
Tetraspanin 5 (TSPAN5), a Novel Gatekeeper of the Tumor Suppressor DLC1 and Myocardin-Related Transcription Factors (MRTFs), Controls HCC Growth and Senescence

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Transfections and Reagents
2.2. Cell Proliferation Assay
2.3. Senescence-Associated ß-Galactosidase Staining
2.4. Scratch Wound Assay
2.5. Invasion Assay
2.6. Spheroid Assay
2.7. RNA Extraction, cDNA Synthesis and Quantitative Real-Time PCR Analysis (qRT-PCR)
2.8. Primer Synthesis
2.9. Microarray Analysis
2.10. Chromatin Immunoprecipitation (ChIP)
2.11. RhoA Assay
2.12. Immunoblotting
2.13. Immunofluorescence Staining
2.14. Measuring the Focal Adhesion Length
2.15. In Situ Proximity Ligation Assay (PLA)
2.16. Chicken Chorioallantoic Membrane Assay (CAM Assay)
2.17. Immunohistochemistry (IHC)
2.18. Scanning of IHC Sections
2.19. Human Liver Samples
2.20. Subcutaneous Tumor Xenograft
2.21. Statistical Analysis
3. Results
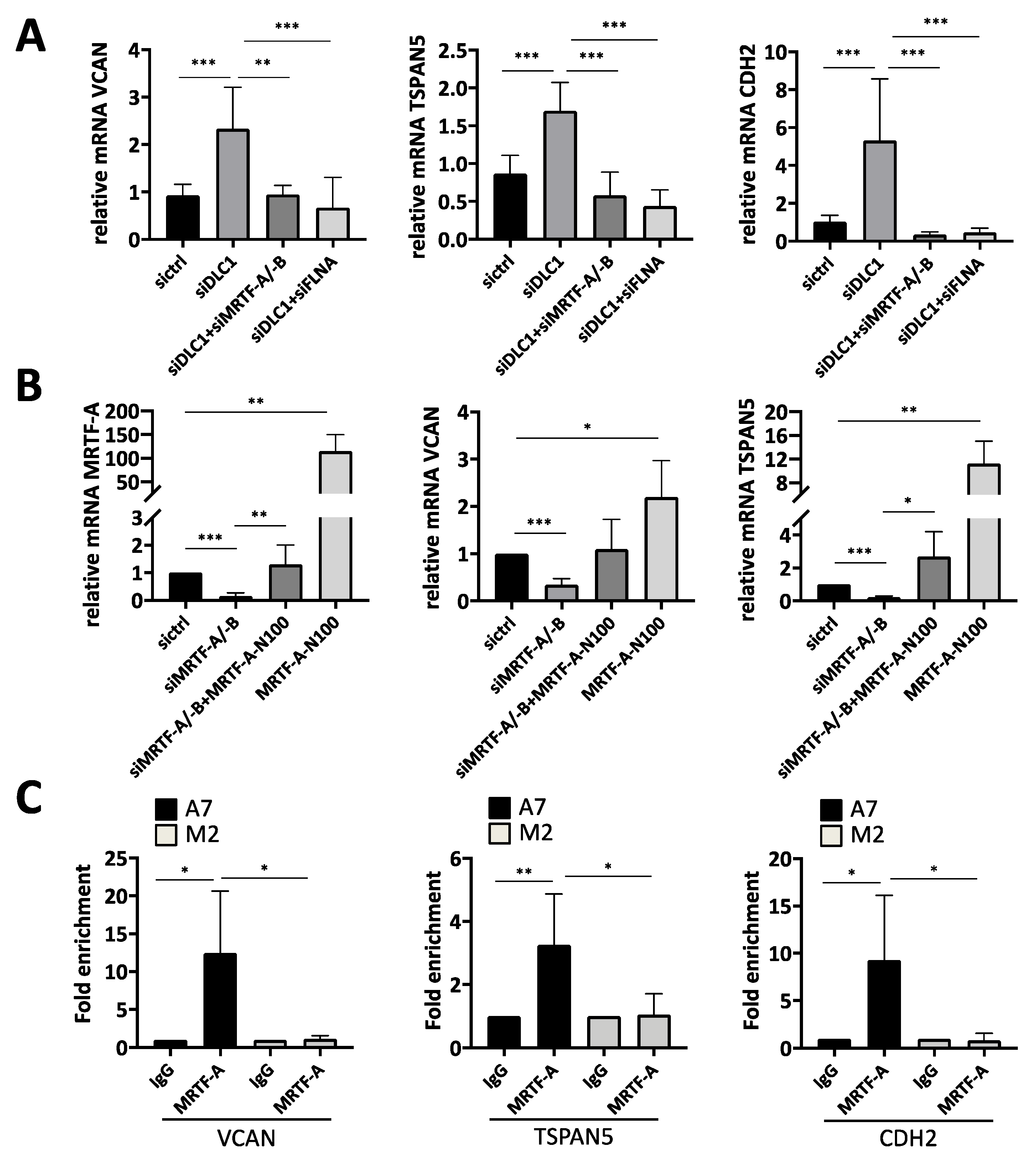
3.1. Gene Expression Profiling to Identify Novel Target Genes Induced by DLC1 Loss
3.2. Transcriptional Regulation of the Newly Identified DLC1-Dependent Genes
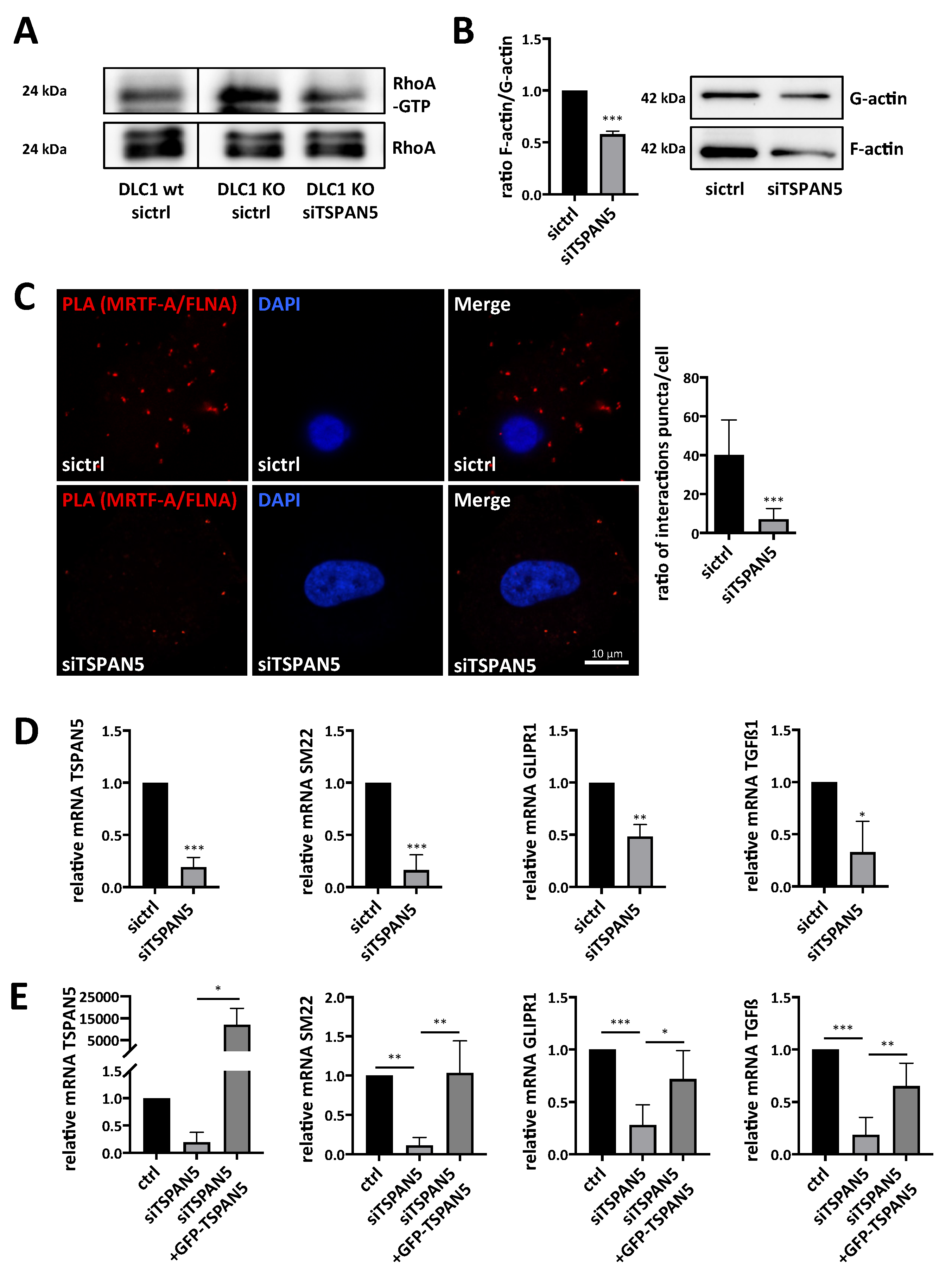
3.3. Requirement of TSPAN5 for HCC Proliferation, Migration, Invasion and Spheroid Formation
3.4. Requirement of TSPAN5 for Oncogene-Induced Senescence
3.5. Depletion of TSPAN5 Affects the Actin/MRTF Signaling Axis
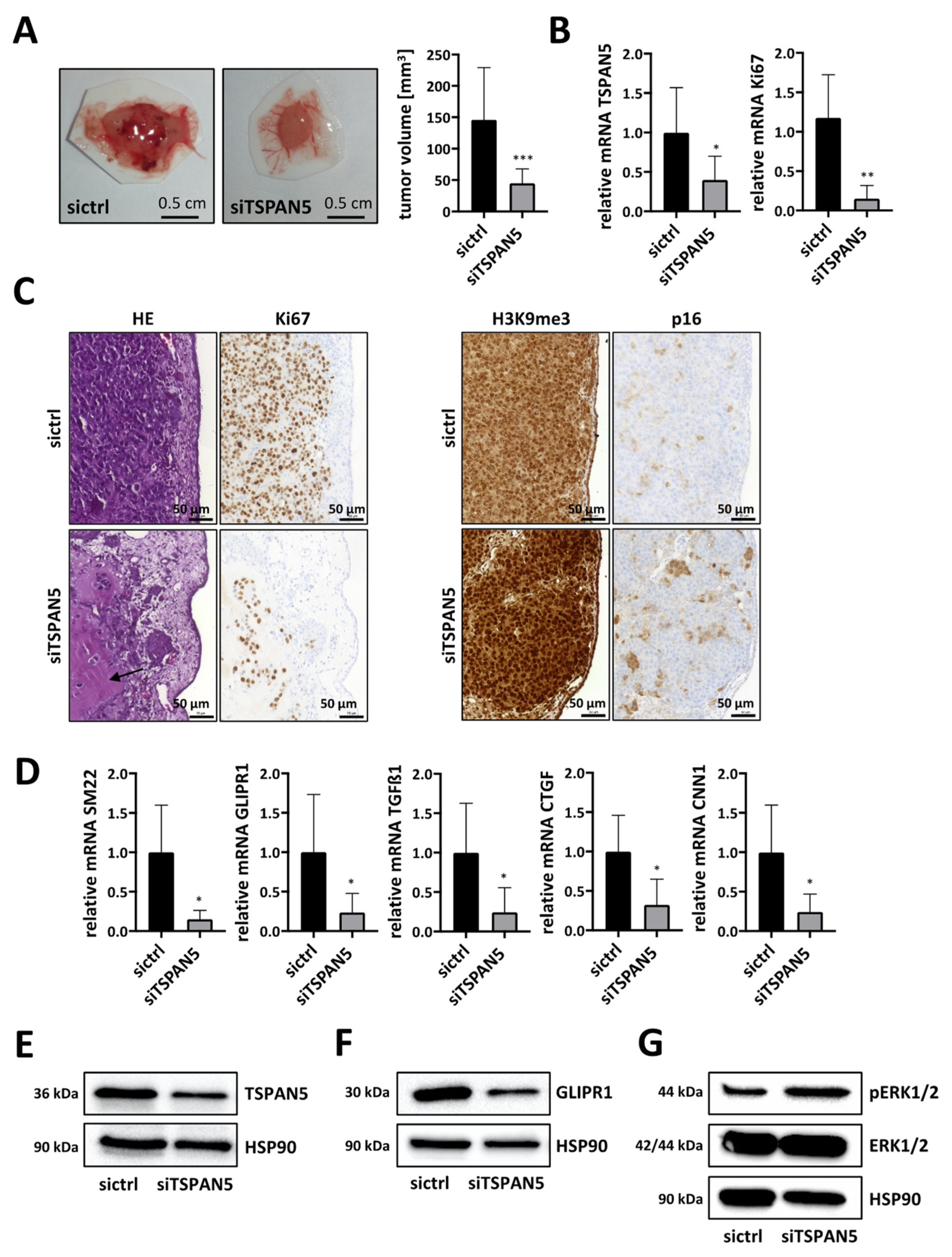
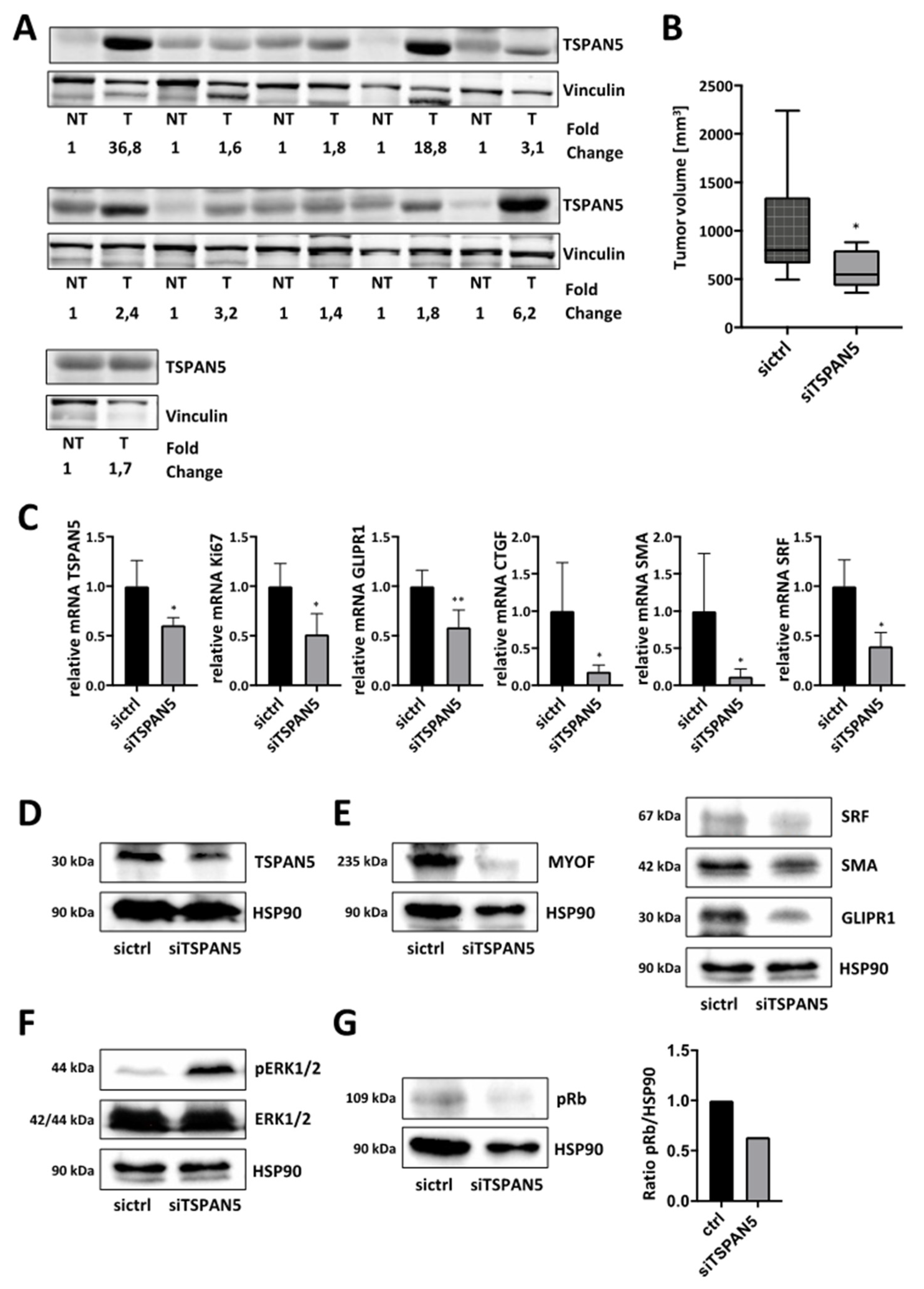
3.6. Anti-Tumor Effects of TSPAN5 Depletion in the Chorioallantoic Membrane (CAM) Assay
3.7. Anti-Tumor Effects of TSPAN5 Depletion in an HCC Xenograft Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Badvie, S. Hepatocellular carcinoma. Postgrad. Med. J. 2000, 76, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanti, F.; Villani, R.; Tamborra, R.; Blonda, M.; Iannelli, G.; di Bello, G.; Facciorusso, A.; Poli, G.; Iuliano, L.; Avolio, C.; et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2018, 15, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A. The influence of diabetes in the pathogenesis and the clinical course of hepatocellular carcinoma: Recent findings and new perspectives. Curr. Diabetes Rev. 2013, 9, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.Z.; Miller, M.J.; Keck, C.L.; Zimonjic, D.B.; Thorgeirsson, S.S.; Popescu, N.C. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998, 58, 2196–2199. [Google Scholar] [PubMed]
- Hampl, V.; Martin, C.; Aigner, A.; Hoebel, S.; Singer, S.; Frank, N.; Sarikas, A.; Ebert, O.; Prywes, R.; Gudermann, T.; et al. Depletion of the transcriptional coactivators megakaryoblastic leukaemia 1 and 2 abolishes hepatocellular carcinoma xenograft growth by inducing oncogene-induced senescence. EMBO Mol. Med. 2013, 5, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Kitadokoro, K.; Bordo, D.; Galli, G.; Petracca, R.; Falugi, F.; Abrignani, S.; Grandi, G.; Bolognesi, M. CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. EMBO J. 2001, 20, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Mó, M.; Barreiro, O.; Gordon, M.; Sala-Valdés, M.; Sánchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef]
- Peñas, P.F.; García-Díez, A.; Sánchez-Madrid, F.; Yáñez-Mó, M. Tetraspanins are localized at motility-related structures and involved in normal human keratinocyte wound healing migration. J. Investig. Dermatol. 2000, 114, 1126–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Rev. Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, C.; Hampl, V.; Holzer, K.; Aigner, A.; Penkava, J.; Frank, N.; Martin, D.E.; Maier, K.C.; Waldburger, N.; Roessler, S.; et al. The novel MKL target gene myoferlin modulates expansion and senescence of hepatocellular carcinoma. Oncogene 2017, 36, 3464–3476. [Google Scholar] [CrossRef] [PubMed]
- Voringer, S.; Schreyer, L.; Nadolni, W.; Meier, M.A.; Woerther, K.; Mittermeier, C.; Ferioli, S.; Singer, S.; Holzer, K.; Zierler, S.; et al. Inhibition of TRPM7 blocks MRTF/SRF-dependent transcriptional and tumorigenic activity. Oncogene 2020, 39, 2328–2344. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod. Toxicol. 2017, 70, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Muenzner, J.K.; Kunze, P.; Lindner, P.; Polaschek, S.; Menke, K.; Eckstein, M.; Geppert, C.I.; Chanvorachote, P.; Baeuerle, T.; Hartmann, A.; et al. Generation and characterization of hepatocellular carcinoma cell lines with enhanced cancer stem cell potential. J. Cell Mol. Med. 2018, 22, 6238–6248. [Google Scholar] [CrossRef] [Green Version]
- Aigner, A. Delivery Systems for the Direct Application of siRNAs to Induce RNA Interference (RNAi) In Vivo. J. Biomed. Biotechnol. 2006, 2006, 71659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, J.; Höbel, S.; Bakowsky, U.; Aigner, A. Liposome-polyethylenimine complexes for enhanced DNA and siRNA delivery. Biomaterials 2010, 31, 6892–6900. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grünweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchardt, H.; Schulz, A.; Datta, K.; Muders, M.H.; Aigner, A. Silencing of Neuropilins and GIPC1 in pancreatic ductal adenocarcinoma exerts multiple cellular and molecular antitumor effects. Sci. Rep. 2019, 9, 15471. [Google Scholar] [CrossRef] [Green Version]
- Borchardt, H.; Ewe, A.; Morawski, M.; Weirauch, U.; Aigner, A. miR24-3p activity after delivery into pancreatic carcinoma cell lines exerts profound tumor-inhibitory effects through distinct pathways of apoptosis and autophagy induction. Cancer Lett. 2021, 503, 174–184. [Google Scholar] [CrossRef]
- Muehlich, S.; Hampl, V.; Khalid, S.; Singer, S.; Frank, N.; Breuhahn, K.; Gudermann, T.; Prywes, R. The transcriptional coactivators megakaryoblastic leukemia 1/2 mediate the effects of loss of the tumor suppressor deleted in liver cancer 1. Oncogene 2012, 31, 3913–3923. [Google Scholar] [CrossRef] [Green Version]
- Kircher, P.; Hermanns, C.; Nossek, M.; Drexler, M.K.; Grosse, R.; Fischer, M.; Sarikas, A.; Penkava, J.; Lewis, T.; Prywes, R.; et al. Filamin A interacts with the coactivator MKL1 to promote the activity of the transcription factor SRF and cell migration. Sci. Signal. 2015, 8, ra112. [Google Scholar] [CrossRef] [Green Version]
- Nishio, K.; Inoue, A. Senescence-associated alterations of cytoskeleton: Extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem. Cell Biol. 2005, 123, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C. Survive or thrive: Tradeoff strategy for cellular senescence. Exp. Mol. Med. 2017, 49, e342. [Google Scholar] [CrossRef]
- Crews, C.M.; Erikson, R.L. Extracellular signals and reversible protein phosphorylation: What to Mek of it all. Cell 1993, 74, 215–217. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Zuffo, R.D.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Braumüller, H.; Wieder, T.; Brenner, E.; Aßmann, S.C.Y.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.M.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferbeyre, G.; De Stanchina, E.; Querido, E.; Baptiste, N.; Prives, C.; Lowe, S.W. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000, 14, 2015–2027. [Google Scholar]
- Bischof, O.; Kirsh, O.; Pearson, M.; Itahana, K.; Pelicci, P.G.; Dejean, A. Deconstructing PML-induced premature senescence. EMBO J. 2002, 21, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Derycke, L.D.; Bracke, M.E. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 2004, 48, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Cleton-Jansen, A.M.; Strumane, K.; De Leeuw, W.J.; Nollet, F.; Van Roy, F.; Cornelisse, C. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 1996, 13, 1919–1925. [Google Scholar]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-cadherin cell surface regulation in cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Werth, S.; Urban-Klein, B.; Dai, L.; Höbel, S.; Grzelinski, M.; Bakowsky, U.; Czubayko, F.; Aigner, A. A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. J. Control Release 2006, 112, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Höbel, S.; Aigner, A. Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Krasnitz, A.; Lucito, R.; Sordella, R.; Vanaelst, L.; Cordon-Cardo, C.; Singer, S.; Kuehnel, F.; Wigler, M.; Powers, S.; et al. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008, 22, 1439–1444. [Google Scholar] [CrossRef] [Green Version]
- Boucheix, C.; Duc, G.H.T.; Jasmin, C.; Rubinstein, E. Tetraspanins and malignancy. Expert Rev. Mol. Med. 2001, 2001, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Baye, T.M.; Carless, M.; Wallace, J.; Komorowski, R.; Kleiner, D.E.; Andris, D.; Makladi, B.; Cole, R.; Charlton, M.; et al. Hepatic gene networks in morbidly obese patients with nonalcoholic fatty liver disease. Obes. Surg. 2010, 20, 1698–1709. [Google Scholar] [CrossRef]
- Qi, Y.; Li, H.; Lv, J.; Qi, W.; Shen, L.; Liu, S.; Ding, A.; Wang, G.; Sun, L.; Qiu, W. Expression and function of transmembrane 4 superfamily proteins in digestive system cancers. Cancer Cell Int. 2020, 20, 314. [Google Scholar] [CrossRef] [PubMed]
- Hobel, S.; Koburger, I.; John, M.; Czubayko, F.; Hadwiger, P.; Vornlocher, H.-P.; Aigner, A. Polyethylenimine/small interfering RNA-mediated knockdown of vascular endothelial growth factor in vivo exerts anti-tumor effects synergistically with Bevacizumab. J. Gene Med. 2010, 12, 287–300. [Google Scholar] [PubMed]
- Heo, K.; Lee, S. TSPAN8 as a Novel Emerging Therapeutic Target in Cancer for Monoclonal Antibody Therapy. Biomolecules 2020, 10, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Pol, J.; Billard, M.; Dornier, E.; Eschenbrenner, E.; Danglot, L.; Boucheix, C.; Charrin, S.; Rubinstein, E. New insights into the tetraspanin Tspan5 using novel monoclonal antibodies. J. Biol. Chem. 2017, 292, 9551–9566. [Google Scholar] [CrossRef] [Green Version]
- Vernier, M.; Bourdeau, V.; Gaumont-Leclerc, M.-F.; Moiseeva, O.; Bégin, V.; Saad, F.; Mes-Masson, A.-M.; Ferbeyre, G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011, 25, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Zöller, M. Exosome target cell selection and the importance of exosomal tetraspanins: A hypothesis. Biochem. Soc. Trans. 2011, 39, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreyer, L.; Mittermeier, C.; Franz, M.J.; Meier, M.A.; Martin, D.E.; Maier, K.C.; Huebner, K.; Schneider-Stock, R.; Singer, S.; Holzer, K.; et al. Tetraspanin 5 (TSPAN5), a Novel Gatekeeper of the Tumor Suppressor DLC1 and Myocardin-Related Transcription Factors (MRTFs), Controls HCC Growth and Senescence. Cancers 2021, 13, 5373. https://doi.org/10.3390/cancers13215373
Schreyer L, Mittermeier C, Franz MJ, Meier MA, Martin DE, Maier KC, Huebner K, Schneider-Stock R, Singer S, Holzer K, et al. Tetraspanin 5 (TSPAN5), a Novel Gatekeeper of the Tumor Suppressor DLC1 and Myocardin-Related Transcription Factors (MRTFs), Controls HCC Growth and Senescence. Cancers. 2021; 13(21):5373. https://doi.org/10.3390/cancers13215373
Chicago/Turabian StyleSchreyer, Laura, Constanze Mittermeier, Miriam J. Franz, Melanie A. Meier, Dietmar E. Martin, Kerstin C. Maier, Kerstin Huebner, Regine Schneider-Stock, Stephan Singer, Kerstin Holzer, and et al. 2021. "Tetraspanin 5 (TSPAN5), a Novel Gatekeeper of the Tumor Suppressor DLC1 and Myocardin-Related Transcription Factors (MRTFs), Controls HCC Growth and Senescence" Cancers 13, no. 21: 5373. https://doi.org/10.3390/cancers13215373
APA StyleSchreyer, L., Mittermeier, C., Franz, M. J., Meier, M. A., Martin, D. E., Maier, K. C., Huebner, K., Schneider-Stock, R., Singer, S., Holzer, K., Fischer, D., Ribback, S., Liebl, B., Gudermann, T., Aigner, A., & Muehlich, S. (2021). Tetraspanin 5 (TSPAN5), a Novel Gatekeeper of the Tumor Suppressor DLC1 and Myocardin-Related Transcription Factors (MRTFs), Controls HCC Growth and Senescence. Cancers, 13(21), 5373. https://doi.org/10.3390/cancers13215373