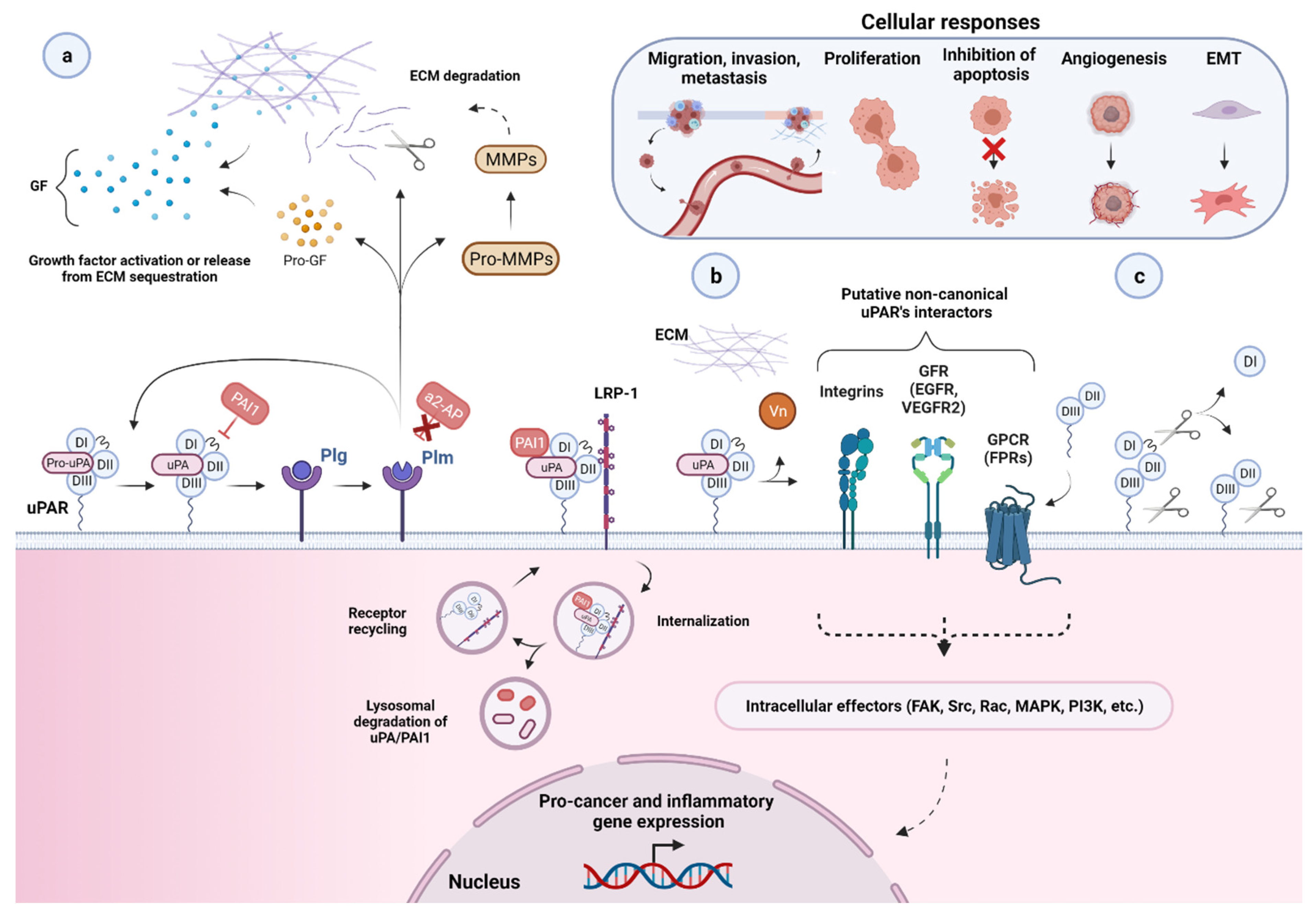
2. Biology of the Urokinase Receptor
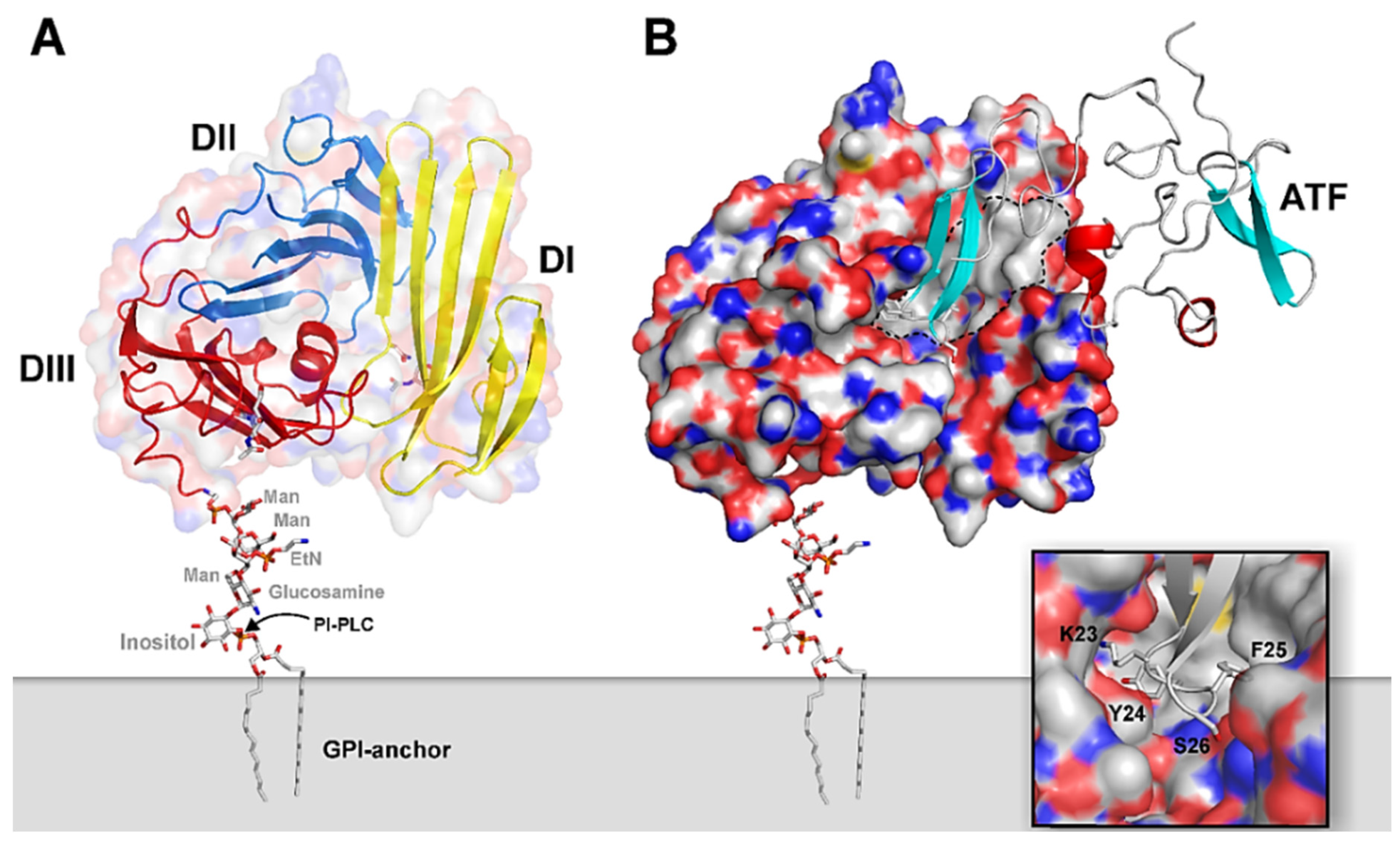
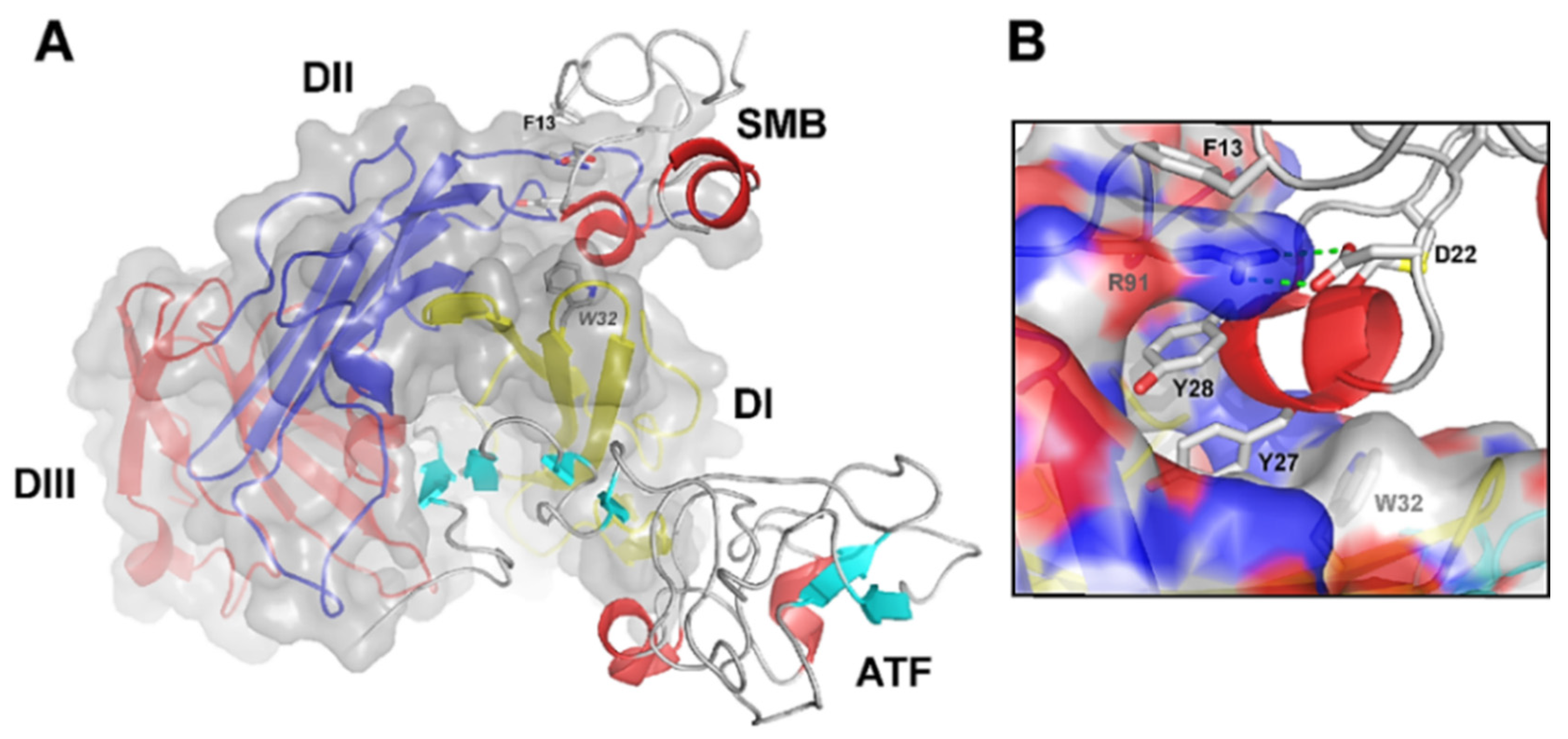
2.1. uPAR: A Flexible Multidomain Receptor
2.2. Biological Functions of uPAR

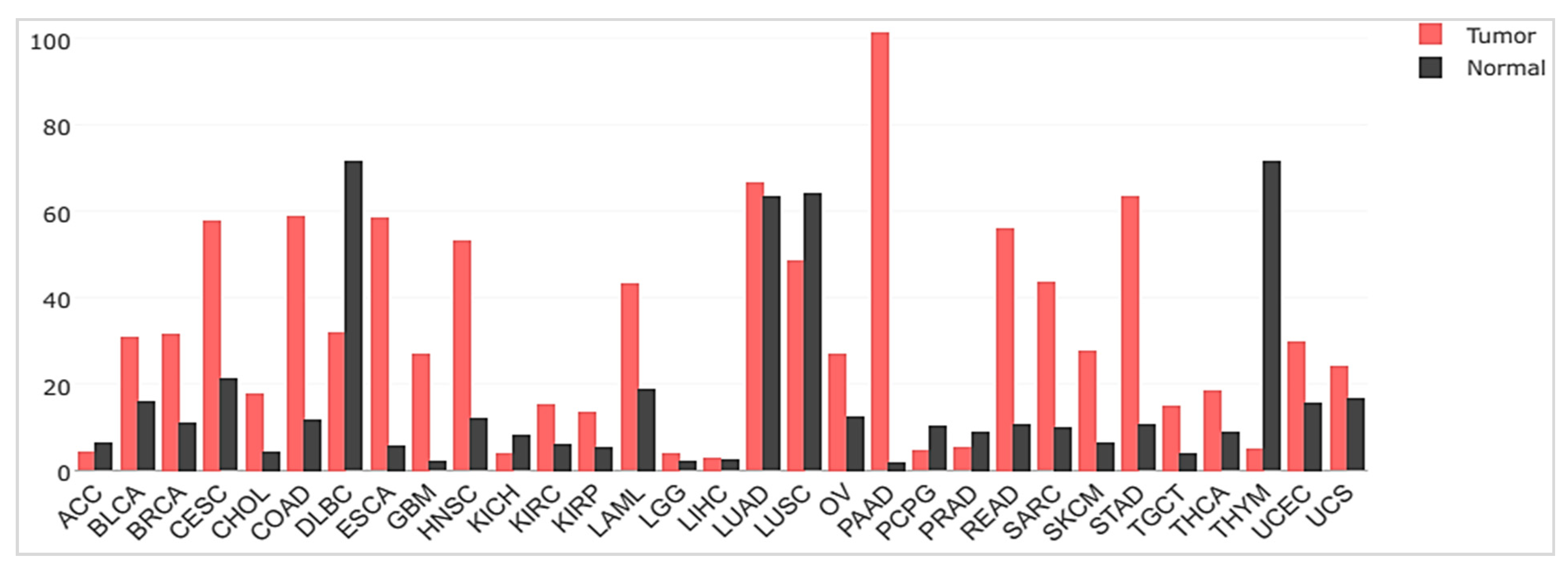
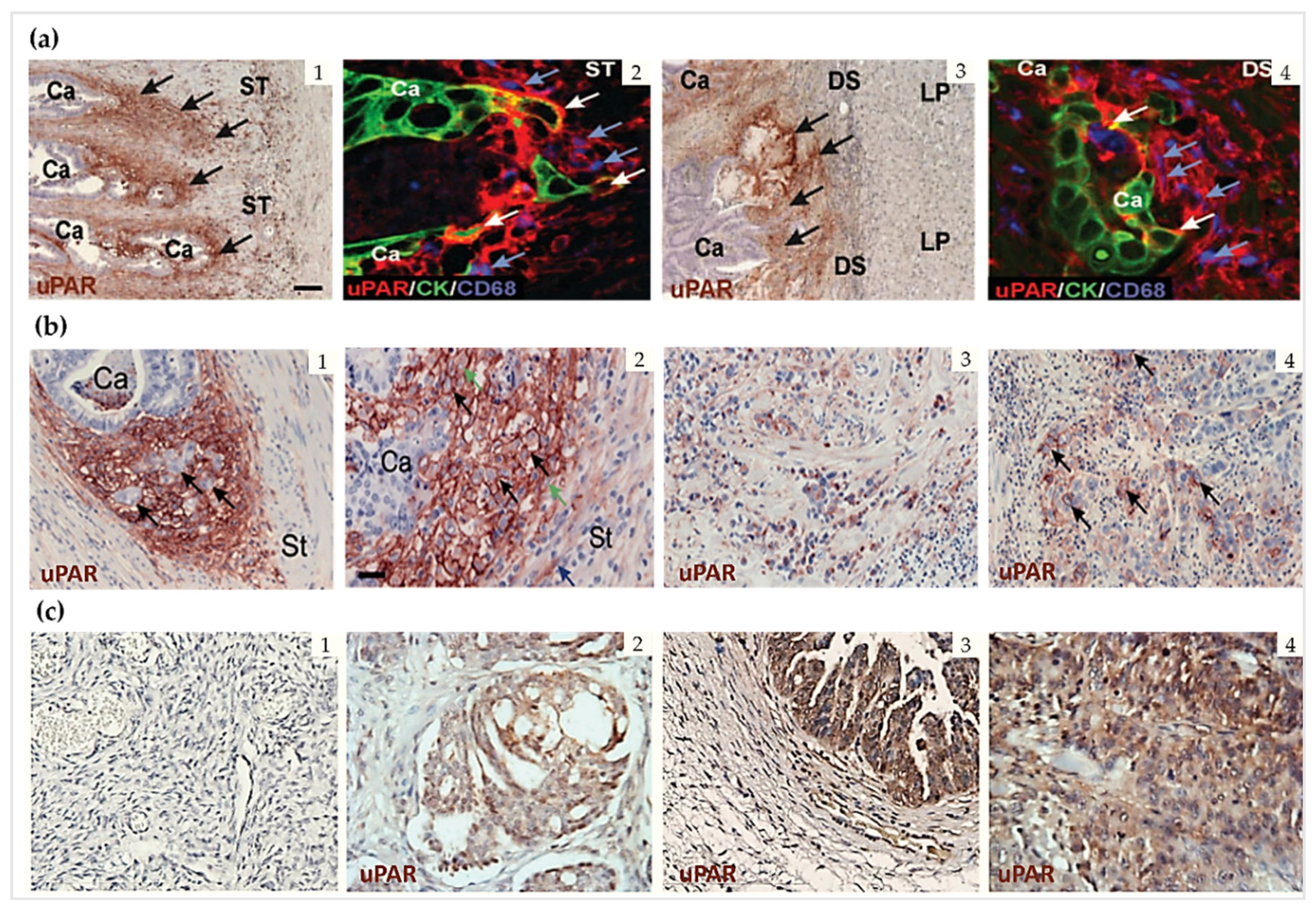
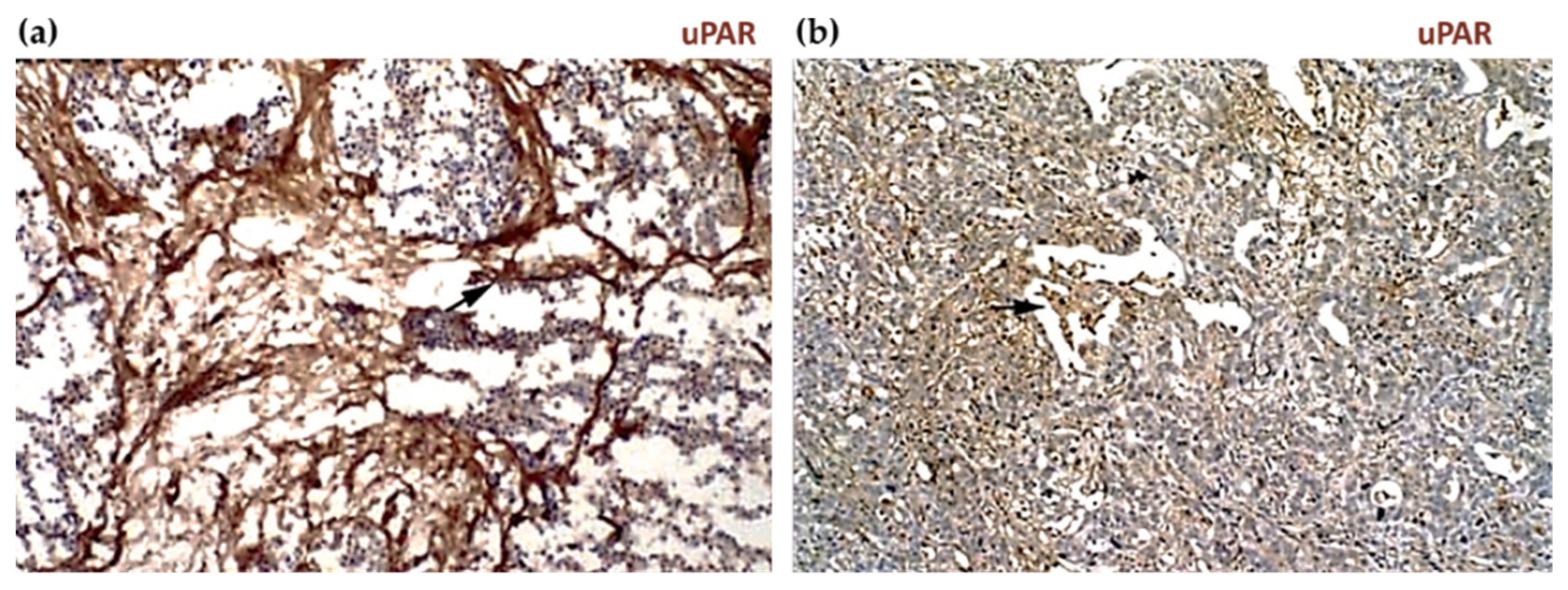
2.3. uPAR Expression during Normal Physiology and in Cancer
3. uPAR: A Potential “Gateway” for Cytotoxic Cancer Therapy
3.1. uPAR-Targeted Radionuclide Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sequence/Structure of the uPAR-Targeting Moiety | Radionuclide | Application | Model System | Ref. |
|---|---|---|---|---|---|
| [Bi3+]-DOTA–(linker4-AE105)2 |  | 213Bi | Preclinical | Ovarian cancer cells and related xenograft mouse models | [162] |
| [Lu2+]-DOTA-AE105 |  | 177Lu | Preclinical | Human prostate and colorectal cancer cells and relative xenograft mouse models | [165] |
| [Lu2+]-2G10 | Recombinant human anti-uPAR IgG identified from a human phage display library | 177Lu | Preclinical | Triple negative breast cancer cells and relative xenograft mouse models | [166] |
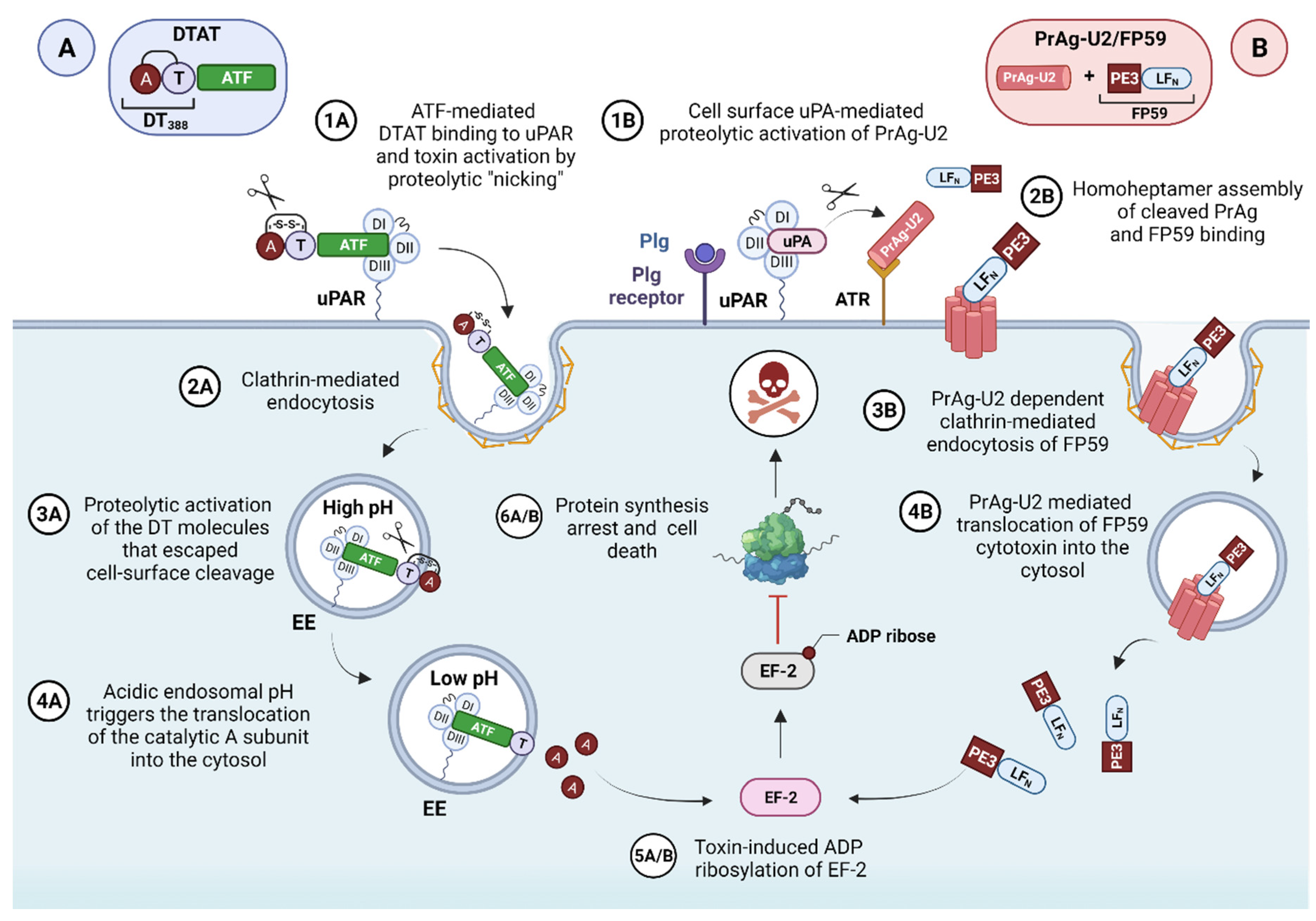
3.2. uPAR-Targeted Recombinant Fusion Toxins, Protease-Activated Prodrugs, and Antibody-Drug Conjugates
3.2.1. uPAR-Targeted Recombinant Fusion Toxins
3.2.2. uPA-Activated Prodrugs
| LT Name | Toxin | Origin of the Toxin | Additional Surface Target | Application | Model System | Ref. |
|---|---|---|---|---|---|---|
| PrAg-U2/FP59 | AT | Bacillus anthracis | / | Preclinical | Human and mice uPAR-overexpressing malignant cell lines; syngeneic mouse and human xenograft cancer models of diverse origin; Pilot POC study of canine OMM | [82,83,213,214,215,217,218] |
| PrAg-U2-R200A/PrAg-L1-I210A | LF/FP59 | Bacillus anthracis | / | Preclinical | Syngeneic mouse models of diverse origin | [216] |
| DT388GMSF | DT | Corynebacterium diphtheriae | GMCSFR | Preclinical | Human AML cell lines | [219] |
| ALA | AGAP | B. martensii Karsch | / | Preclinical | Human breast cancer cells | [223] |
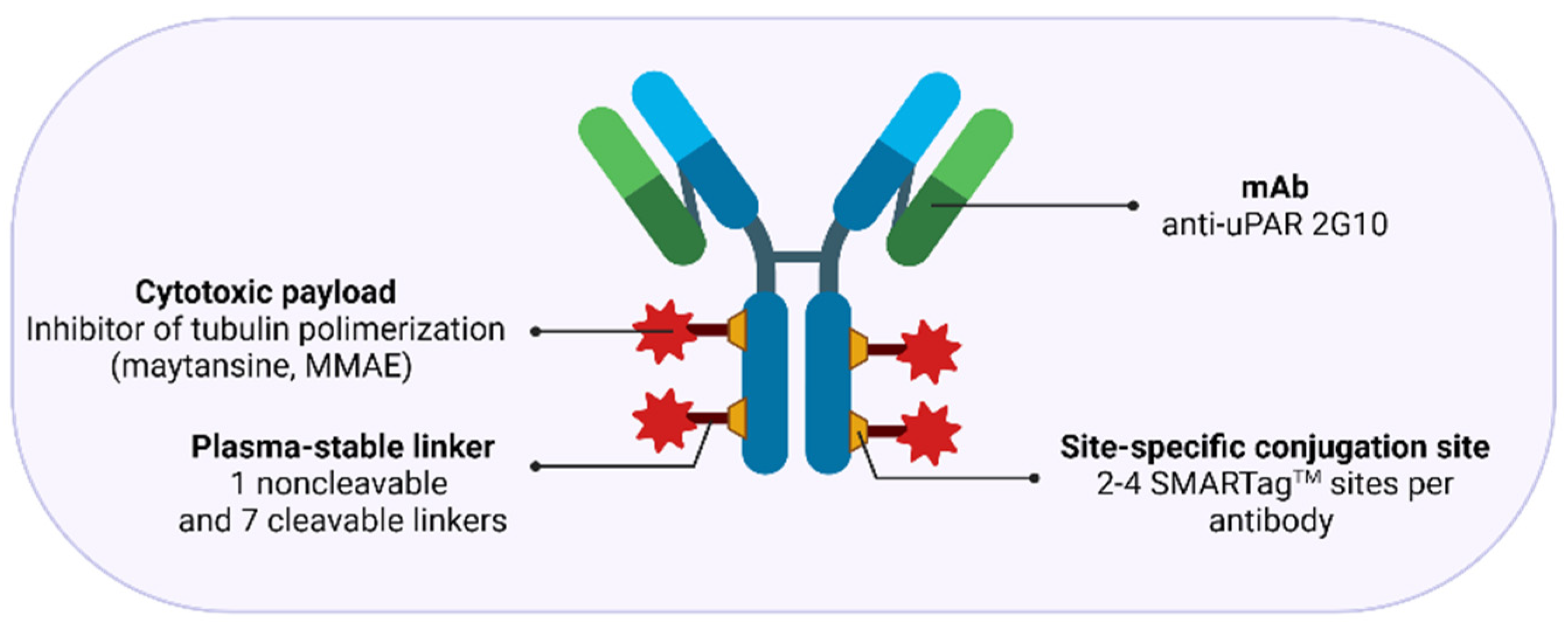
3.2.3. uPAR: A Novel Molecular Target for Antibody-Drug Conjugates (ADCs)
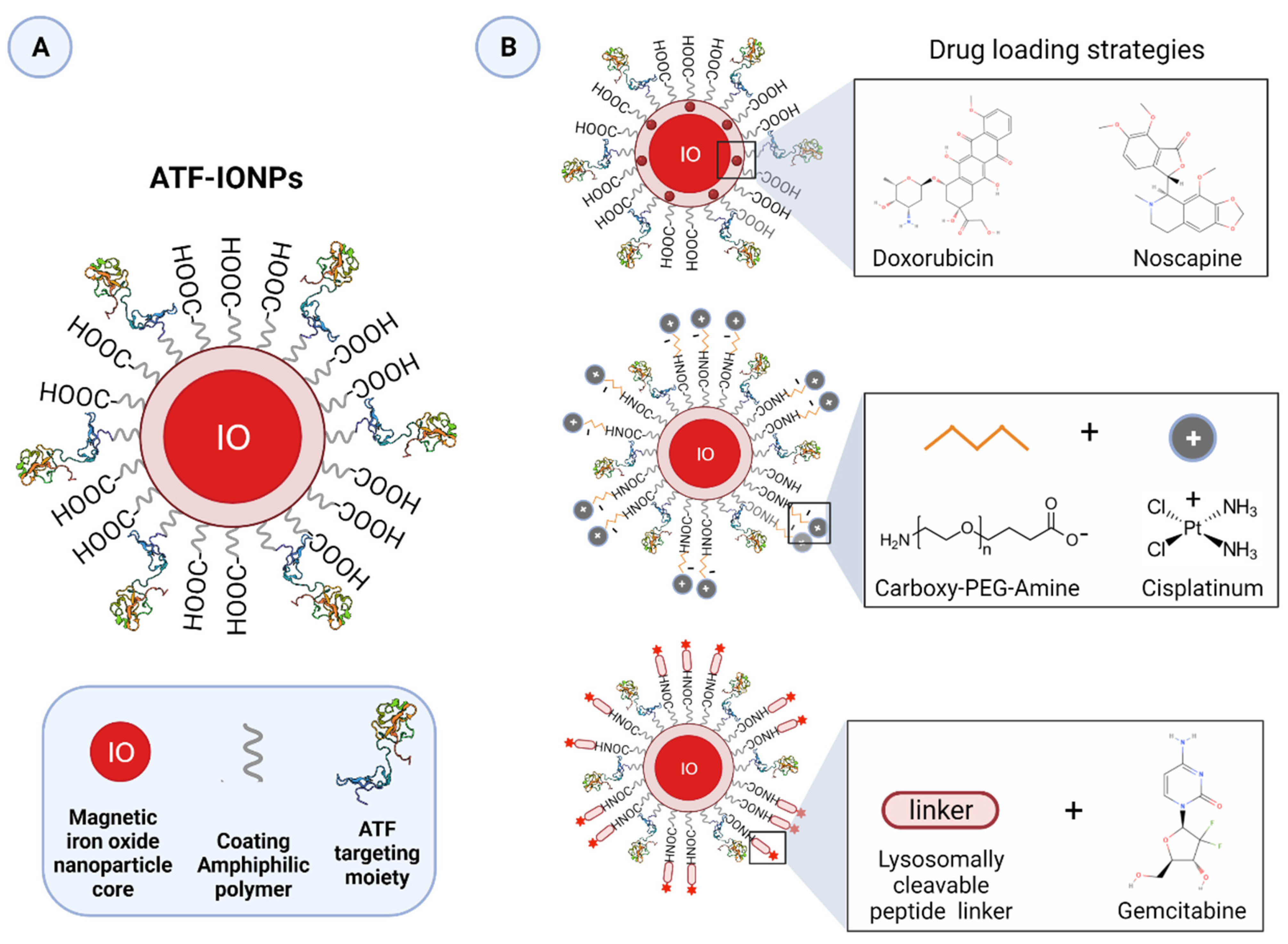
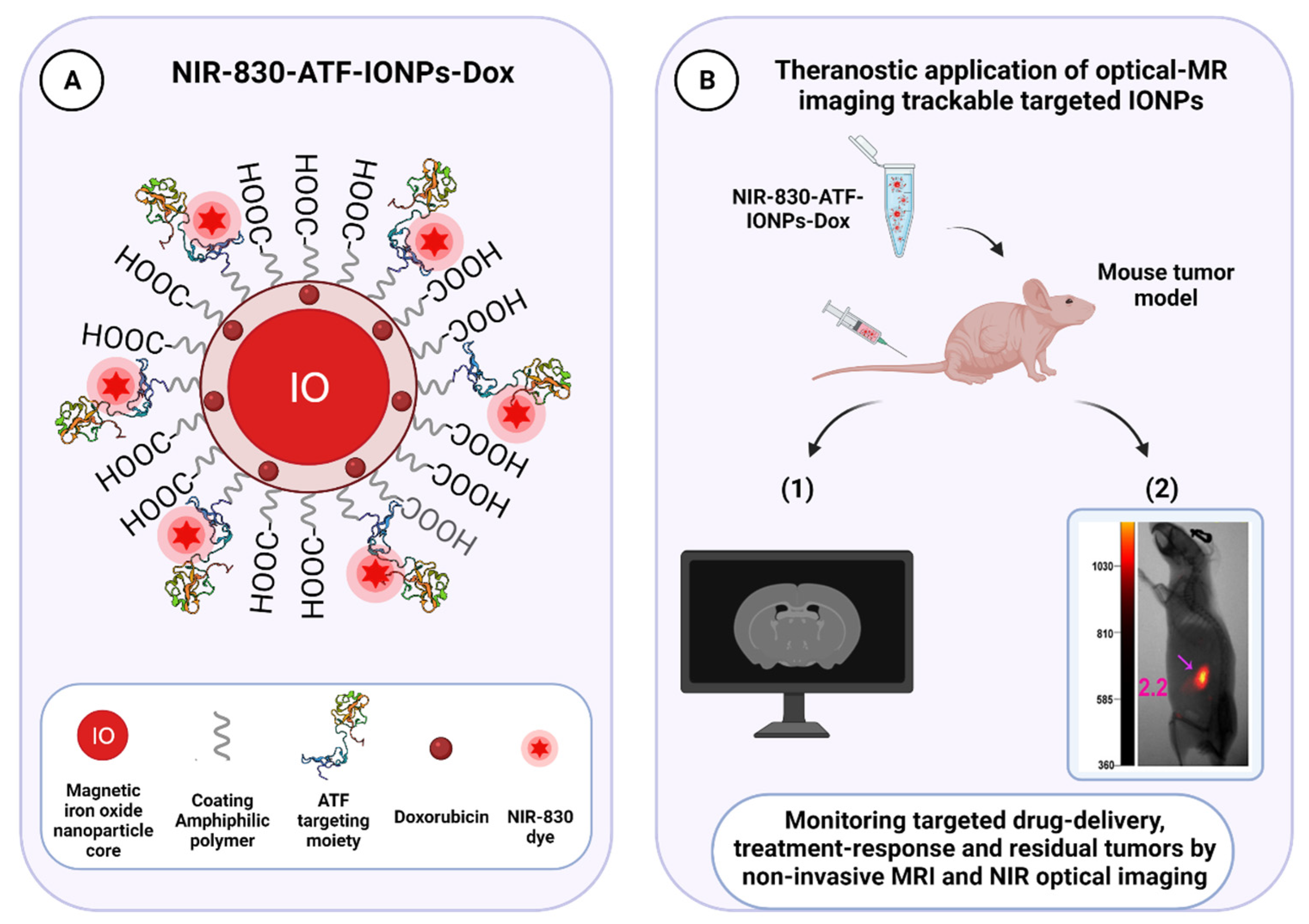
3.3. uPAR-Targeted Nanoparticles as Potential Anti-Tumor Theranostic Platforms
| Nanoparticle Composition | Cytotoxic Payload | Drug Release Mechanism | Imaging Modalities | Application | Model System | Ref. |
|---|---|---|---|---|---|---|
| Amphiphilic polymer-coated ATF-PEG-IONPs | Dox | pH-sensitive | MRI | Preclinical | Human breast cancer cells | [237] |
| Amphiphilic polymer-coated NIR-830-ATF-PEG-IONPs | Dox | pH-sensitive | MRI and NIR | Preclinical | Orthotopic human breast and pancreatic xenograft mouse models | [238] |
| Amphiphilic polymer-coated ATF-IONPs | Gem | pH- and lysosomal enzyme-dependent | MRI | Preclinical | Human pancreatic cancer cells; orthotopic human pancreatic xenograft mouse model | [236] |
| Amphiphilic polymer-coated NIR-830-ATF-PEG-IONPs | Dox; Cis | pH-sensitive | MRI and NIR | Preclinical | Mouse pancreatic cancer cells; subcutaneous and orthotopic human pancreatic xenograft mouse models; | [239] |
| Amphiphilic polymer-coated Cy5.5-ATF-IONPs | Nos | pH-sensitive | MRI and NIR | Preclinical | Human prostate cancer cells | [241] |
| Amphiphilic polymer-coated LHRH-AE105-IONPs | PTX | pH-sensitive | MRI | Preclinical | Human prostate cancer cells | [243] |
| Dual drugs co-encapsulated U11-NPs | Dox and Cur | pH-sensitive | / | Preclinical | Human lung cancer cells; human lung xenograft mouse model | [219] |
| AE147-PEG-Lipo | DTX | n/s | FI | Preclinical | Human breast cancer cells; human breast xenograft mouse model | [247] |
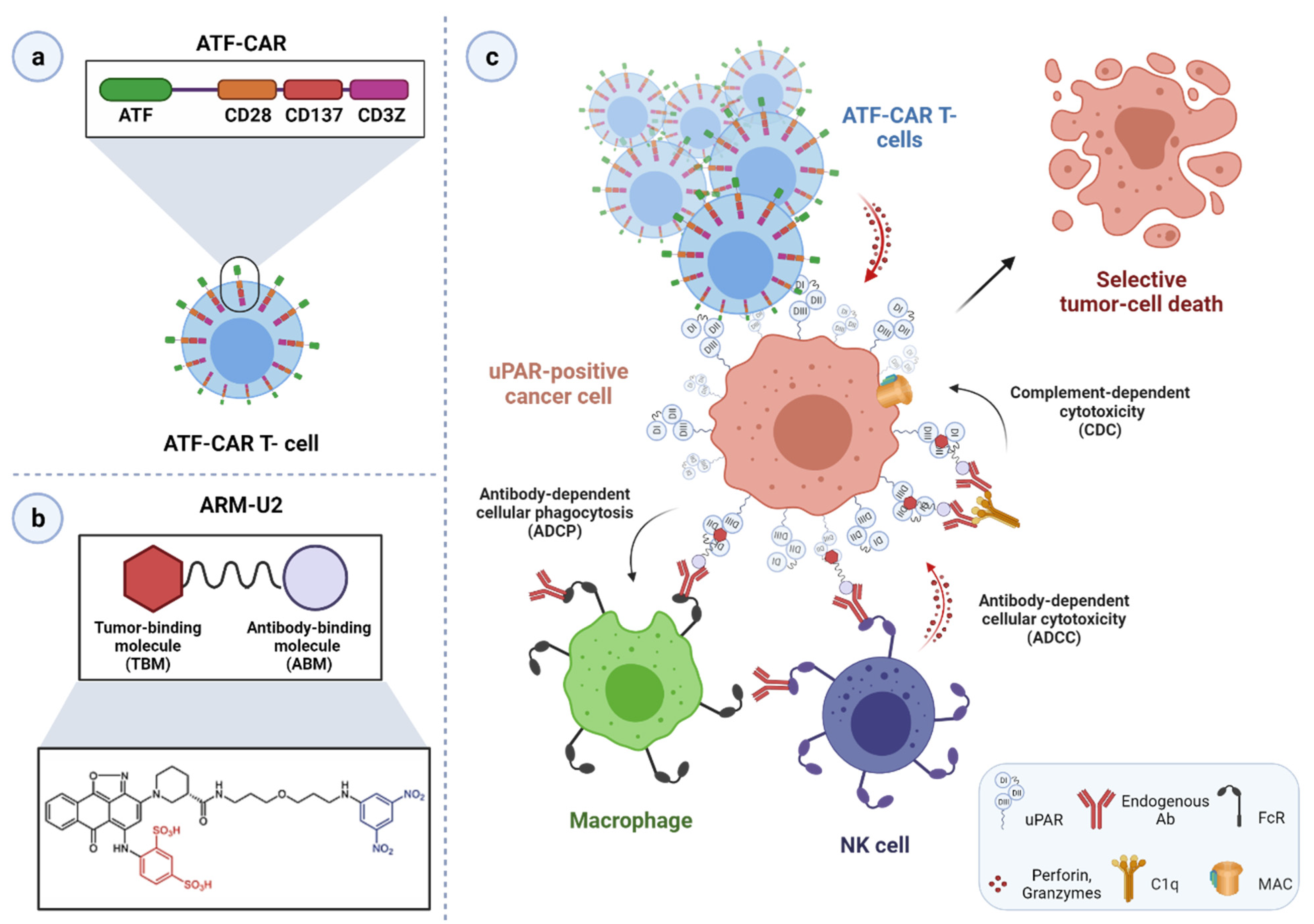
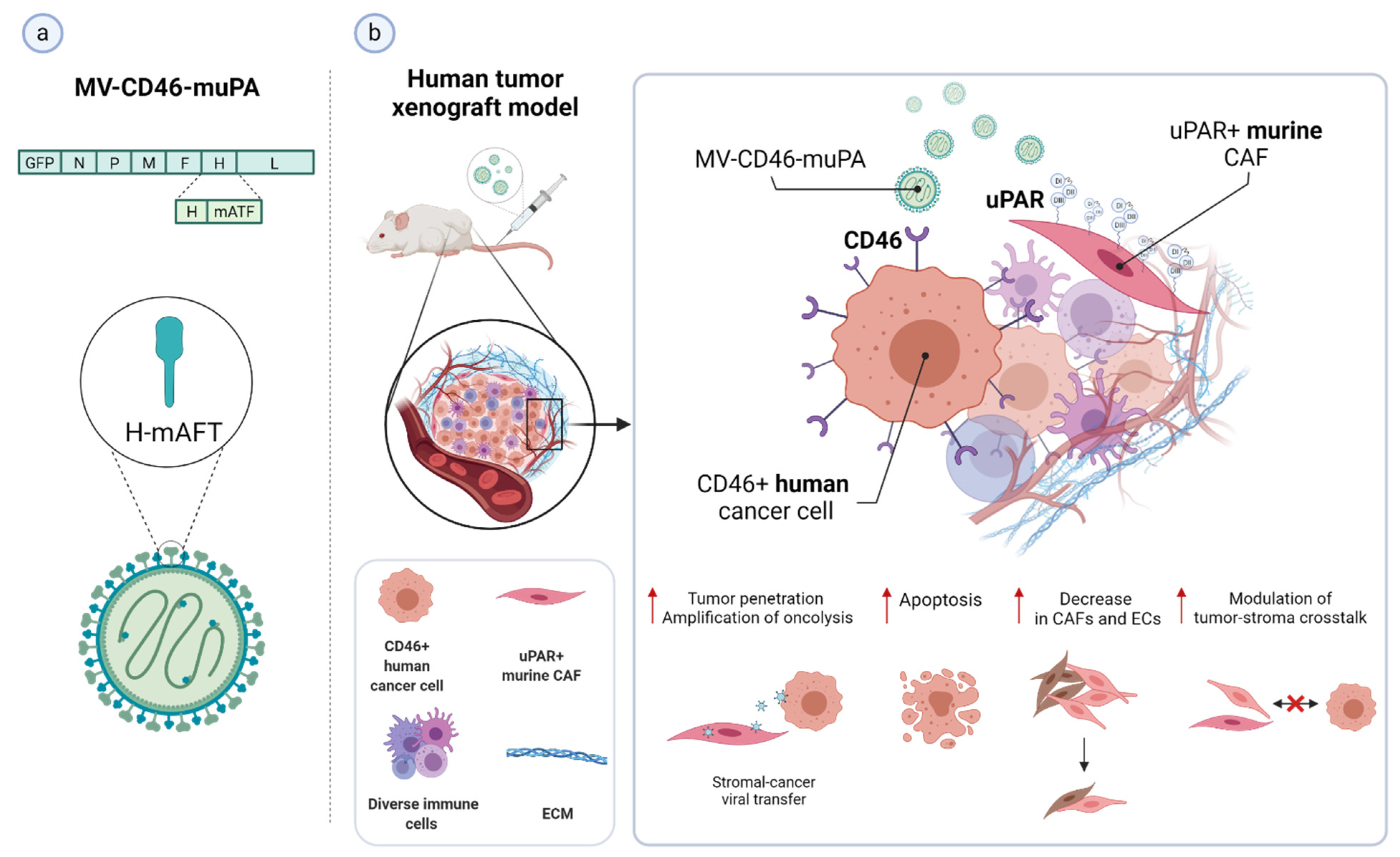
3.4. uPAR: A Novel Target for Cancer Immuno- and Virotherapy

| Approach | Application | Model System | Ref. |
|---|---|---|---|
| ATF-CAR T cells | Preclinical | Ovarian cancer cells | [251] |
| ARM-U2 | Preclinical | Glioblastoma cells; mouse melanoma allograft model | [257] |
| Oncolytic MV-m-uPA, MV-h-uPA | Preclinical | Murine and human colon and breast cancer cells, CAFs. HUVECs and murine EC; breast and colon cancer xenograft models | [261,262,263,265] |
| Dual-targeted oncolytic MV-CD46-muPA | Preclinical | CD46-positive colon tumor cells, murine uPAR-expressing CAFs; human colon tumor xenograft; | [264] |
4. Conclusions, Challenges, and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bailly, C. Cell-targeted cytotoxics: A new generation of cytotoxic agents for cancer treatment. Phytochem. Rev. 2014, 13, 171–181. [Google Scholar] [CrossRef]
- Everts, A.; Bergeman, M.; McFadden, G.; Kemp, V. Simultaneous Tumor and Stroma Targeting by Oncolytic Viruses. Biomedicines 2020, 8, 474. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Ragno, P. Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front. Biosci. 2009, 14, 2494–2503. [Google Scholar] [CrossRef]
- Conese, M.; Nykjaer, A.; Petersen, C.M.; Cremona, O.; Pardi, R.; Andreasen, P.A.; Gliemann, J.; Christensen, E.I.; Blasi, F. alpha-2 Macroglobulin receptor/Ldl receptor-related protein (Lrp)-dependent internalization of the urokinase receptor. J. Cell Biol. 1995, 131, 1609–1622. [Google Scholar] [CrossRef]
- Cubellis, M.V.; Wun, T.C.; Blasi, F. Receptor-mediated internalization and degradation of urokinase is caused by its specific inhibitor PAI-1. EMBO J. 1990, 9, 1079–1085. [Google Scholar] [CrossRef]
- Cortese, K.; Sahores, M.; Madsen, C.D.; Tacchetti, C.; Blasi, F. Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS ONE 2008, 3, e3730. [Google Scholar] [CrossRef]
- Nykjaer, A.; Conese, M.; Christensen, E.I.; Olson, D.; Cremona, O.; Gliemann, J.; Blasi, F. Recycling of the urokinase receptor upon internalization of the uPA:serpin complexes. EMBO J. 1997, 16, 2610–2620. [Google Scholar] [CrossRef]
- Hildenbrand, R.; Niedergethmann, M.; Marx, A.; Belharazem, D.; Allgayer, H.; Schleger, C.; Ströbel, P. Amplification of the urokinase-type plasminogen activator receptor (uPAR) gene in ductal pancreatic carcinomas identifies a clinically high-risk group. Am. J. Pathol. 2009, 174, 2246–2253. [Google Scholar] [CrossRef]
- Lund, I.K.; Illemann, M.; Thurison, T.; Christensen, I.J.; Høyer-Hansen, G. uPAR as anti-cancer target: Evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets 2011, 12, 1744–1760. [Google Scholar] [CrossRef]
- Mazar, A.P. The urokinase plasminogen activator receptor (uPAR) as a target for the diagnosis and therapy of cancer. Anticancer Drugs 2001, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl. Med. UniSa 2016, 15, 15–21. [Google Scholar]
- Ulisse, S.; Baldini, E.; Sorrenti, S.; D’Armiento, M. The urokinase plasminogen activator system: A target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9, 32–71. [Google Scholar] [CrossRef] [PubMed]
- Mazar, A.P.; Ahn, R.W.; O’Halloran, T.V. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr. Pharm. Des. 2011, 17, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Li Santi, A.; Napolitano, F.; Montuori, N.; Ragno, P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 4111. [Google Scholar] [CrossRef]
- Kriegbaum, M.C.; Persson, M.; Haldager, L.; Alpízar-Alpízar, W.; Jacobsen, B.; Gårdsvoll, H.; Kjær, A.; Ploug, M. Rational targeting of the urokinase receptor (uPAR): Development of antagonists and non-invasive imaging probes. Curr. Drug Targets 2011, 12, 1711–1728. [Google Scholar] [CrossRef]
- Ngo, J.C.; Jiang, L.; Lin, Z.; Yuan, C.; Chen, Z.; Zhang, X.; Yu, H.; Wang, J.; Lin, L.; Huang, M. Structural basis for therapeutic intervention of uPA/uPAR system. Curr. Drug Targets 2011, 12, 1729–1743. [Google Scholar] [CrossRef]
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics targeting the fibrinolytic system. Exp. Mol. Med. 2020, 52, 367–379. [Google Scholar] [CrossRef]
- Pillay, V.; Dass, C.R.; Choong, P.F. The urokinase plasminogen activator receptor as a gene therapy target for cancer. Trends Biotechnol. 2007, 25, 33–39. [Google Scholar] [CrossRef]
- Rockway, T.W.; Nienaber, V.; Giranda, V.L. Inhibitors of the protease domain of urokinase-type plasminogen activator. Curr. Pharm. Des. 2002, 8, 2541–2558. [Google Scholar] [CrossRef] [PubMed]
- Baart, V.M.; Houvast, R.D.; de Geus-Oei, L.F.; Quax, P.H.A.; Kuppen, P.J.K.; Vahrmeijer, A.L.; Sier, C.F.M. Molecular imaging of the urokinase plasminogen activator receptor: Opportunities beyond cancer. EJNMMI Res. 2020, 10, 87. [Google Scholar] [CrossRef]
- Ploug, M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist. Theranostics 2013, 3, 467–476. [Google Scholar] [CrossRef]
- Skovgaard, D.; Persson, M.; Kjaer, A. PET imaging of urokinase-type plasminogen activator receptor (uPAR) in prostate cancer: Current status and future perspectives. Clin. Transl. Imaging 2016, 4, 457–465. [Google Scholar] [CrossRef]
- Persson, M.; Kjaer, A. Urokinase-type plasminogen activator receptor (uPAR) as a promising new imaging target: Potential clinical applications. Clin. Physiol. Funct Imaging 2013, 33, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Leth, J.M.; Ploug, M. Targeting the Urokinase-Type Plasminogen Activator Receptor (uPAR) in Human Diseases with a View to Non-invasive Imaging and Therapeutic Intervention. Front. Cell Dev. Biol. 2021, 9, 732015. [Google Scholar] [CrossRef]
- Fosbøl, M.; Kurbegovic, S.; Johannesen, H.H.; Røder, M.A.; Hansen, A.E.; Mortensen, J.; Loft, A.; Petersen, P.M.; Madsen, J.; Brasso, K.; et al. Urokinase-Type Plasminogen Activator Receptor (uPAR) PET/MRI of Prostate Cancer for Noninvasive Evaluation of Aggressiveness: Comparison with Gleason Score in a Prospective Phase 2 Clinical Trial. J. Nucl. Med. 2021, 62, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Skovgaard, D.; Brandt-Larsen, M.; Christensen, C.; Madsen, J.; Nielsen, C.H.; Thurison, T.; Klausen, T.L.; Holm, S.; Loft, A.; et al. First-in-human uPAR PET: Imaging of Cancer Aggressiveness. Theranostics 2015, 5, 1303–1316. [Google Scholar] [CrossRef]
- Roldan, A.L.; Cubellis, M.V.; Masucci, M.T.; Behrendt, N.; Lund, L.R.; Danø, K.; Appella, E.; Blasi, F. Cloning and expression of the receptor for human urokinase plasminogen activator, a central molecule in cell surface, plasmin dependent proteolysis. EMBO J. 1990, 9, 467–474. [Google Scholar] [CrossRef]
- Stoppelli, M.P.; Corti, A.; Soffientini, A.; Cassani, G.; Blasi, F.; Assoian, R.K. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 4939–4943. [Google Scholar] [CrossRef]
- Vassalli, J.D.; Baccino, D.; Belin, D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J. Cell Biol. 1985, 100, 86–92. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Gilquin, B.; Le Du, M.H.; Ménèz, A.; Jørgensen, T.J.; Ploug, M. Characterization of the functional epitope on the urokinase receptor. Complete alanine scanning mutagenesis supplemented by chemical cross-linking. J. Biol. Chem. 2006, 281, 19260–19272. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Ploug, M. Mapping of the vitronectin-binding site on the urokinase receptor: Involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 2007, 282, 13561–13572. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Danø, K.; Ploug, M. Mapping part of the functional epitope for ligand binding on the receptor for urokinase-type plasminogen activator by site-directed mutagenesis. J. Biol. Chem. 1999, 274, 37995–38003. [Google Scholar] [CrossRef] [PubMed]
- Magdolen, V.; Rettenberger, P.; Koppitz, M.; Goretzki, L.; Kessler, H.; Weidle, U.H.; König, B.; Graeff, H.; Schmitt, M.; Wilhelm, O. Systematic mutational analysis of the receptor-binding region of the human urokinase-type plasminogen activator. Eur. J. Biochem. 1996, 237, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 2006, 311, 656–659. [Google Scholar] [CrossRef]
- Huai, Q.; Zhou, A.; Lin, L.; Mazar, A.P.; Parry, G.C.; Callahan, J.; Shaw, D.E.; Furie, B.; Furie, B.C.; Huang, M. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 2008, 15, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Llinas, P.; Le Du, M.H.; Gårdsvoll, H.; Danø, K.; Ploug, M.; Gilquin, B.; Stura, E.A.; Ménez, A. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005, 24, 1655–1663. [Google Scholar] [CrossRef]
- Xu, X.; Gårdsvoll, H.; Yuan, C.; Lin, L.; Ploug, M.; Huang, M. Crystal structure of the urokinase receptor in a ligand-free form. J. Mol. Biol. 2012, 416, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Mertens, H.D.; Kjaergaard, M.; Mysling, S.; Gårdsvoll, H.; Jørgensen, T.J.; Svergun, D.I.; Ploug, M. A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 2012, 287, 34304–34315. [Google Scholar] [CrossRef]
- Barinka, C.; Parry, G.; Callahan, J.; Shaw, D.E.; Kuo, A.; Bdeir, K.; Cines, D.B.; Mazar, A.; Lubkowski, J. Structural basis of interaction between urokinase-type plasminogen activator and its receptor. J. Mol. Biol. 2006, 363, 482–495. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Hansen, L.V.; Jacobsen, B.; Gardsvoll, H.; Ploug, M. Structure and ligand interactions of the urokinase receptor (uPAR). Front. Biosci. 2008, 13, 5441–5461. [Google Scholar] [CrossRef]
- Ploug, M. Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr. Pharm. Des. 2003, 9, 1499–1528. [Google Scholar] [CrossRef]
- Ploug, M.; Ellis, V. Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett. 1994, 349, 163–168. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Werner, F.; Søndergaard, L.; Danø, K.; Ploug, M. Characterization of low-glycosylated forms of soluble human urokinase receptor expressed in Drosophila Schneider 2 cells after deletion of glycosylation-sites. Protein Expr. Purif. 2004, 34, 284–295. [Google Scholar] [CrossRef]
- Ploug, M.; Rahbek-Nielsen, H.; Nielsen, P.F.; Roepstorff, P.; Dano, K. Glycosylation profile of a recombinant urokinase-type plasminogen activator receptor expressed in Chinese hamster ovary cells. J. Biol. Chem 1998, 273, 13933–13943. [Google Scholar] [CrossRef] [PubMed]
- Ploug, M.; Rønne, E.; Behrendt, N.; Jensen, A.L.; Blasi, F.; Danø, K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J. Biol. Chem 1991, 266, 1926–1933. [Google Scholar] [CrossRef]
- Leth, J.M.; Leth-Espensen, K.Z.; Kristensen, K.K.; Kumari, A.; Lund Winther, A.M.; Young, S.G.; Ploug, M. Evolution and Medical Significance of LU Domain-Containing Proteins. Int. J. Mol. Sci 2019, 20, 2760. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Gårdsvoll, H.; Huai, Q.; Huang, M.; Ploug, M. Structure-based Engineering of Species Selectivity in the Interaction between Urokinase and Its Receptor: Implication for Preclinical Cancer Therapy. J. Biol. Chem. 2010, 285, 10982–10992. [Google Scholar] [CrossRef] [PubMed]
- Leth, J.M.; Mertens, H.D.T.; Leth-Espensen, K.Z.; Jørgensen, T.J.D.; Ploug, M. Did evolution create a flexible ligand-binding cavity in the urokinase receptor through deletion of a plesiotypic disulfide bond? J. Biol. Chem. 2019, 294, 7403–7418. [Google Scholar] [CrossRef]
- Huang, M.; Mazar, A.P.; Parry, G.; Higazi, A.A.; Kuo, A.; Cines, D.B. Crystallization of soluble urokinase receptor (suPAR) in complex with urokinase amino-terminal fragment (1-143). Acta Cryst. D Biol. Cryst. 2005, 61, 697–700. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Jacobsen, B.; Kriegbaum, M.C.; Behrendt, N.; Engelholm, L.; Østergaard, S.; Ploug, M. Conformational regulation of urokinase receptor function: Impact of receptor oc.ccupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J. Biol. Chem. 2011, 286, 33544–33556. [Google Scholar] [CrossRef] [PubMed]
- Gårdsvoll, H.; Kjaergaard, M.; Jacobsen, B.; Kriegbaum, M.C.; Huang, M.; Ploug, M. Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor. J. Biol. Chem. 2011, 286, 43515–43526. [Google Scholar] [CrossRef] [PubMed]
- Waltz, D.A.; Chapman, H.A. Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy. J. Biol. Chem. 1994, 269, 14746–14750. [Google Scholar] [CrossRef]
- Blasi, F.; Sidenius, N. The urokinase receptor: Focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010, 584, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; D’Alessio, S. Chapter 12-The urokinase plasminogen activator receptor as a target for cancer therapy. In The Cancer Degradome; Edwards, D., Høyer-Hansen, G., Blasi, F., Sloane, B.F., Eds.; Springer: New York, NY, USA, 2008; pp. 223–243. [Google Scholar]
- Montuori, N.; Visconte, V.; Rossi, G.; Ragno, P. Soluble and cleaved forms of the urokinase-receptor: Degradation products or active molecules? Thromb. Haemost. 2005, 93, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Rasch, M.G.; Lund, I.K.; Almasi, C.E.; Hoyer-Hansen, G. Intact and cleaved uPAR forms: Diagnostic and prognostic value in cancer. Front. Biosci. 2008, 13, 6752–6762. [Google Scholar] [CrossRef]
- Madunić, J. The Urokinase Plasminogen Activator System in Human Cancers: An Overview of Its Prognostic and Predictive Role. Thromb. Haemost. 2018, 118, 2020–2036. [Google Scholar] [CrossRef]
- Boonstra, M.C.; Verspaget, H.W.; Ganesh, S.; Kubben, F.J.; Vahrmeijer, A.L.; van de Velde, C.J.; Kuppen, P.J.; Quax, P.H.; Sier, C.F. Clinical applications of the urokinase receptor (uPAR) for cancer patients. Curr. Pharm. Des. 2011, 17, 1890–1910. [Google Scholar] [CrossRef]
- Yuan, C.; Guo, Z.; Yu, S.; Jiang, L.; Huang, M. Development of inhibitors for uPAR: Blocking the interaction of uPAR with its partners. Drug Discov. Today 2021, 26, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F. The urokinase receptor. A cell surface, regulated chemokine. Apmis 1999, 107, 96–101. [Google Scholar] [CrossRef]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef]
- Bifulco, K.; Votta, G.; Ingangi, V.; Di Carluccio, G.; Rea, D.; Losito, S.; Montuori, N.; Ragno, P.; Stoppelli, M.P.; Arra, C.; et al. Urokinase receptor promotes ovarian cancer cell dissemination through its 84–95 sequence. Oncotarget 2014, 5, 4154–4169. [Google Scholar] [CrossRef][Green Version]
- de Paulis, A.; Montuori, N.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Visconte, V.; Rossi, G.; Marone, G.; Ragno, P. Urokinase Induces Basophil Chemotaxis through a Urokinase Receptor Epitope That Is an Endogenous Ligand for Formyl Peptide Receptor-Like 1 and -Like 2. J. Immunol. 2004, 173, 5739–5748. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, K.; Longanesi-Cattani, I.; Gala, M.; Di Carluccio, G.; Masucci, M.T.; Pavone, V.; Lista, L.; Arra, C.; Stoppelli, M.P.; Carriero, M.V. The soluble form of urokinase receptor promotes angiogenesis through its Ser⁸⁸-Arg-Ser-Arg-Tyr⁹2 chemotactic sequence. J. Thromb. Haemost. 2010, 8, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Resnati, M.; Guttinger, M.; Valcamonica, S.; Sidenius, N.; Blasi, F.; Fazioli, F. Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO J. 1996, 15, 1572–1582. [Google Scholar] [CrossRef]
- Fazioli, F.; Resnati, M.; Sidenius, N.; Higashimoto, Y.; Appella, E.; Blasi, F. A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotactic activity. EMBO J. 1997, 16, 7279–7286. [Google Scholar] [CrossRef]
- Ellis, V.; Behrendt, N.; Danø, K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J. Biol. Chem. 1991, 266, 12752–12758. [Google Scholar] [CrossRef]
- Ellis, V.; Scully, M.F.; Kakkar, V.V. Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J. Biol. Chem. 1989, 264, 2185–2188. [Google Scholar] [CrossRef]
- Stephens, R.W.; Pöllänen, J.; Tapiovaara, H.; Leung, K.C.; Sim, P.S.; Salonen, E.M.; Rønne, E.; Behrendt, N.; Danø, K.; Vaheri, A. Activation of pro-urokinase and plasminogen on human sarcoma cells: A proteolytic system with surface-bound reactants. J. Cell Biol. 1989, 108, 1987–1995. [Google Scholar] [CrossRef]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The urokinase receptor interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef]
- Bugge, T.H.; Flick, M.J.; Danton, M.J.; Daugherty, C.C.; Romer, J.; Dano, K.; Carmeliet, P.; Collen, D.; Degen, J.L. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc. Natl. Acad. Sci. USA 1996, 93, 5899–5904. [Google Scholar] [CrossRef]
- Bugge, T.H.; Suh, T.T.; Flick, M.J.; Daugherty, C.C.; Rømer, J.; Solberg, H.; Ellis, V.; Danø, K.; Degen, J.L. The receptor for urokinase-type plasminogen activator is not essential for mouse development or fertility. J. Biol. Chem. 1995, 270, 16886–16894. [Google Scholar] [CrossRef]
- Dewerchin, M.; Nuffelen, A.V.; Wallays, G.; Bouché, A.; Moons, L.; Carmeliet, P.; Mulligan, R.C.; Collen, D. Generation and characterization of urokinase receptor-deficient mice. J. Clin. Investig. 1996, 97, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Bugge, T.H.; Flick, M.J.; Daugherty, C.C.; Degen, J.L. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995, 9, 794–807. [Google Scholar] [CrossRef]
- Carmeliet, P.; Moons, L.; Dewerchin, M.; Rosenberg, S.; Herbert, J.M.; Lupu, F.; Collen, D. Receptor-independent role of urokinase-type plasminogen activator in pericellular plasmin and matrix metalloproteinase proteolysis during vascular wound healing in mice. J. Cell Biol. 1998, 140, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Nichols, A.; Meda, P.; Vassalli, J.D. Urokinase-type plasminogen activator and its receptor synergize to promote pathogenic proteolysis. EMBO J. 2000, 19, 4817–4826. [Google Scholar] [CrossRef]
- Bolon, I.; Zhou, H.-M.; Charron, Y.; Wohlwend, A.; Vassalli, J.-D. Plasminogen mediates the pathological effects of urokinase-type plasminogen activator overexpression. Am. J. Pathol. 2004, 164, 2299–2304. [Google Scholar] [CrossRef]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef]
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of Tumor Cells by Cell Surface Urokinase Plasminogen Activator-dependent Anthrax Toxin. J. Biol. Chem. 2001, 276, 17976–17984. [Google Scholar] [CrossRef]
- Wang, Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Med. Res. Rev. 2001, 21, 146–170. [Google Scholar] [CrossRef]
- Allgayer, H. Translational research on u-PAR. Eur. J. Cancer 2010, 46, 1241–1251. [Google Scholar] [CrossRef]
- Almholt, K.; Lærum, O.D.; Nielsen, B.S.; Lund, I.K.; Lund, L.R.; Rømer, J.; Jögi, A. Spontaneous lung and lymph node metastasis in transgenic breast cancer is independent of the urokinase receptor uPAR. Clin. Exp. Metastasis 2015, 32, 543–554. [Google Scholar] [CrossRef]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.D.; Jo, M.; Montel, V.; Takimoto, S.; Gonias, S.L. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J. Cell Biol. 2007, 178, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Gonias, S.L.; Hu, J. Urokinase receptor and resistance to targeted anticancer agents. Front. Pharm. 2015, 6, 154. [Google Scholar] [CrossRef]
- Jo, M.; Lester, R.D.; Montel, V.; Eastman, B.; Takimoto, S.; Gonias, S.L. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J. Biol. Chem. 2009, 284, 22825–22833. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef]
- Yu, W.; Kim, J.; Ossowski, L. Reduction in surface urokinase receptor forces malignant cells into a protracted state of dormancy. J. Cell Biol. 1997, 137, 767–777. [Google Scholar] [CrossRef]
- Jo, M.; Eastman, B.M.; Webb, D.L.; Stoletov, K.; Klemke, R.; Gonias, S.L. Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res. 2010, 70, 8948–8958. [Google Scholar] [CrossRef]
- Ragno, P. The urokinase receptor: A ligand or a receptor? Story of a sociable molecule. Cell Mol. Life Sci. 2006, 63, 1028–1037. [Google Scholar] [CrossRef]
- Wei, Y.; Waltz, D.A.; Rao, N.; Drummond, R.J.; Rosenberg, S.; Chapman, H.A. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J. Biol. Chem. 1994, 269, 32380–32388. [Google Scholar] [CrossRef]
- Ferraris, G.M.S.; Schulte, C.; Buttiglione, V.; De Lorenzi, V.; Piontini, A.; Galluzzi, M.; Podestà, A.; Madsen, C.D.; Sidenius, N. The interaction between uPAR and vitronectin triggers ligand-independent adhesion signalling by integrins. EMBO J. 2014, 33, 2458–2472. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.D.; Ferraris, G.M.; Andolfo, A.; Cunningham, O.; Sidenius, N. uPAR-induced cell adhesion and migration: Vitronectin provides the key. J. Cell Biol. 2007, 177, 927–939. [Google Scholar] [CrossRef]
- Madsen, C.D.; Sidenius, N. The interaction between urokinase receptor and vitronectin in cell adhesion and signalling. Eur. J. Cell Biol. 2008, 87, 617–629. [Google Scholar] [CrossRef]
- Hillig, T.; Engelholm, L.H.; Ingvarsen, S.; Madsen, D.H.; Gårdsvoll, H.; Larsen, J.K.; Ploug, M.; Danø, K.; Kjøller, L.; Behrendt, N. A composite role of vitronectin and urokinase in the modulation of cell morphology upon expression of the urokinase receptor. J. Biol. Chem. 2008, 283, 15217–15223. [Google Scholar] [CrossRef]
- Kjøller, L.; Hall, A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J. Cell Biol. 2001, 152, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Salasznyk, R.M.; Zappala, M.; Zheng, M.; Yu, L.; Wilkins-Port, C.; McKeown-Longo, P.J. The uPA receptor and the somatomedin B region of vitronectin direct the localization of uPA to focal adhesions in microvessel endothelial cells. Matrix Biol. 2007, 26, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Wei, Y.; Chapman, H.A. Urokinase receptor and integrin interactions. Curr. Pharm. Des. 2003, 9, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Hong, S.; Huang, S. Role of urokinase receptor in tumor progression and development. Theranostics 2013, 3, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Degryse, B.; Resnati, M.; Czekay, R.P.; Loskutoff, D.J.; Blasi, F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: Generation of a new integrin inhibitor. J. Biol. Chem. 2005, 280, 24792–24803. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, P.; Aguirre-Ghiso, J.A.; Liang, O.D.; Gardsvoll, H.; Ploug, M.; Ossowski, L. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J. Biol. Chem. 2006, 281, 14852–14863. [Google Scholar] [CrossRef]
- Tang, C.H.; Hill, M.L.; Brumwell, A.N.; Chapman, H.A.; Wei, Y. Signaling through urokinase and urokinase receptor in lung cancer cells requires interactions with beta1 integrins. J. Cell Sci. 2008, 121, 3747–3756. [Google Scholar] [CrossRef]
- Ossowski, L.; Aguirre-Ghiso, J.A. Urokinase receptor and integrin partnership: Coordination of signaling for cell adhesion, migration and growth. Curr. Opin. Cell Biol. 2000, 12, 613–620. [Google Scholar] [CrossRef]
- Solberg, H.; Ploug, M.; Høyer-Hansen, G.; Nielsen, B.S.; Lund, L.R. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J. Histochem. Cytochem. 2001, 49, 237–246. [Google Scholar] [CrossRef]
- Jacobsen, B.; Ploug, M. The urokinase receptor and its structural homologue C4.4A in human cancer: Expression, prognosis and pharmacological inhibition. Curr. Med. Chem. 2008, 15, 2559–2573. [Google Scholar] [CrossRef]
- Danø, K.; Behrendt, N.; Brunner, N.; Ellis, V.; Ploug, M.; Pyke, C. The urokinase receptor. Protein structure and role in plasminogen activation and cancer invasion. Fibrinolysis 1994, 8, 189–203. [Google Scholar] [CrossRef]
- Tjwa, M.; Sidenius, N.; Moura, R.; Jansen, S.; Theunissen, K.; Andolfo, A.; De Mol, M.; Dewerchin, M.; Moons, L.; Blasi, F.; et al. Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. J. Clin. Investig. 2009, 119, 1008–1018. [Google Scholar] [CrossRef]
- Rømer, J.; Lund, L.R.; Eriksen, J.; Pyke, C.; Kristensen, P.; Danø, K. The receptor for urokinase-type plasminogen activator is expressed by keratinocytes at the leading edge during re-epithelialization of mouse skin wounds. J. Investig. Dermatol. 1994, 102, 519–522. [Google Scholar] [CrossRef]
- Multhaupt, H.A.; Mazar, A.; Cines, D.B.; Warhol, M.J.; McCrae, K.R. Expression of urokinase receptors by human trophoblast. A histochemical and ultrastructural analysis. Lab. Investig. 1994, 71, 392–400. [Google Scholar] [PubMed]
- Baker, S.K.; Strickland, S. A critical role for plasminogen in inflammation. J. Exp. Med. 2020, 217, e20191865. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, M.; Fibbi, G.; Pucci, M.; Margheri, F.; Serrati, S. The plasminogen activation system in inflammation. Front. Biosci. 2008, 13, 4667–4686. [Google Scholar] [CrossRef]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Bulle, A.; Lim, K.-H. Beyond just a tight fortress: Contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 249. [Google Scholar] [CrossRef]
- de Geus, S.W.; Baart, V.M.; Boonstra, M.C.; Kuppen, P.J.; Prevoo, H.A.; Mazar, A.P.; Bonsing, B.A.; Morreau, H.; van de Velde, C.J.; Vahrmeijer, A.L.; et al. Prognostic Impact of Urokinase Plasminogen Activator Receptor Expression in Pancreatic Cancer: Malignant Versus Stromal Cells. Biomark. Insights 2017, 12, 15443. [Google Scholar] [CrossRef]
- Cantero, D.; Friess, H.; Deflorin, J.; Zimmermann, A.; Bründler, M.A.; Riesle, E.; Korc, M.; Büchler, M.W. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br. J. Cancer 1997, 75, 388–395. [Google Scholar] [CrossRef]
- Shiomi, H.; Eguchi, Y.; Tani, T.; Kodama, M.; Hattori, T. Cellular distribution and clinical value of urokinase-type plasminogen activator, its receptor, and plasminogen activator inhibitor-2 in esophageal squamous cell carcinoma. Am. J. Pathol. 2000, 156, 567–575. [Google Scholar] [CrossRef]
- Yamashita, K.; Tanaka, Y.; Mimori, K.; Inoue, H.; Mori, M. Differential expression of MMP and uPA systems and prognostic relevance of their expression in esophageal squamous cell carcinoma. Int. J. Cancer 2004, 110, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sawaya, R.; Mohanam, S.; Rao, V.H.; Bruner, J.M.; Nicolson, G.L.; Rao, J.S. Expression and localization of urokinase-type plasminogen activator receptor in human gliomas. Cancer Res. 1994, 54, 5016–5020. [Google Scholar]
- Salajegheh, M.; Rudnicki, A.; Smith, T.W. Expression of urokinase-type plasminogen activator receptor (uPAR) in primary central nervous system neoplasms. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 184–189. [Google Scholar] [CrossRef]
- O’Halloran, T.V.; Ahn, R.; Hankins, P.; Swindell, E.; Mazar, A.P. The many spaces of uPAR: Delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics 2013, 3, 496–506. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rømer, J.; Pyke, C.; Lund, L.R.; Danø, K.; Ralfkiær, E. Cancer Cell Expression of Urokinase-Type Plasminogen Activator Receptor mRNA in Squamous Cell Carcinomas of th.he Skin. J. Investig. Dermatol. 2001, 116, 353–358. [Google Scholar] [CrossRef]
- Danø, K.; Rømer, J.; Nielsen, B.S.; Bjørn, S.; Pyke, C.; Rygaard, J.; Lund, L.R. Cancer invasion and tissue remodeling--cooperation of protease systems and cell types. Apmis 1999, 107, 120–127. [Google Scholar] [CrossRef]
- Christensen, L.; Wiborg Simonsen, A.C.; Heegaard, C.W.; Moestrup, S.K.; Andersen, J.A.; Andreasen, P.A. Immunohistochemical localization of urokinase-type plasminogen activator, type-1 plasminogen-activator inhibitor, urokinase receptor and alpha (2)-macroglobulin receptor in human breast carcinomas. Int. J. Cancer 1996, 66, 441–452. [Google Scholar] [CrossRef]
- Illemann, M.; Bird, N.; Majeed, A.; Lærum, O.D.; Lund, L.R.; Danø, K.; Nielsen, B.S. Two distinct expression patterns of urokinase, urokinase receptor and plasminogen activator inhibitor-1 in colon cancer liver metastases. Int. J. Cancer 2009, 124, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Illemann, M.; Laerum, O.D.; Hasselby, J.P.; Thurison, T.; Høyer-Hansen, G.; Nielsen, H.J.; Christensen, I.J. Urokinase-type plasminogen activator receptor (uPAR) on tumor-associated macrophages is a marker of poor prognosis in colorectal cancer. Cancer Med. 2014, 3, 855–864. [Google Scholar] [CrossRef]
- Alpízar-Alpízar, W.; Christensen, I.J.; Santoni-Rugiu, E.; Skarstein, A.; Ovrebo, K.; Illemann, M.; Laerum, O.D. Urokinase plasminogen activator receptor on invasive cancer cells: A prognostic factor in distal gastric adenocarcinoma. Int. J. Cancer 2012, 131, E329–E336. [Google Scholar] [CrossRef] [PubMed]
- Laerum, O.D.; Ovrebo, K.; Skarstein, A.; Christensen, I.J.; Alpízar-Alpízar, W.; Helgeland, L.; Danø, K.; Nielsen, B.S.; Illemann, M. Prognosis in adenocarcinomas of lower oesophagus, gastro-oesophageal junction and cardia evaluated by uPAR-immunohistochemistry. Int. J. Cancer 2012, 131, 558–569. [Google Scholar] [CrossRef]
- Akahane, T.; Ishii, M.; Ohtani, H.; Nagura, H.; Toyota, T. Stromal expression of urokinase-type plasminogen activator receptor (uPAR) is associated with invasive growth in primary liver cancer. Liver 1998, 18, 414–419. [Google Scholar] [CrossRef]
- Dubuisson, L.; Monvoisin, A.; Nielsen, B.S.; Le Bail, B.; Bioulac-Sage, P.; Rosenbaum, J. Expression and cellular localization of the urokinase-type plasminogen activator and its receptor in human hepatocellular carcinoma. J. Pathol. 2000, 190, 190–195. [Google Scholar] [CrossRef]
- Giannopoulou, I.; Mylona, E.; Kapranou, A.; Mavrommatis, J.; Markaki, S.; Zoumbouli, C.; Keramopoulos, A.; Nakopoulou, L. The prognostic value of the topographic distribution of uPAR expression in invasive breast carcinomas. Cancer Lett. 2007, 246, 262–267. [Google Scholar] [CrossRef]
- Hildenbrand, R.; Glienke, W.; Magdolen, V.; Graeff, H.; Stutte, H.J.; Schmitt, M. Urokinase receptor localization in breast cancer and benign lesions assessed by in situ hybridization and immunohistochemistry. Histochem. Cell Biol. 1998, 110, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Pyke, C.; Graem, N.; Ralfkiaer, E.; Rønne, E.; Høyer-Hansen, G.; Brünner, N.; Danø, K. Receptor for urokinase is present in tumor-associated macrophages in ductal breast carcinoma. Cancer Res. 1993, 53, 1911–1915. [Google Scholar] [PubMed]
- Usher, P.A.; Thomsen, O.F.; Iversen, P.; Johnsen, M.; Brünner, N.; Høyer-Hansen, G.; Andreasen, P.; Danø, K.; Nielsen, B.S. Expression of urokinase plasminogen activator, its receptor and type-1 inhibitor in malignant and benign prostate tissue. Int. J. Cancer 2005, 113, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Dohn, L.H.; Illemann, M.; Høyer-Hansen, G.; Christensen, I.J.; Hostmark, J.; Litlekalsoy, J.; von der Maase, H.; Pappot, H.; Laerum, O.D. Urokinase-type plasminogen activator receptor (uPAR) expression is associated with T-stage and survival in urothelial carcinoma of the bladder. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 165.e115–165.e124. [Google Scholar] [CrossRef]
- Dohn, L.H.; Pappot, H.; Iversen, B.R.; Illemann, M.; Høyer-Hansen, G.; Christensen, I.J.; Thind, P.; Salling, L.; von der Maase, H.; Laerum, O.D. uPAR Expression Pattern in Patients with Urothelial Carcinoma of the Bladder--Possible Clinical Implications. PLoS ONE 2015, 10, e0135824. [Google Scholar] [CrossRef]
- Serpa, M.S.; Mafra, R.P.; Queiroz, S.; Silva, L.P.D.; Souza, L.B.; Pinto, L.P. Expression of urokinase-type plasminogen activator and its receptor in squamous cell carcinoma of the oral tongue. Braz. Oral Res. 2018, 32, e93. [Google Scholar] [CrossRef]
- Lindberg, P.; Larsson, A.; Nielsen, B.S. Expression of plasminogen activator inhibitor-1, urokinase receptor and laminin gamma-2 chain is an early coordinated event in incipient oral squamous cell carcinoma. Int. J. Cancer 2006, 118, 2948–2956. [Google Scholar] [CrossRef]
- Nozaki, S.; Endo, Y.; Kawashiri, S.; Nakagawa, K.; Yamamoto, E.; Yonemura, Y.; Sasaki, T. Immunohistochemical localization of a urokinase-type plasminogen activator system in squamous cell carcinoma of the oral cavity: Association with mode of invasion and lymph node metastasis. Oral Oncol. 1998, 34, 58–62. [Google Scholar] [CrossRef]
- Al-Hassan, N.N.; Behzadian, A.; Caldwell, R.; Ivanova, V.S.; Syed, V.; Motamed, K.; Said, N.A. Differential Roles of uPAR in Peritoneal Ovarian Carcinomatosis. Neoplasia 2012, 14, 259-IN252. [Google Scholar] [CrossRef]
- He, Y.; Liu, X.D.; Chen, Z.Y.; Zhu, J.; Xiong, Y.; Li, K.; Dong, J.H.; Li, X. Interaction between cancer cells and stromal fibroblasts is required for activation of the uPAR-uPA-MMP-2 cascade in pancreatic cancer metastasis. Clin. Cancer Res. 2007, 13, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.G.; Holloway, R.W.; Miller, V.A.; Waisman, D.M. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838. [Google Scholar] [CrossRef]
- Hu, J.; Jo, M.; Eastman, B.M.; Gilder, A.S.; Bui, J.D.; Gonias, S.L. uPAR induces expression of transforming growth factor β and interleukin-4 in cancer cells to promote tumor-permissive conditioning of macrophages. Am. J. Pathol. 2014, 184, 3384–3393. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, Y.; Medcalf, R.L.; Muñoz-Cánoves, P. Transcriptional and posttranscriptional regulation of the plasminogen activator system. Thromb. Haemost. 2005, 93, 661–675. [Google Scholar] [CrossRef]
- Pierga, J.Y.; Bonneton, C.; Magdelénat, H.; Vincent-Salomon, A.; Nos, C.; Boudou, E.; Pouillart, P.; Thiery, J.P.; de Cremoux, P. Real-time quantitative PCR determination of urokinase-type plasminogen activator receptor (uPAR) expression of isolated micrometastatic cells from bone marrow of breast cancer patients. Int. J. Cancer 2005, 114, 291–298. [Google Scholar] [CrossRef]
- Heiss, M.M.; Simon, E.H.; Beyer, B.C.; Gruetzner, K.U.; Tarabichi, A.; Babic, R.; Schildberg, F.W.; Allgayer, H. Minimal residual disease in gastric cancer: Evidence of an independent prognostic relevance of urokinase receptor expression by disseminated tumor cells in the bone marrow. J. Clin. Oncol. 2002, 20, 2005–2016. [Google Scholar] [CrossRef]
- Mimori, K.; Kataoka, A.; Yamaguchi, H.; Masuda, N.; Kosaka, Y.; Ishii, H.; Ohno, S.; Mori, M. Preoperative u-PAR Gene Expression in Bone Marrow Indicates the Potential Power of Recurrence in Breast Cancer Cases. Ann. Surg. Oncol. 2009, 16, 2035. [Google Scholar] [CrossRef]
- Allgayer, H.; Aguirre-Ghiso, J.A. The urokinase receptor (u-PAR)—A link between tumor cell dormancy and minimal residual disease in bone marrow? APMIS Acta Pathol. Microbiol. Et Immunol. Scand. 2008, 116, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Indira Chandran, V.; Eppenberger-Castori, S.; Venkatesh, T.; Vine, K.L.; Ranson, M. HER2 and uPAR cooperativity contribute to metastatic phenotype of HER2-positive breast cancer. Oncoscience 2015, 2, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Bourgeois, M.; Bailly, C.; Frindel, M.; Guerard, F.; Chérel, M.; Faivre-Chauvet, A.; Kraeber-Bodéré, F.; Bodet-Milin, C. Radioimmunoconjugates for treating cancer: Recent advances and current opportunities. Expert Opin Biol. 2017, 17, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Strosberg, J. Radionuclide Therapy for Neuroendocrine Tumors. Curr. Oncol. Rep. 2017, 19, 9. [Google Scholar] [CrossRef]
- Li, D.; Liu, S.; Shan, H.; Conti, P.; Li, Z. Urokinase plasmi.inogen activator receptor (uPAR) targeted nuclear imaging and radionuclide therapy. Theranostics 2013, 3, 507–515. [Google Scholar] [CrossRef][Green Version]
- Ploug, M.; Østergaard, S.; Gårdsvoll, H.; Kovalski, K.; Holst-Hansen, C.; Holm, A.; Ossowski, L.; Danø, K. Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 2001, 40, 12157–12168. [Google Scholar] [CrossRef]
- Knör, S.; Sato, S.; Huber, T.; Morgenstern, A.; Bruchertseifer, F.; Schmitt, M.; Kessler, H.; Senekowitsch-Schmidtke, R.; Magdolen, V.; Seidl, C. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 53–64. [Google Scholar] [CrossRef]
- Luo, Y.; Pan, Q.; Li, F. Decreased 68Ga-NOTA-exendin-4 renal uptake in patients pretreated with Gelofusine infusion: A randomized controlled study. J. Pancreatol. 2020, 3, 1968–1976. [Google Scholar] [CrossRef]
- Melis, M.; Bijster, M.; de Visser, M.; Konijnenberg, M.W.; de Swart, J.; Rolleman, E.J.; Boerman, O.C.; Krenning, E.P.; de Jong, M. Dose-response effect of Gelofusine on renal uptake and retention of radiolabelled octreotate in rats with CA20948 tumours. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1968–1976. [Google Scholar] [CrossRef]
- Persson, M.; Juhl, K.; Rasmussen, P.; Brandt-Larsen, M.; Madsen, J.; Ploug, M.; Kjaer, A. uPAR targeted radionuclide therapy with (177) Lu-DOTA-AE105 inhibits dissemination of metastatic prostate cancer. Mol. Pharm. 2014, 11, 2796–2806. [Google Scholar] [CrossRef] [PubMed]
- Duriseti, S.; Goetz, D.H.; Hostetter, D.R.; LeBeau, A.M.; Wei, Y.; Craik, C.S. Antagonistic anti-urokinase plasminogen activator receptor (uPAR) antibodies significantly inhibit uPAR-mediated cellular signaling and migration. J. Biol. Chem. 2010, 285, 26878–26888. [Google Scholar] [CrossRef] [PubMed]
- LeBeau, A.M.; Duriseti, S.; Murphy, S.T.; Pepin, F.; Hann, B.; Gray, J.W.; VanBrocklin, H.F.; Craik, C.S. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013, 73, 2070–2081. [Google Scholar] [CrossRef]
- Antignani, A.; Ho, E.C.H.; Bilotta, M.T.; Qiu, R.; Sarnvosky, R.; FitzGerald, D.J. Targeting Receptors on Cancer Cells with Protein Toxins. Biomolecules 2020, 10, 1331. [Google Scholar] [CrossRef]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef]
- Johannes, L.; Decaudin, D. Protein toxins: Intracellular trafficking for targeted therapy. Gene Ther. 2005, 12, 1360–1368. [Google Scholar] [CrossRef]
- Li, M.; Liu, Z.-S.; Liu, X.-L.; Hui, Q.; Lu, S.-Y.; Qu, L.-L.; Li, Y.-S.; Zhou, Y.; Ren, H.-L.; Hu, P. Clinical targeting recombinant immunotoxins for cancer therapy. Onco. Targets Ther. 2017, 10, 3645–3665. [Google Scholar] [CrossRef]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Chandramohan, V.; Sampson, J.H.; Pastan, I.H.; Bigner, D.D. Chapter 10-Immunotoxin Therapy for Brain Tumors. In Translational Immunotherapy of Brain Tumors; Sampson, J.H., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 227–260. [Google Scholar]
- Wang, Z.; Zheng, Q.; Zhang, H.; Bronson, R.T.; Madsen, J.C.; Sachs, D.H.; Huang, C.A.; Wang, Z. Ontak-like human IL-2 fusion toxin. J. Immunol. Methods 2017, 448, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Appella, E.; Robinson, E.A.; Ullrich, S.J.; Stoppelli, M.P.; Corti, A.; Cassani, G.; Blasi, F. The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J. Biol. Chem. 1987, 262, 4437–4440. [Google Scholar] [CrossRef]
- de Virgilio, M.; Silvestris, F. Urokinase receptor (uPAR) ligand based recombinant toxins for human cancer therapy. Curr. Pharm. Des. 2011, 17, 1979–1983. [Google Scholar] [CrossRef] [PubMed]
- Rustamzadeh, E.; Low, W.C.; Vallera, D.A.; Hall, W.A. Immunotoxin therapy for CNS tumor. J. Neurooncol. 2003, 64, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Li, C.; Jin, N.; Panoskaltsis-Mortari, A.; Hall, W.A. Targeting urokinase-type plasminogen activator receptor on human glioblastoma tumors with diphtheria toxin fusion protein DTAT. J. Natl. Cancer Inst. 2002, 94, 597–606. [Google Scholar] [CrossRef]
- Fan, F.; Schimming, A.; Jaeger, D.; Podar, K. Targeting th.he tumor microenvironment: Focus on angiogenesis. J. Oncol. 2012, 2012, 281261. [Google Scholar] [CrossRef]
- Todhunter, D.A.; Hall, W.A.; Rustamzadeh, E.; Shu, Y.; Doumbia, S.O.; Vallera, D.A. A bispecific immunotoxin (DTAT13) targeting human IL-13 receptor (IL-13R) and urokinase-type plasminogen activator receptor (uPAR) in a mouse xenograft model. Protein Eng. Des. Sel. 2004, 17, 157–164. [Google Scholar] [CrossRef]
- Hall, W.A.; Vallera, D.A. Efficacy of antiangiogenic targeted toxins against glioblastoma multiforme. Neurosurg. Focus 2006, 20, E23. [Google Scholar] [CrossRef]
- Rustamzadeh, E.; Vallera, D.A.; Todhunter, D.A.; Low, W.C.; Panoskaltsis-Mortari, A.; Hall, W.A. Immunotoxin pharmacokinetics: A comparison of the anti-glioblastoma bi-specific fusion protein (DTAT13) to DTAT and DTIL13. J. Neurooncol. 2006, 77, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Rustamzadeh, E.; Hall, W.A.; Todhunter, D.A.; Vallera, V.D.; Low, W.C.; Liu, H.; Panoskaltsis-Mortari, A.; Vallera, D.A. Intracranial therapy of glioblastoma with the fusion protein DTAT in immunodeficient mice. Int. J. Cancer 2007, 120, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: A side effect of immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef]
- Mehta, A.M.; Sonabend, A.M.; Bruce, J.N. Convection-Enhanced Delivery. Neurotherapeutics 2017, 14, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.M.; Massague, J.; Sicheneder, A.; Vallera, D.A.; Hall, W.A. Intracerebral infusion of the bispecific targeted toxin DTATEGF in a mouse xenograft model of a human metastatic non-small cell lung cancer. J. Neurooncol. 2012, 109, 229–238. [Google Scholar] [CrossRef]
- Mazor, R.; King, E.M.; Pastan, I. Strategies to Reduce the Immunogenicity of Recombinant Immunotoxins. Am. J. Pathol. 2018, 188, 1736–1743. [Google Scholar] [CrossRef]
- Griswold, K.E.; Bailey-Kellogg, C. Design and engineering of deimmunized biotherapeutics. Curr. Opin. Struct. Biol. 2016, 39, 79–88. [Google Scholar] [CrossRef]
- Oh, F.; Modiano, J.F.; Bachanova, V.; Vallera, D.A. Bispecific Targeting of EGFR and Urokinase Receptor (uPAR) Using Ligand-Targeted Toxins in Solid Tumors. Biomolecules 2020, 10, 956. [Google Scholar] [CrossRef]
- Tsai, A.K.; Oh, S.; Chen, H.; Shu, Y.; Ohlfest, J.R.; Vallera, D.A. A novel bispecific ligand-directed toxin designed to simultaneously target EGFR on human glioblastoma cells and uPAR on tumor neovasculature. J. Neurooncol. 2011, 103, 255–266. [Google Scholar] [CrossRef]
- Oh, S.; Tsai, A.K.; Ohlfest, J.R.; Panoskaltsis-Mortari, A.; Vallera, D.A. Evaluation of a bispecific biological drug designed to simultaneously target glioblastoma and its neovasculature in the brain. J. Neurosurg. 2011, 114, 1662–1671. [Google Scholar] [CrossRef]
- Onda, M.; Nagata, S.; FitzGerald, D.J.; Beers, R.; Fisher, R.J.; Vincent, J.J.; Lee, B.; Nakamura, M.; Hwang, J.; Kreitman, R.J.; et al. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J. Immunol. 2006, 177, 8822–8834. [Google Scholar] [CrossRef]
- Nagata, S.; Pastan, I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 977–985. [Google Scholar] [CrossRef]
- Waldron, N.N.; Oh, S.; Vallera, D.A. Bispecific targeting of EGFR and uPAR in a mouse model of head and neck squamous cell carcinoma. Oral Oncol. 2012, 48, 1202–1207. [Google Scholar] [CrossRef]
- Pilbeam, K.; Wang, H.; Taras, E.; Bergerson, R.J.; Ettestad, B.; DeFor, T.; Borgatti, A.; Vallera, D.A.; Verneris, M.R. Targeting pediatric sarcoma with a bispecific ligand immunotoxin targeting urokinase and epidermal growth factor receptors. Oncotarget 2017, 9, 11938–11947. [Google Scholar] [CrossRef]
- Oh, F.; Todhunter, D.; Taras, E.; Vallera, D.A.; Borgatti, A. Targeting EGFR and uPAR on human rhabdomyosarcoma, osteosarcoma, and ovarian adenocarcinoma with a bispecific ligand-directed toxin. Clin. Pharmacol. 2018, 10, 113–121. [Google Scholar] [CrossRef]
- Harandi, A.; Zaidi, A.S.; Stocker, A.M.; Laber, D.A. Clinical Efficacy and Toxicity of Anti-EGFR Therapy in Common Cancers. J. Oncol. 2009, 2009, 567486. [Google Scholar] [CrossRef]
- Borgatti, A.; Koopmeiners, J.S.; Sarver, A.L.; Winter, A.L.; Stuebner, K.; Todhunter, D.; Rizzardi, A.E.; Henriksen, J.C.; Schmechel, S.; Forster, C.L.; et al. Safe and Effective Sarcoma Therapy through Bispecific Targeting of EGFR and uPAR. Mol. Cancer Ther. 2017, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; del Vecchio, A.; Lappi, D.A.; Soria, M.R. A conjugate between human urokinase and saporin, a type-1 ribosome-inactivating protein, is selectively cytotoxic to urokinase receptor-expressing cells. J. Biol. Chem. 1993, 268, 23186–23190. [Google Scholar] [CrossRef]
- Polito, L.; Bortolotti, M.; Mercatelli, D.; Battelli, M.G.; Bolognesi, A. Saporin-S6: A useful tool in cancer therapy. Toxins 2013, 5, 1698–1722. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Nykjaer, A.; Nielsen, M.; Soria, M.R. Alpha 2-macroglobulin receptor mediates binding and cytotoxicity of plant ribosome-inactivating proteins. Eur. J. Biochem. 1995, 232, 165–171. [Google Scholar] [CrossRef]
- Fabbrini, M.S.; Carpani, D.; Bello-Rivero, I.; Soria, M.R. The amino-terminal fragment of human urokinase directs a recombinant chimeric toxin to target cells: Internalization is toxin mediated. FASEB J. 1997, 11, 1169–1176. [Google Scholar] [CrossRef]
- Ippoliti, R.; Lendaro, E.; Benedetti, P.A.; Torrisi, M.R.; Belleudi, F.; Carpani, D.; Soria, M.R.; Fabbrini, M.S. Endocytosis of a chimera between human pro-urokinase and the plant toxin saporin: An unusual internalization mechanism. FASEB J. 2000, 14, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Bursomanno, S.; Lopardo, T.; Traini, R.; Colombatti, M.; Ippoliti, R.; Flavell, D.J.; Flavell, S.U.; Ceriotti, A.; Fabbrini, M.S. Pichia pastoris as a host for secretion of toxic saporin chimeras. FASEB J. 2010, 24, 253–265. [Google Scholar] [CrossRef]
- Errico Provenzano, A.; Posteri, R.; Giansanti, F.; Angelucci, F.; Flavell, S.U.; Flavell, D.J.; Fabbrini, M.S.; Porro, D.; Ippoliti, R.; Ceriotti, A.; et al. Optimization of construct design and fermentation strategy for the production of bioactive ATF-SAP, a saporin based anti-tumoral uPAR-targeted chimera. Microb. Cell Fact. 2016, 15, 194. [Google Scholar] [CrossRef] [PubMed]
- Zuppone, S.; Assalini, C.; Minici, C.; Bertagnoli, S.; Branduardi, P.; Degano, M.; Fabbrini, M.S.; Montorsi, F.; Salonia, A.; Vago, R. The anti-tumoral potential of the saporin-based uPAR-targeting chimera ATF-SAP. Sci. Rep. 2020, 10, 2521. [Google Scholar] [CrossRef]
- Atkinson, J.M.; Siller, C.S.; Gill, J.H. Tumour endoproteases: The cutting edge of cancer drug delivery? Br. J. Pharmacol. 2008, 153, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Yizhen LIU, S.-Y.L. Urokinase-targeted recombinant bacterial protein toxins–A rationally designed and engineered anticancer agent for cancer therapy. Front. Biol. 2009, 4, 1–6. [Google Scholar] [CrossRef]
- Arora, N.; Leppla, S.H. Residues 1-254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J. Biol. Chem. 1993, 268, 3334–3341. [Google Scholar] [CrossRef]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef]
- Zdanovsky, A.G.; Chiron, M.; Pastan, I.; FitzGerald, D.J. Mechanism of action of Pseudomonas exotoxin. Identification of a rate-limiting step. J. Biol. Chem. 1993, 268, 21791–21799. [Google Scholar] [CrossRef]
- Abi-Habib, R.J.; Singh, R.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Frankel, A.E. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol. Cancer Ther. 2006, 5, 2556–2562. [Google Scholar] [CrossRef]
- Su, Y.; Ortiz, J.; Liu, S.; Bugge, T.H.; Singh, R.; Leppla, S.H.; Frankel, A.E. Systematic urokinase-activated anthrax toxin therapy produces regressions of subcutaneous human non-small cell lung tumor in athymic nude mice. Cancer Res. 2007, 67, 3329–3336. [Google Scholar] [CrossRef]
- Rønø, B.; Rømer, J.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Kristjansen, P.E. Antitumor efficacy of a urokinase activation-dependent anthrax toxin. Mol. Cancer Ther. 2006, 5, 89–96. [Google Scholar] [CrossRef]
- Liu, S.; Redeye, V.; Kuremsky, J.G.; Kuhnen, M.; Molinolo, A.; Bugge, T.H.; Leppla, S.H. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol 2005, 23, 725–730. [Google Scholar] [CrossRef]
- Schafer, J.M.; Peters, D.E.; Morley, T.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP-activated engineered anthrax toxin. PLoS ONE 2011, 6, e20532. [Google Scholar] [CrossRef]
- Nishiya, A.T.; Nagamine, M.K.; Fonseca, I.I.; Miraldo, A.C.; Villar Scattone, N.; Guerra, J.L.; Xavier, J.G.; Santos, M.; Massoco de Salles Gomes, C.O.; Ward, J.M.; et al. Inhibitory Effects of a Reengineered Anthrax Toxin on Canine Oral Mucosal Melanomas. Toxins 2020, 12, 157. [Google Scholar] [CrossRef]
- Abi-Habib, R.J.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Frankel, A.E. A urokinase-activated recombinant diphtheria toxin targeting the granulocyte-macrophage colony-stimulating factor receptor is selectively cytotoxic to human acute myeloid leukemia blasts. Blood 2004, 104, 2143–2148. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Powell, B.L.; Hall, P.D.; Case, L.D.; Kreitman, R.J. Phase I trial of a novel diphtheria toxin/granulocyte macrophage colony-stimulating factor fusion protein (DT388GMCSF) for refractory or relapsed acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 1004–1013. [Google Scholar]
- Jardí, M.; Inglés-Esteve, J.; Burgal, M.; Azqueta, C.; Velasco, F.; López-Pedrera, C.; Miles, L.A.; Félez, J. Distinct patterns of urokinase receptor (uPAR) expression by leukemic cells and peripheral blood cells. Thromb. Haemost. 1996, 76, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; O’Donovan, N.; Crown, J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat. Rev. 2011, 37, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Sunchen, S.; Liu, M.; Shen, C.; Wu, J.; Zhao, W.; Yu, B.; Liu, J. A novel tumor-activated ALA fusion protein for specific inhibition on the growth and invasion of breast cancer cells MDA-MB-231. Drug Deliv. 2017, 24, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 94. [Google Scholar] [CrossRef]
- Harel, E.T.; Drake, P.M.; Barfield, R.M.; Lui, I.; Farr-Jones, S.; Van’t Veer, L.; Gartner, Z.J.; Green, E.M.; Lourenço, A.L.; Cheng, Y.; et al. Antibody-Drug Conjugates Targeting the Urokinase Receptor (uPAR) as a Possible Treatment of Aggressive Breast Cancer. Antibodies 2019, 8, 54. [Google Scholar] [CrossRef]
- Mudshinge, S.R.; Deore, A.B.; Patil, S.; Bhalgat, C.M. Nanoparticles: Emerging carriers for drug delivery. Saudi Pharm. J. 2011, 19, 129–141. [Google Scholar] [CrossRef]
- Zavaleta, C.; Ho, D.; Chung, E.J. Theranostic Nanoparticles for Tracking and Monitoring Disease State. SLAS Technol. 2018, 23, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.-H.; Qian, X.; Mao, H.; Wang, A.Y.; Chen, Z.G.; Nie, S.; Shin, D.M. Targeted magnetic iron oxide nanoparticles for tumor imaging and therapy. IJN 2008, 3, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, F.F. Using Nanoparticles in Medicine for Liver Cancer Imaging. Oman Med. J. 2017, 32, 269–274. [Google Scholar] [CrossRef]
- Taruno, K.; Kurita, T.; Kuwahata, A.; Yanagihara, K.; Enokido, K.; Katayose, Y.; Nakamura, S.; Takei, H.; Sekino, M.; Kusakabe, M. Multicenter clinical trial on sentinel lymph node biopsy using superparamagnetic iron oxide nanoparticles and a novel handheld magnetic probe. J. Surg. Oncol. 2019, 120, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kotani, Y.; Suzuki, A.; Takaya, H.; Nakai, H.; Matsuki, M.; Sato, T.; Mandai, M.; Matsumura, N. Superparamagnetic iron oxide as a tracer for sentinel lymph node detection in uterine cancer: A pilot study. Sci. Rep. 2020, 10, 7945. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Peng, X.-H.; Wang, Y.A.; Wang, X.; Cao, Z.; Ni, C.; Karna, P.; Zhang, X.; Wood, W.C.; Gao, X.; et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4722–4732. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Qian, W.P.; Wang, L.; Wang, Y.A.; Staley, C.A.; Satpathy, M.; Nie, S.; Mao, H.; Yang, L. Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano 2013, 7, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cao, Z.; Sajja, H.K.; Mao, H.; Wang, L.; Geng, H.; Xu, H.; Jiang, T.; Wood, W.C.; Nie, S.; et al. Development of Receptor Targeted Magnetic Iron Oxide Nanoparticles for Efficient Drug Delivery and Tumor Imaging. J. Biomed. Nanotechnol. 2008, 4, 439–449. [Google Scholar] [CrossRef]
- Yang, L.; Sajja, H.K.; Cao, Z.; Qian, W.; Bender, L.; Marcus, A.I.; Lipowska, M.; Wood, W.C.; Wang, Y.A. uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection. Theranostics 2013, 4, 106–118. [Google Scholar] [CrossRef]
- Gao, N.; Bozeman, E.N.; Qian, W.; Wang, L.; Chen, H.; Lipowska, M.; Staley, C.A.; Wang, Y.A.; Mao, H.; Yang, L. Tumor Penetrating Theranostic Nanoparticles for Enhancement of Targeted and Image-guided Drug Delivery into Peritoneal Tumors following Intraperitoneal Delivery. Theranostics 2017, 7, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Deirram, N.; Zhang, C.; Kermaniyan, S.S.; Johnston, A.P.R.; Such, G.K. pH-Responsive Polymer Nanoparticles for Drug Delivery. Macromol. Rapid Commun. 2019, 40, 1800917. [Google Scholar] [CrossRef]
- Abdalla, M.O.; Karna, P.; Sajja, H.K.; Mao, H.; Yates, C.; Turner, T.; Aneja, R. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J. Control. Release 2011, 149, 314–322. [Google Scholar] [CrossRef]
- Wang, N.; Cheng, X.; Li, N.; Wang, H.; Chen, H. Nanocarriers and Their Loading Strategies. Adv. Health Mater. 2019, 8, 1801002. [Google Scholar] [CrossRef]
- Ahmed, M.S.U.; Salam, A.B.; Yates, C.; Willian, K.; Jaynes, J.; Turner, T.; Abdalla, M.O. Double-receptor-targeting multifunctional iron oxide nanoparticles drug delivery system for the treatment and imaging of prostate cancer. Int. J. Nanomed. 2017, 12, 6973–6984. [Google Scholar] [CrossRef]
- Hong, Y.; Che, S.; Hui, B.; Yang, Y.; Wang, X.; Zhang, X.; Qiang, Y.; Ma, H. Lung cancer therapy using doxorubicin and curcumin combination: Targeted prodrug based, pH sensitive nanomedicine. Biomed. Pharm. 2019, 112, 108614. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Löwik, D.W.; Miller, A.D.; Thanou, M. Targeting the urokinase plasminogen activator receptor with synthetic self-assembly nanoparticles. Bioconjug. Chem. 2009, 20, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Larsen, E.K.; Nielsen, E.H.; Iversen, F.; Liu, Z.; Thomsen, K.; Pedersen, M.; Skrydstrup, T.; Nielsen, N.C.; Ploug, M.; et al. Targeting of peptide conjugated magnetic nanoparticles to urokinase plasminogen activator receptor (uPAR) expressing cells. Nanoscale 2013, 5, 8192–8201. [Google Scholar] [CrossRef]
- Park, J.Y.; Shin, Y.; Won, W.R.; Lim, C.; Kim, J.C.; Kang, K.; Husni, P.; Lee, E.S.; Youn, Y.S.; Oh, K.T. Development of AE147 Peptide-Conjugated Nanocarriers for Targeting uPAR-Overexpressing Cancer Cells. IJN 2021, 16, 5437–5449. [Google Scholar] [CrossRef]
- Li, Z.; Wang, C.; Chen, J.; Lian, X.; Xiong, C.; Tian, R.; Hu, L.; Xiong, X.; Tian, J. uPAR targeted phototheranostic metal-organic framework nanoprobes for MR/NIR-II imaging-guided therapy and surgical resection of glioblastoma. Mater. Des. 2021, 198, 109386. [Google Scholar] [CrossRef]
- Hu, Y.; Chi, C.; Wang, S.; Wang, L.; Liang, P.; Liu, F.; Shang, W.; Wang, W.; Zhang, F.; Li, S.; et al. A Comparative Study of Clinical Intervention and Interventional Photothermal Therapy for Pancreatic Cancer. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, R.; Zhao, L.; Zhang, X.; Xu, T.; Cui, M. Basing on uPAR-binding fragment to design chimeric antigen receptors triggers antitumor efficacy against uPAR expressing ovarian cancer cells. Biomed. Pharmacother. 2019, 117, 109173. [Google Scholar] [CrossRef] [PubMed]
- Murad, J.M.; Graber, D.J.; Sentman, C.L. Advances in the use of natural receptor- or ligand-based chimeric antigen receptors (CARs) in haematologic malignancies. Best Pr. Res. Clin. Haematol 2018, 31, 176–183. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Achilli, S.; Berthet, N.; Renaudet, O. Antibody recruiting molecules (ARMs): Synthetic immunotherapeutics to fight cancer. RSC Chem. Biol. 2021, 2, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Jakobsche, C.E.; McEnaney, P.J.; Zhang, A.X.; Spiegel, D.A. Reprogramming Urokinase into an Antibody-Recruiting Anticancer Agent. ACS Chem. Biol. 2012, 7, 316–321. [Google Scholar] [CrossRef]
- Rullo, A.F.; Fitzgerald, K.J.; Muthusamy, V.; Liu, M.; Yuan, C.; Huang, M.; Kim, M.; Cho, A.E.; Spiegel, D.A. Re-engineering the Immune Response to Metastatic Cancer: Antibody-Recruiting Small Molecules Targeting the Urokinase Receptor. Angew Chem. Int. Ed. Engl. 2016, 55, 3642–3646. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses with Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Jing, Y.; Tong, C.; Zhang, J.; Nakamura, T.; Iankov, I.; Russell, S.J.; Merchan, J.R. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009, 69, 1459–1468. [Google Scholar] [CrossRef]
- Jing, Y.; Chavez, V.; Ban, Y.; Acquavella, N.; El-Ashry, D.; Pronin, A.; Chen, X.; Merchan, J.R. Molecular Effects of Stromal-Selective Targeting by uPAR-Retargeted Oncolytic V.Virus in Breast Cancer. Mol. Cancer Res. 2017, 15, 1410–1420. [Google Scholar] [CrossRef]
- Jing, Y.; Zaias, J.; Duncan, R.; Russell, S.J.; Merchan, J.R. In vivo safety, biodistribution and antitumor effects of uPAR retargeted oncolytic measles virus in syngeneic cancer models. Gene Ther. 2014, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Chavez, V.; Khatwani, N.; Ban, Y.; Espejo, A.P.; Chen, X.; Merchan, J.R. In vivo antitumor activity by dual stromal and tumor-targeted oncolytic measles viruses. Cancer Gene Ther. 2020, 27, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Bejarano, M.T.; Zaias, J.; Merchan, J.R. In vivo anti-metastatic effects of uPAR retargeted measles virus in syngeneic and xenograft models of mammary cancer. Breast Cancer Res. Treat. 2015, 149, 99–108. [Google Scholar] [CrossRef] [PubMed]










| LT Name | Toxin | Origin of the Toxin | Additional Surface Target | Application | Model System | Ref. |
|---|---|---|---|---|---|---|
| DTAT | DT | Corynebacterium diphtheriae | / | Preclinical | Human GBM and HUVECs cells; Human GBM SC and IC xenograft models | [179,182] |
| DTAT13 | DT | Corynebacterium diphtheriae | IL-13Rα2 | Preclinical | Human GBM and HUVECs cells; Human GBM SC and IC xenograft models | [181,183] |
| DTATEGF | DT | Corynebacterium diphtheriae | EGFR | Preclinical | Human NSCLC cells; Human metastatic NSCLC IC xenograft model | [187] |
| eBAT | PE38 | Pseudomonas aeruginosa | / | Preclinical | Human GBM, HUVECs, HNSCC, breast, ovarian, sarcoma and pediatric sarcoma cell lines; GBM SC and IC xenograft models; Adaptive dose-finding, phase I–II clinical trial for canine HSA | [191,192,195,196,197,199] |
| ATF-SAP | Saporin | Saponaria officinalis | / | Preclinical | Human bladder and triple negative breast cancer cell lines; bladder cancer SC xenograft models | [203,204,206,207] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metrangolo, V.; Ploug, M.; Engelholm, L.H. The Urokinase Receptor (uPAR) as a “Trojan Horse” in Targeted Cancer Therapy: Challenges and Opportunities. Cancers 2021, 13, 5376. https://doi.org/10.3390/cancers13215376
Metrangolo V, Ploug M, Engelholm LH. The Urokinase Receptor (uPAR) as a “Trojan Horse” in Targeted Cancer Therapy: Challenges and Opportunities. Cancers. 2021; 13(21):5376. https://doi.org/10.3390/cancers13215376
Chicago/Turabian StyleMetrangolo, Virginia, Michael Ploug, and Lars H. Engelholm. 2021. "The Urokinase Receptor (uPAR) as a “Trojan Horse” in Targeted Cancer Therapy: Challenges and Opportunities" Cancers 13, no. 21: 5376. https://doi.org/10.3390/cancers13215376
APA StyleMetrangolo, V., Ploug, M., & Engelholm, L. H. (2021). The Urokinase Receptor (uPAR) as a “Trojan Horse” in Targeted Cancer Therapy: Challenges and Opportunities. Cancers, 13(21), 5376. https://doi.org/10.3390/cancers13215376