
TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy

 , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumour Sample Collection and Patient Population
2.2. Clinical Data Collection
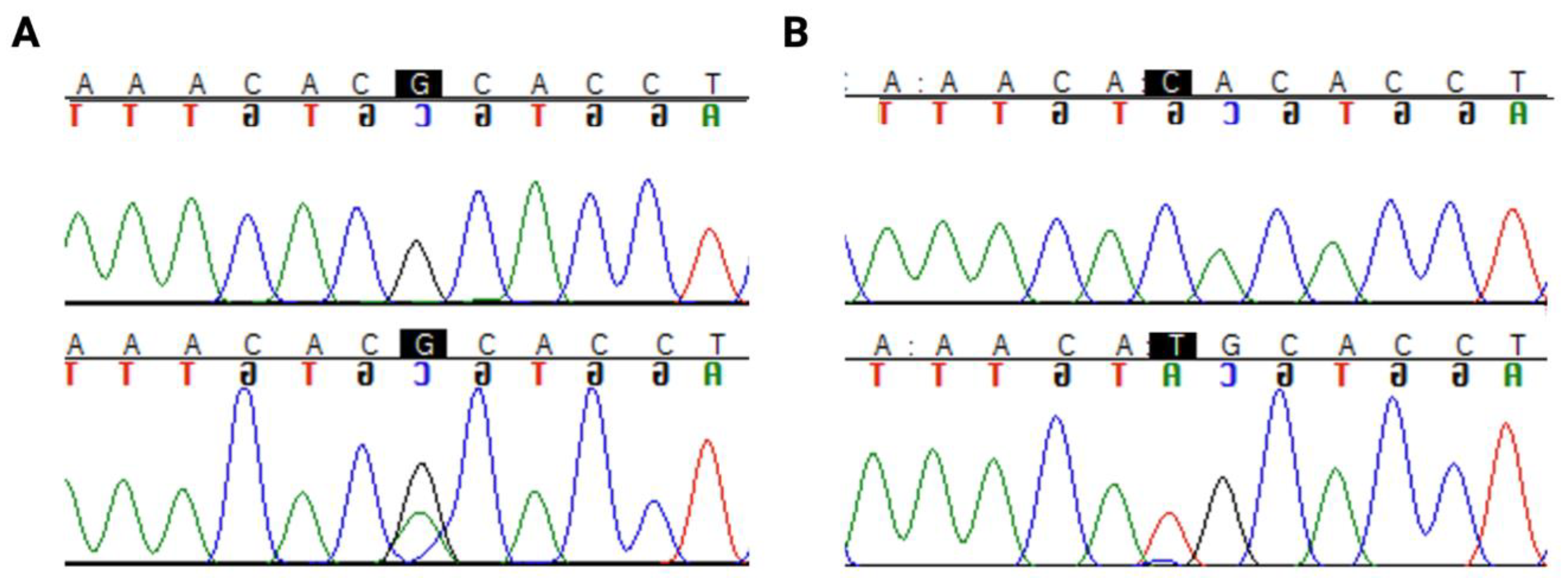
2.3. TP53 Sequencing
2.4. IDH1 Sequencing
2.5. Determination of Pathogenic Mutations
2.6. Survival Analysis
2.7. Survival Studies with Public Cancer Datasets
2.8. Determination of Prognostic Effects of Hippo Signalling Pathway Genes in TCGA Cancers and Their Correlation with TP53
2.9. Differential Gene Expression, Gene Ontology and Pathway Analysis
2.10. Protein–Protein Interaction Analysis
2.11. Statistical Analysis
3. Results
3.1. Tumour Cohort Characteristics
3.2. Population Survival Characteristics
3.3. TP53 Mutation Types and Prevalence in LGG
3.4. Correlation of TP53 Mutation with Other Clinicopathological Features
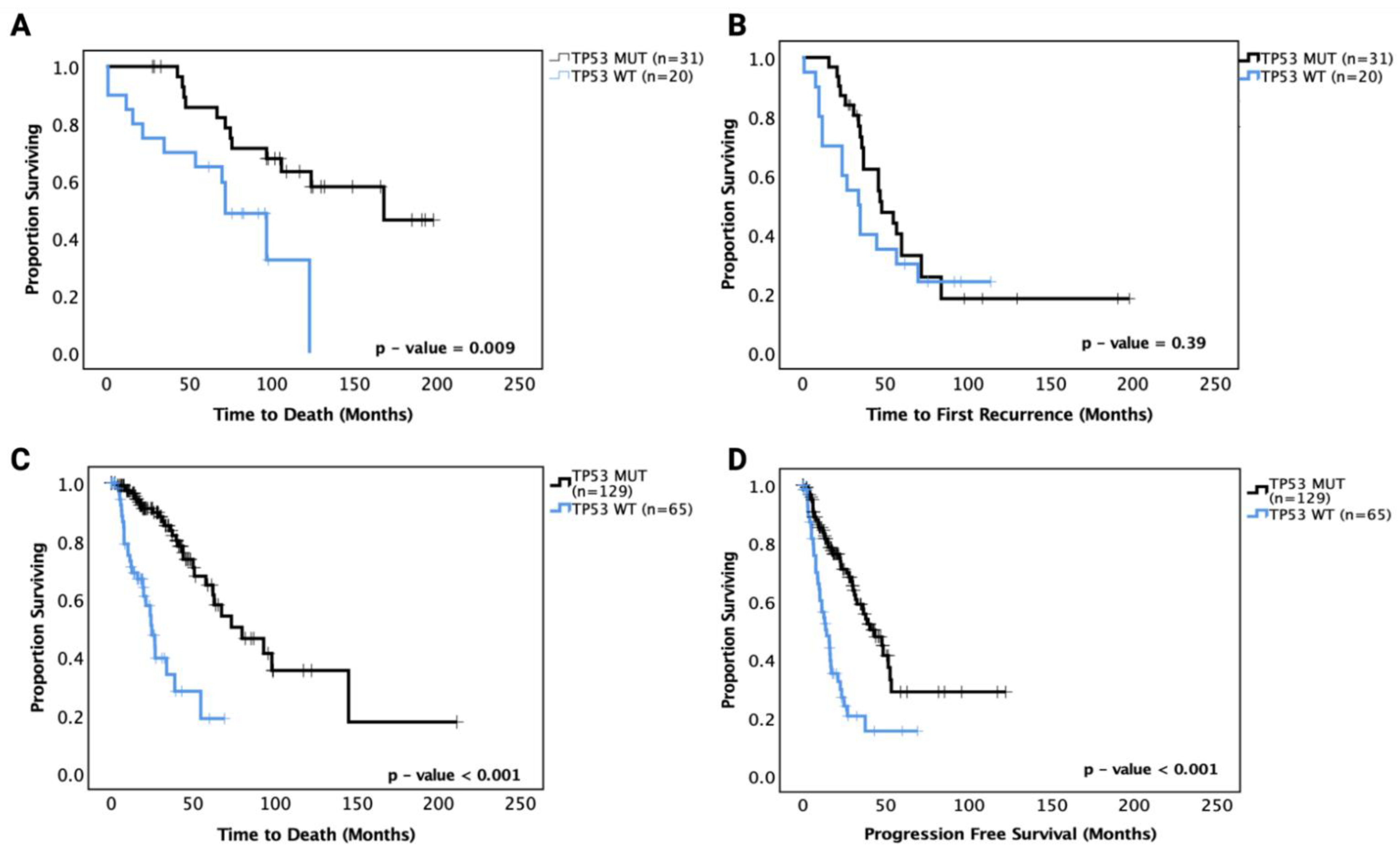
3.5. Prognostic Effects of TP53 Mutations in LGG
3.5.1. Prognostic Effects of TP53 Mutations in Astrocytoma
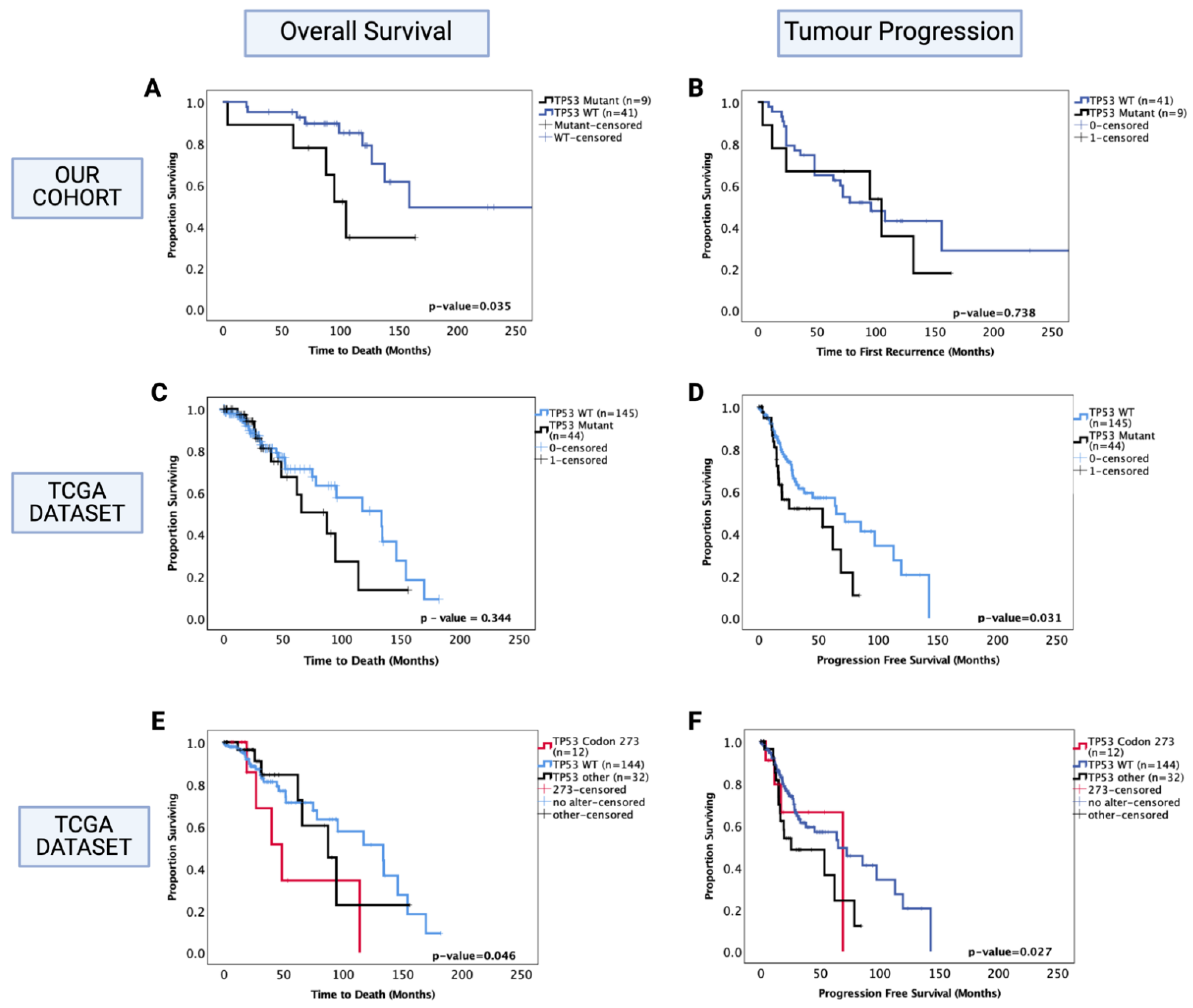
3.5.2. Prognostic Effects of TP53 Mutations in Oligodendroglioma
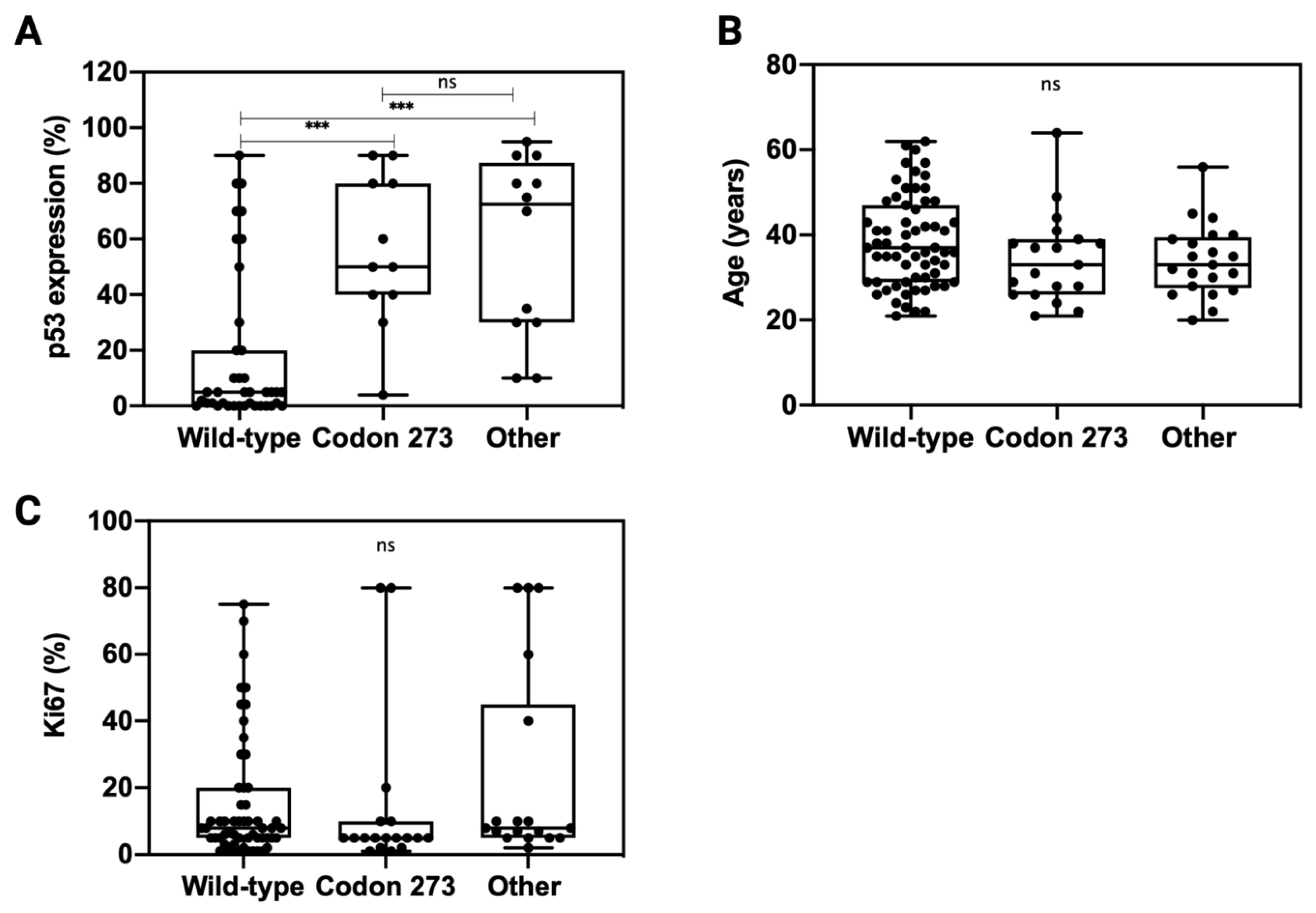
3.6. TP53 Mutation Is Associated with Elevated p53 Expression
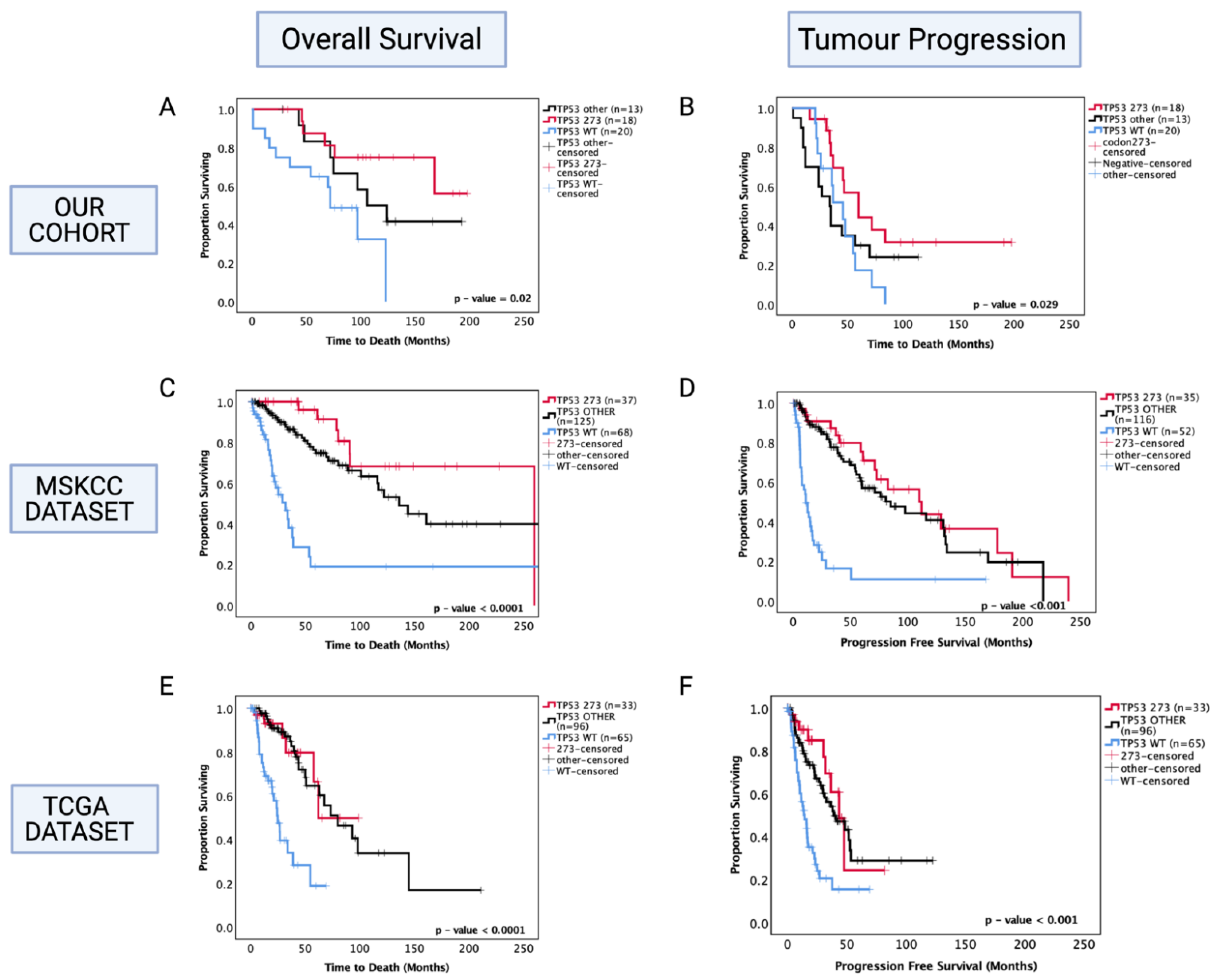
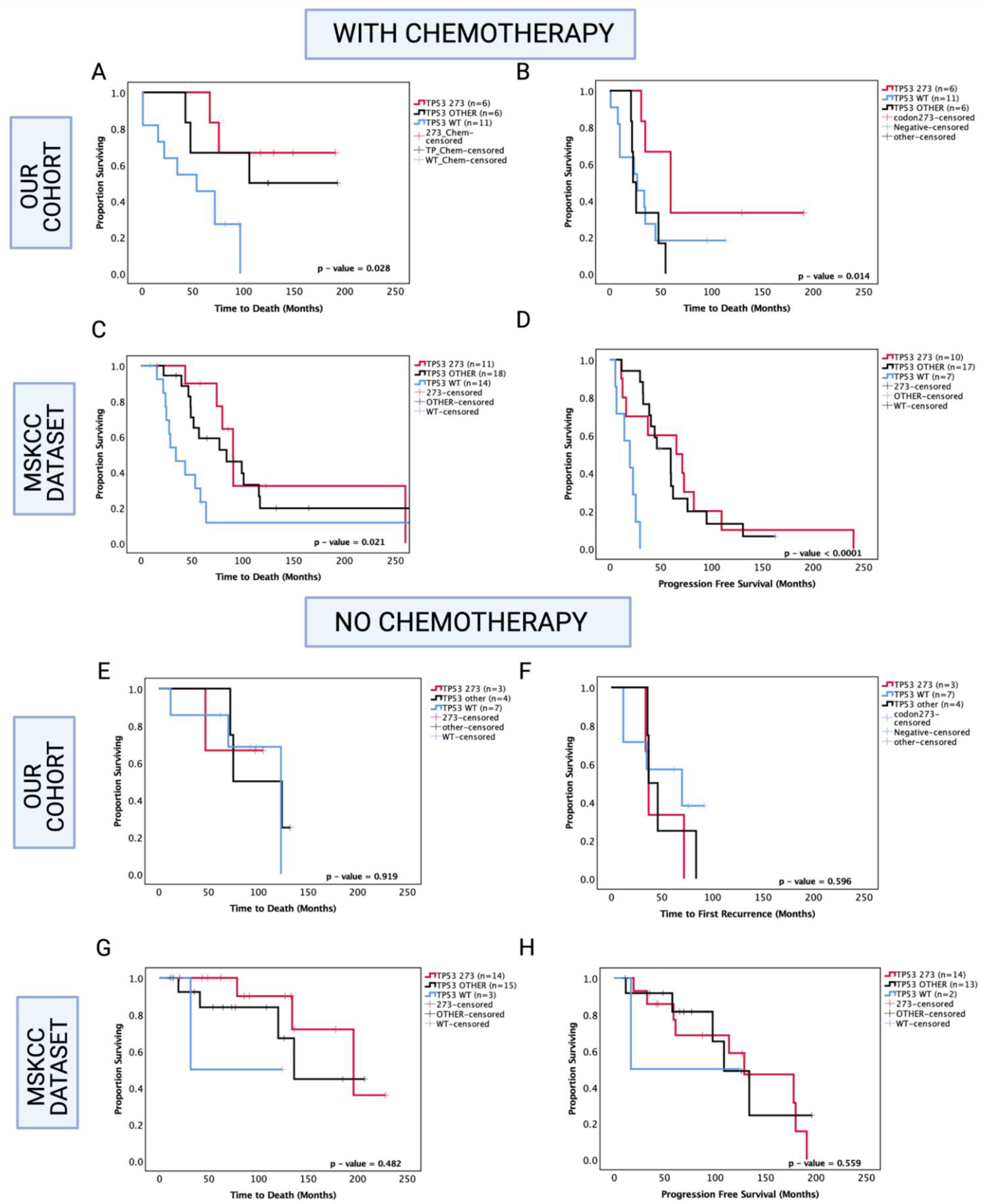
3.7. Enhanced Chemosensitivity of Codon 273 Mutant Astrocytoma May Lead to Improved Overall and Progression-Free Survival
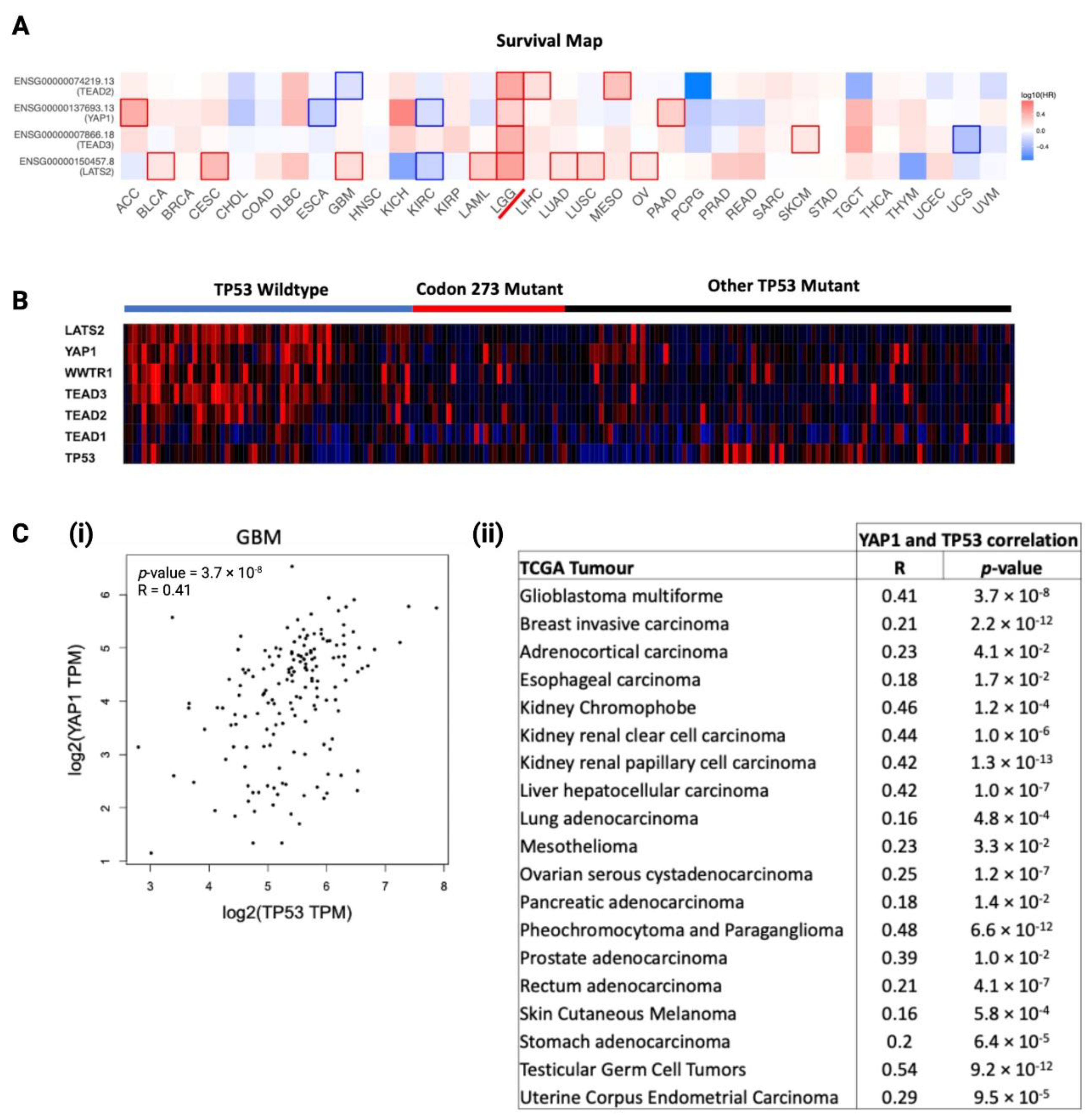
3.8. YAP1 mRNA Expression Is Associated with TP53, and Hippo Signalling Pathway Genes Are Prognostic Factors in LGG
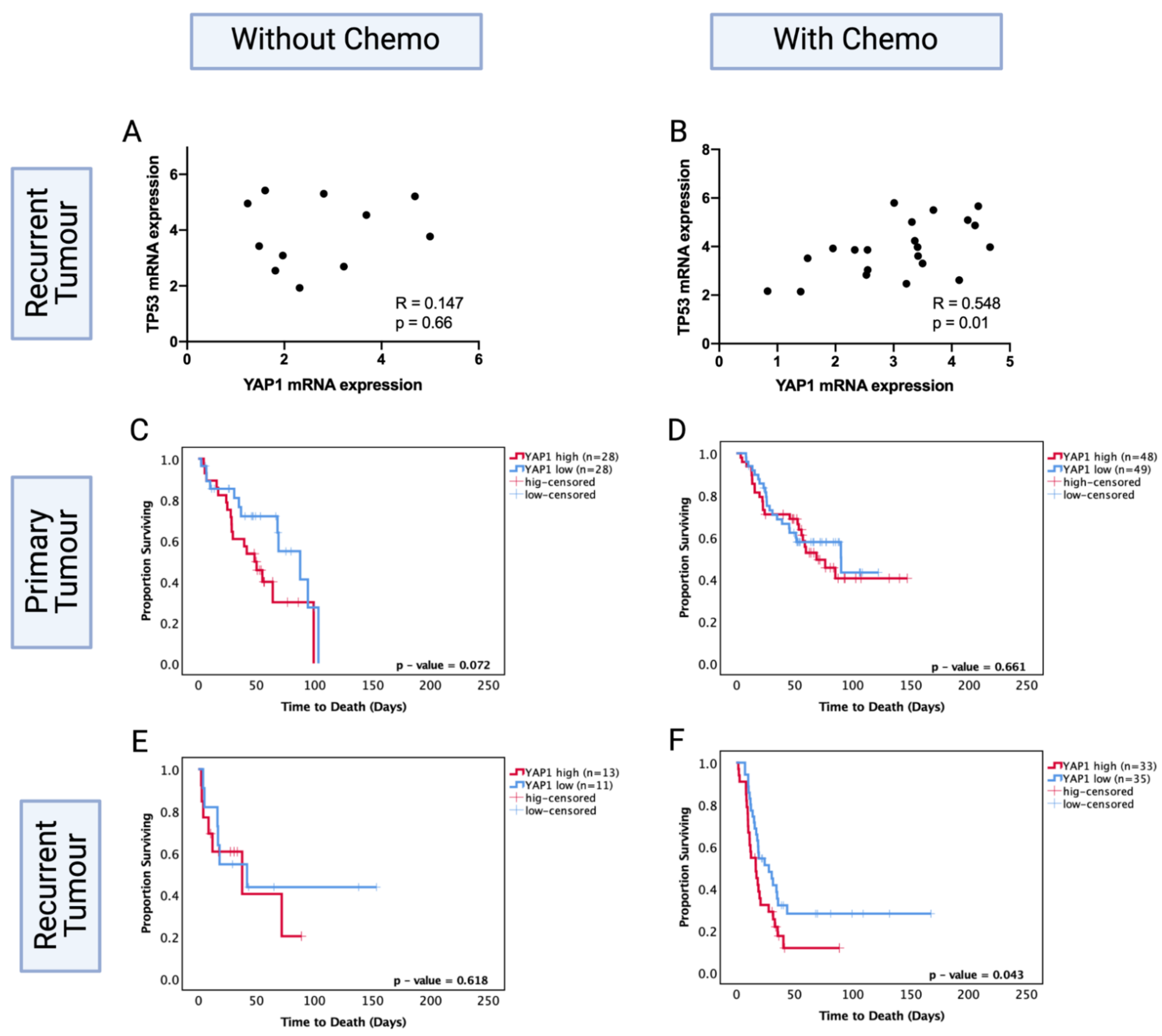
3.9. YAP1 mRNA Expression Levels Are Prognostic in Only Chemotherapy-Treated Recurrent Astrocytoma
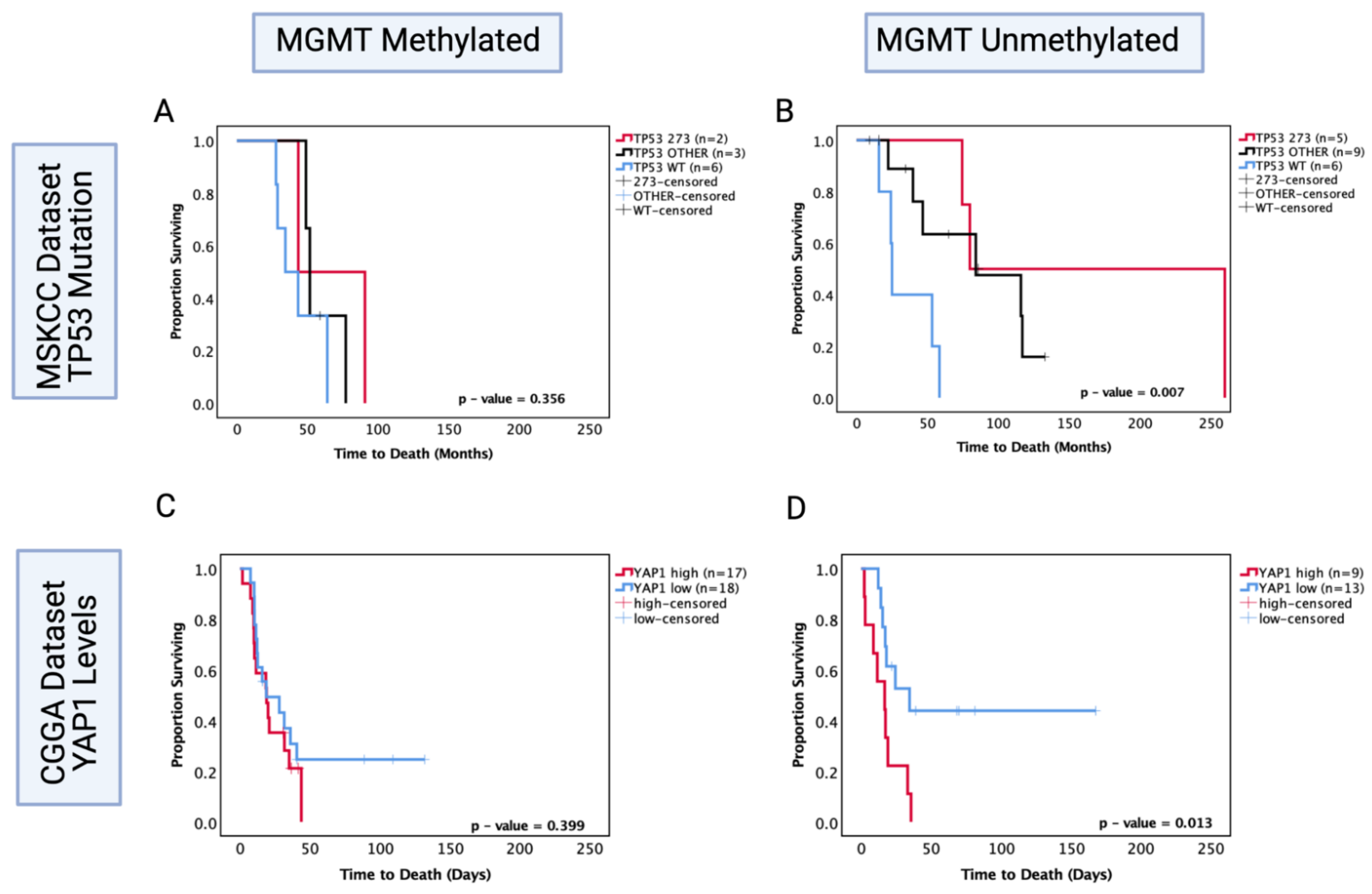
3.10. O6-Methylguanine-DNA Methyltransferase (MGMT) Methylation Status of Astrocytoma Is a Factor in TP53 Mutation/YAP1-Mediated Chemosensitivity
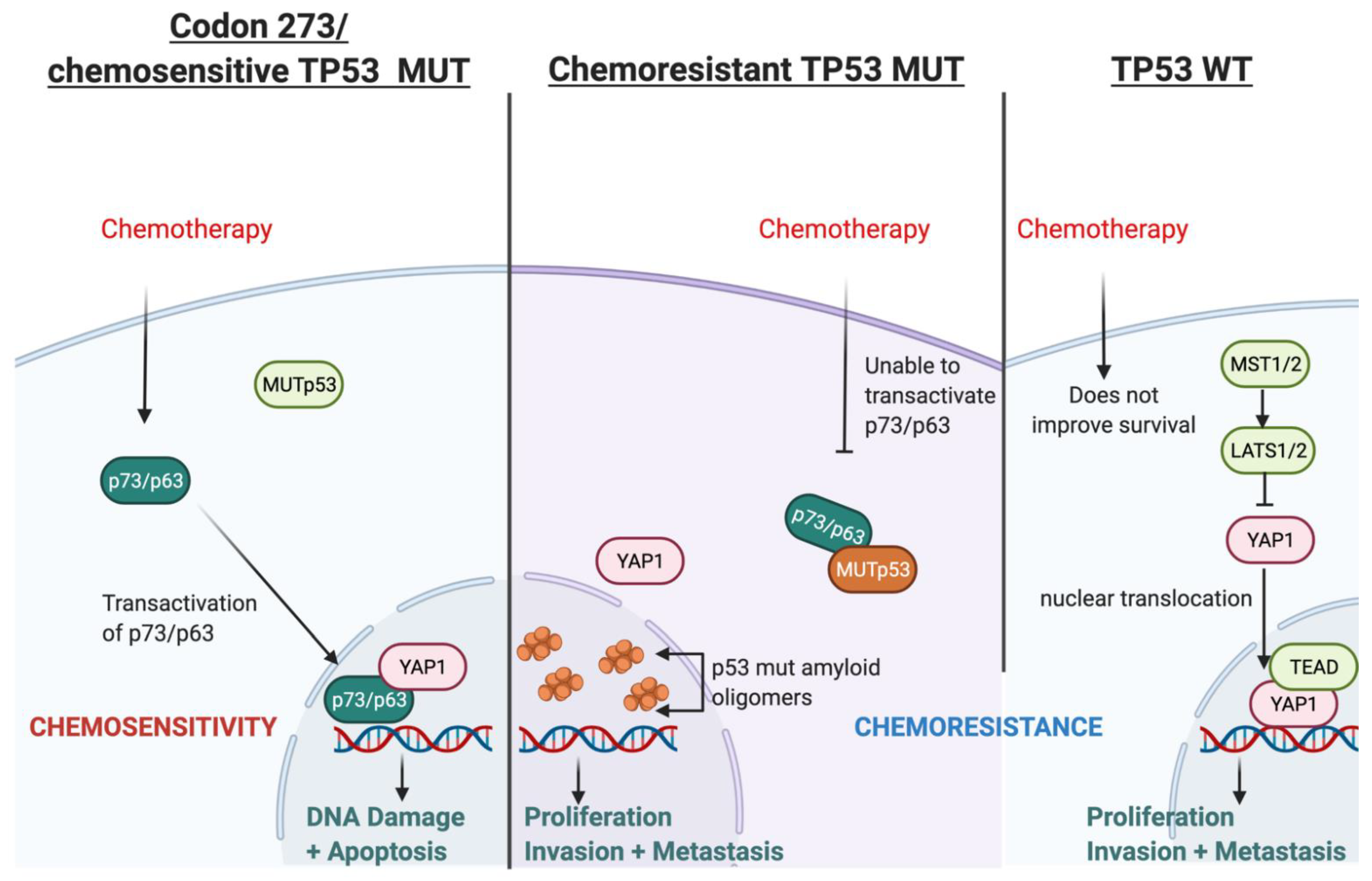
3.11. Possible Mechanism for TP53-Mutation-Dependent Chemosensitivity in Astrocytoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, J.J.; Wen, P.Y. Emerging targeted therapies for glioma. Expert Opin. Emerg. Drugs 2016, 21, 441–452. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Claus, E.B.; Walsh, K.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.S.; Jenkins, R.B.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015, 38, E6. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in Malignant Glioma: Standard of Care and Future Directions. J. Clin. Oncol. 2007, 25, 4127–4136. [Google Scholar] [CrossRef]
- Bush, N.A.O.; Chang, S. Treatment Strategies for Low-Grade Glioma in Adults. J. Oncol. Pr. 2016, 12, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Touat, M.; Maher, E.; De La Fuente, M.; Cloughesy, T.F.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Huang, R.; et al. ACTR-46. AG-120, A FIRST-IN-class Mutant IDH1 Inhibitor in Patients with Recurrent or Progressive IDH1 Mutant Glioma: Updated Results from the Phase 1 Non-Enhancing Glioma Population. Neuro-Oncology 2017, 19, vi10–vi11. [Google Scholar] [CrossRef]
- Kopinja, J.; Sevilla, R.S.; Levitan, D.; Dai, D.; Vanko, A.; Spooner, E.; Ware, C.; Forget, R.; Hu, K.; Kral, A.; et al. A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit. Sci. Rep. 2017, 7, 13853. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, H.; Machida, Y.; Nakagawa, M.; Yamaguchi, M.; Ogawara, Y.; Shima, Y.; Yamagata, K.; Katsumoto, T.; Hattori, A.; Itoh, M.; et al. Characterization of a novel BBB-permeable mutant IDH1 inhibitor, DS-1001b. Ann. Oncol. 2019, 30, v145–v146. [Google Scholar] [CrossRef]
- Natsume, A.; Wakabayashi, T.; Miyakita, Y.; Narita, Y.; Mineharu, Y.; Arakawa, Y.; Yamasaki, F.; Sugiyama, K.; Hata, N.; Muragaki, Y.; et al. Phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001b in patients with recurrent or progressive IDH1 mutant gliomas. J. Clin. Oncol. 2019, 37, 2004. [Google Scholar] [CrossRef]
- National Cancer Genome Atlas Research; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Takami, H.; Yoshida, A.; Fukushima, S.; Arita, H.; Matsushita, Y.; Nakamura, T.; Ohno, M.; Miyakita, Y.; Shibui, S.; Narita, Y.; et al. Revisiting TP53 Mutations and Immunohistochemistry-A Comparative Study in 157 Diffuse Gliomas. Brain Pathol. 2014, 25, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Binayke, A.; Mishra, S.; Suman, P.; DAS, S.; Chander, H. Awakening the “guardian of genome”: Reactivation of mutant p53. Cancer Chemother. Pharmacol. 2019, 83, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Van Meyel, D.J.; Ramsay, D.A.; Casson, A.G.; Keeney, M.; Chambers, A.F.; Cairncross, J.G. p53 Mutation, Expression, and DNA Ploidy in Evolving Gliomas: Evidence for Two Pathways of Progression. J. Natl. Cancer Inst. 1994, 86, 1011–1017. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Wang, M.; Wu, Z.; Li, N.; Zhang, J.; Yang, C. EZH2-, CHD4-, and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J. Mol. Cell Biol. 2017, 9, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Gillet, E.; Alentorn, A.; Doukouré, B.; Mundwiller, E.; Van Thuij, H.; Reijneveld, J.C.; Medina, J.A.M.; Liou, A.; Marie, Y.; Mokhtari, K.; et al. TP53 and p53 statuses and their clinical impact in diffuse low grade gliomas. J. Neuro-Oncol. 2014, 118, 131–139. [Google Scholar] [CrossRef]
- Okamoto, Y.; Di Patre, P.-L.; Burkhard, C.; Horstmann, S.; Jourde, B.; Fahey, M.; Schüler, D.; Probst-Hensch, N.M.; Yasargil, M.G.; Yonekawa, Y.; et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol. 2004, 108, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Liu, Y.; Li, C.; Zhu, J.-J.; Ye, Z.-R.; Mao, Y.; Wang, Y. Loss of heterozygosity of chromosome 1p/19q and p53 protein expression in oligodendroglioma. Chin. J. Pathol. 2009, 38, 445–450. [Google Scholar]
- Nayak, A.; Ralte, A.M.; Sharma, M.C.; Singh, V.P.; Mahapatra, A.K.; Mehta, V.S.; Sarkar, C. p53 protein alterations in adult astrocytic tumors and oligodendrogliomas. Neurol. India 2004, 52, 228–232. [Google Scholar] [PubMed]
- Hu, X.; Miao, W.; Zou, Y.; Zhang, W.; Zhang, Y.; Liu, H. Expression of p53, epidermal growth factor receptor, Ki-67 and O6-methylguanine-DNA methyltransferase in human gliomas. Oncol. Lett. 2013, 6, 130–134. [Google Scholar] [CrossRef][Green Version]
- Robles, A.; Harris, C.C. Clinical Outcomes and Correlates of TP53 Mutations and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 2, a001016. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Katayama, Y.; Yoshino, A.; Komine, C.; Yokoyama, T. Deregulation of the TP53/p14ARF tumor suppressor pathway in low-grade diffuse astrocytomas and its influence on clinical course. Clin. Cancer Res. 2003, 9, 4884–4890. [Google Scholar] [PubMed]
- Kraus, J.A.; Bolln, C.; Wolf, H.K.; Neumann, J.; Kindermann, D.; Fimmers, R.; Forster, F.; Baumann, A.; Schlegel, U. TP53 alterations and clinical outcome in low grade astrocytomas. Genes Chromosom. Cancer 1994, 10, 143–149. [Google Scholar] [CrossRef] [PubMed]
- James, C.D.; Galanis, E.; Frederick, L.; Kimmel, D.W.; Cunningham, J.M.; Atherton-Skaff, P.J.; O’Fallon, J.R.; Jenkins, R.B.; Buckner, J.C.; Hunter, S.B.; et al. Tumor suppressor gene alterations in malignant gliomas: Histopathological associations and prognostic evaluation. Int. J. Oncol. 1999, 15, 547–600. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Herndon, J.E.; Stenzel, T.T.; Raetz, J.G.M.; Kendelhardt, J.; Friedman, H.S.; Friedman, A.H.; Bigner, D.D.; Bigner, S.H.; McLendon, R.E. Molecular markers of prognosis in astrocytic tumors. Cancer 2002, 94, 2688–2697. [Google Scholar] [CrossRef]
- Arteche-López, A.; Ávila-Fernández, A.; Romero, R.; Riveiro-Álvarez, R.; López-Martínez, M.A.; Giménez-Pardo, A.; Vélez-Monsalve, C.; Gallego-Merlo, J.; García-Vara, I.; Almoguera, B.; et al. Sanger sequencing is no longer always necessary based on a single-center validation of 1109 NGS variants in 825 clinical exomes. Sci. Rep. 2021, 11, 5697. [Google Scholar] [CrossRef]
- The TCGA Legacy. Cell 2018, 173, 281–282. [CrossRef] [PubMed]
- Jonsson, P.; Lin, A.L.; Young, R.J.; DiStefano, N.M.; Hyman, D.M.; Li, B.T.; Berger, M.F.; Zehir, A.; Ladanyi, M.; Solit, D.B.; et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin. Cancer Res. 2019, 25, 5537–5547. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, T.; You, G.; Peng, X.; Chen, C.; You, Y.; Yao, K.; Wu, C.; Ma, J.; Sha, Z.; et al. Localizing seizure-susceptible brain regions associated with low-grade gliomas using voxel-based lesion-symptom mapping. Neuro-Oncology 2014, 17, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.; Qian, Z.; Sun, Z.; Xu, K.; Wang, K.; Liu, S.; Fan, X.; Li, S.; Zhang, Z.; et al. A radiomic signature as a non-invasive predictor of progression-free survival in patients with lower-grade gliomas. NeuroImage Clin. 2018, 20, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef] [PubMed]
- Badouel, C.; McNeill, H. SnapShot: The Hippo Signaling Pathway. Cell 2011, 145, 484–484.e1. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Alentorn, A.; Van Thuijl, H.F.; Marie, Y.; Alshehhi, H.; Carpentier, C.; Boisselier, B.; Laigle-Donadey, F.; Mokhtari, K.; Scheinin, I.; Wesseling, P.; et al. Clinical value of chromosome arms 19q and 11p losses in low-grade gliomas. Neuro-Oncology 2013, 16, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, A.; Pediconi, N.; Narcisi, A.; Guerrieri, F.; Belloni, L.; Fausti, F.; Botti, E.; Levrero, M. TP63andTP73in cancer, an unresolved “family” puzzle of complexity, redundancy and hierarchy. FEBS Lett. 2014, 588, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Strano, S.; Blandino, G. YAP and p73: A Matter of Mutual Specificity in Tumor Suppression. In The Hippo Signaling Pathway and Cancer; Oren, M., Aylon, Y., Eds.; Springer: New York, NY, USA, 2013; Volume 1, pp. 147–172. [Google Scholar] [CrossRef]
- Keshet, R.; Adler, J.; Ricardo-Lax, I.; Shanzer, M.; Porat, Z.; Reuven, N.; Shaul, Y. c-Abl antagonizes the YAP oncogenic function. Cell Death Differ. 2014, 22, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Anderson, K.C.; Tonon, G. Awakening the Hippo co-activator YAP1, a mercurial cancer gene, in hematologic cancers. Mol. Cell. Oncol. 2014, 1, e970055. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A Elicits Apoptosis through an MST2 Pathway Directing Proapoptotic Transcription by the p73 Tumor Suppressor Protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 Are Components of a Proapoptotic Autoregulatory Feedback Loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How the TP53 Family ProteinsTP63andTP73Contribute to Tumorigenesis: Regulators and Effectors. Hum. Mutat. 2014, 35, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Katoh, I.; Aisaki, K.-I.; Kurata, S.-I.; Ikawa, S.; Ikawa, Y. p51A (TAp63γ), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene 2000, 19, 3126–3130. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Schilling, T.; Sayan, A.E.; Kairat, A.; Lorenz, K.; Schulze-Bergkamen, H.; Oren, M.; Koch, A.; Tannapfel, A.; Stremmel, W.; et al. TAp73/ΔNp73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ. 2005, 12, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Rocco, J.W.; Leong, C.-O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.-O.; Vidnovic, N.; Deyoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Investig. 2007, 117, 1370–1380. [Google Scholar] [CrossRef]
- Yamaki, T.; Suenaga, Y.; Iuchi, T.; Alagu, J.; Takatori, A.; Itami, M.; Araki, A.; Ohira, M.; Inoue, M.; Kageyama, H.; et al. Temozolomide suppresses MYC via activation of TAp63 to inhibit progression of human glioblastoma. Sci. Rep. 2013, 3, 1160. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Kondo, K.; Marin, M.C.; Cheng, L.S.; Hahn, W.C.; Kaelin, W.G. Chemosensitivity linked to p73 function. Cancer Cell 2003, 3, 403–410. [Google Scholar] [CrossRef]
- Lunghi, P.; Costanzo, A.; Mazzera, L.; Rizzoli, V.; Levrero, M.; Bonati, A. The p53 Family Protein p73 Provides New Insights into Cancer Chemosensitivity and Targeting. Clin. Cancer Res. 2009, 15, 6495–6502. [Google Scholar] [CrossRef][Green Version]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 Function Is Inhibited by Tumor-Derived p53 Mutants in Mammalian Cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.; Marín, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.; Jacks, T.; Vousden, K.H.; et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nat. Cell Biol. 2000, 407, 645–648. [Google Scholar] [CrossRef]
- Strano, S.; Blandino, G. p73-mediated chemosensitivity: A preferential target of oncogenic mutant p53. Cell Cycle 2003, 2, 345–346. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A Subset of Tumor-Derived Mutant Forms of p53 Down-Regulate p63 and p73 through a Direct Interaction with the p53 Core Domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, D.; Gasco, M.; Hiller, L.; Sullivan, A.; Syed, N.; Trigiante, G.; Yulug, I.; Merlano, M.; Numico, G.; Comino, A.; et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 2003, 3, 387–402. [Google Scholar] [CrossRef]
- Davison, T.S.; Vagner, C.; Kaghad, M.; Ayed, A.; Caput, D.; Arrowsmith, C. p73 and p63 Are Homotetramers Capable of Weak Heterotypic Interactions with Each Other but Not with p53. J. Biol. Chem. 1999, 274, 18709–18714. [Google Scholar] [CrossRef]
- Gomez-Manzano, C.; Fueyo, J.; Kyritsis, A.P.; Steck, P.A.; Levin, V.A.; Yung, W.K.A.; McDonnell, T.J. Characterization of p53 and p21 Functional Interactions in Glioma Cells en Route to Apoptosis. J. Natl. Cancer Inst. 1997, 89, 1036–1044. [Google Scholar] [CrossRef]
- Blough, M.D.; Beauchamp, D.C.; Westgate, M.R.; Kelly, J.J.; Cairncross, J.G. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J. Neuro-Oncol. 2011, 102, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pedrote, M.M.; Motta, M.F.; Ferretti, G.D.; Norberto, D.R.; Spohr, T.C.; Lima, F.R.S.; Gratton, E.; Silva, J.L.; de Oliveira, G.A. Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers. iScience 2020, 23, 100820. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.; Guan, K.-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical Interaction with Yes-associated Protein Enhances p73 Transcriptional Activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2015, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Bam, R. Reciprocal Crosstalk Between YAP1/Hippo Pathway and the p53 Family Proteins: Mechanisms and Outcomes in Cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Zhang, C.; Liang, N.; Zhang, Z.; Chang, A.; Yin, J.; Li, Z.; Li, N.; Tan, X.; Luo, N.; et al. Yes-associated protein (YAP) increases chemosensitivity of hepatocellular carcinoma cells by modulation of p53. Cancer Biol. Ther. 2013, 14, 511–520. [Google Scholar] [CrossRef]
- Furth, N.; Aylon, Y.; Oren, M. p53 shades of Hippo. Cell Death Differ. 2018, 25, 81–92. [Google Scholar] [CrossRef]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The Transcriptional Coactivator Yes-Associated Protein Drives p73 Gene-Target Specificity in Response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Labussiere, M.; Idbaih, A.; Wang, X.-W.; Marie, Y.; Boisselier, B.; Falet, C.; Paris, S.; Laffaire, J.; Carpentier, C.; Criniere, E.; et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010, 74, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Ständer, M.; Peraud, A.; Leroch, B.; Kreth, F.W. Prognostic impact of TP53 mutation status for adult patients with supratentorial World Health Organization Grade II astrocytoma or oligoastrocytoma. Cancer 2004, 101, 1028–1035. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Nobusawa, S.; Mittelbronn, M.; Paulus, W.; Brokinkel, B.; Keyvani, K.; Sure, U.; Wrede, K.; Nakazato, Y.; Tanaka, Y.; et al. Molecular Classification of Low-Grade Diffuse Gliomas. Am. J. Pathol. 2010, 177, 2708–2714. [Google Scholar] [CrossRef] [PubMed]
- Xiu, J.; Piccioni, D.; Juarez, T.; Pingle, S.C.; Hu, J.; Rudnick, J.; Fink, K.; Spetzler, D.B.; Maney, T.; Ghazalpour, A.; et al. Multi-platform molecular profiling of a large cohort of glioblastomas reveals potential therapeutic strategies. Oncotarget 2016, 7, 21556–21569. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Ames, H.; Yuan, M.; Vizcaíno, M.A.; Yu, W.; Rodriguez, F.J. MicroRNA profiling of low-grade glial and glioneuronal tumors shows an independent role for cluster 14q32.31 member miR-487b. Mod. Pathol. 2016, 30, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.; Malta, T.; Sabedot, T.S.; Salama, S.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Jeun, S.S.; Kim, S.H.; Yoo, C.Y.; Baek, H.-M.; Yang, S.H. Metabolic profiling of human gliomas assessed with NMR. J. Clin. Neurosci. 2019, 68, 275–280. [Google Scholar] [CrossRef]
- Chen, B.; Liang, T.; Yang, P.; Wang, H.; Liu, Y.; Yang, F.; You, G. Classifying lower grade glioma cases according to whole genome gene expression. Oncotarget 2016, 7, 74031–74042. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fults, D.; Brockmeyer, D.; Tullous, M.W.; Pedone, C.A.; Cawthon, R.M. p53 mutation and loss of heterozygosity on chromosomes 17 and 10 during human astrocytoma progression. Cancer Res. 1992, 52, 674–679. [Google Scholar]
- Tsuzuki, T.; Tsunoda, S.; Sakaki, T.; Konishi, N.; Hiasa, Y.; Nakamura, M. Alterations of retinoblastoma, p53, p16(CDKN2), and p15 genes in human astrocytomas. Cancer 1996, 78, 287–293. [Google Scholar] [CrossRef]
- Thangnipon, W.; Mizoguchi, M.; Kukita, Y.; Inazuka, M.; Iwaki, T.; Fukui, M.; Hayashi, K. Distinct pattern of PCR–SSCP analysis of p53 mutations in human astrocytomas. Cancer Lett. 1999, 141, 195–201. [Google Scholar] [CrossRef]
- Watanabe, K.; Sato, K.; Biernat, W.; Tachibana, O.; Von Ammon, K.; Ogata, N.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin. Cancer Res. 1997, 3, 523–530. [Google Scholar] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.-C.; Van Meir, E.G. Frequent Co-Alterations of TP53, p16/CDKN2A, p14ARF, PTEN Tumor Suppressor Genes in Human Glioma Cell Lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef]
- Hagel, C.; Laking, G.; Laas, R.; Scheil, S.; Jung, R.; Milde-Langosch, K.; Stavrou, D. Demonstration of p53 protein and TP53 gene mutations in oligodendrogliomas. Eur. J. Cancer 1996, 32, 2242–2248. [Google Scholar] [CrossRef]
- Patt, S.; Gries, H.; Giraldo, M.; Cervos-Navarro, J.; Martin, H.; Jänisch, W.; Brockmoller, J. p53 gene mutations in human astrocytic brain tumors including pilocytic astrocytomas. Hum. Pathol. 1996, 27, 586–589. [Google Scholar] [CrossRef]
- Rasheed, B.K.A.; Wiltshire, R.N.; Bigner, S.H.; Bigner, D.D. Molecular pathogenesis of malignant gliomas. Curr. Opin. Oncol. 1999, 11, 162–167. [Google Scholar] [CrossRef]
- Weber, R.; Sabel, M.; Reifenberger, J.; Sommer, C.; Oberstrass, J.; Reifenberger, G.; Kiessling, M.; Cremer, T. Characterization of genomic alterations associated with glioma progression by comparative genomic hybridization. Oncogene 1996, 13, 983–994. [Google Scholar] [PubMed]
- Kraus, J.A.; Koopmann, J.; Kaskel, P.; Maintz, D.; Brandner, S.; Schramm, J.; Louis, D.N.; Wiestler, O.D.; von Deimling, A. Shared Allelic Losses on Chromosomes 1p and 19q Suggest a Common Origin of Oligodendroglioma and Oligoastrocytoma. J. Neuropathol. Exp. Neurol. 1995, 54, 91–95. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Gerges, N.; Korshunov, A.; Sabha, N.; Khuong-Quang, D.-A.; Fontebasso, A.M.; Fleming, A.; Hadjadj, D.; Schwartzentruber, J.; Majewski, J.; et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012, 124, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Hakin-Smith, V.; Teo, M.; Xinarianos, G.E.; Jellinek, D.A.; Carroll, T.; McDowell, D.; Macfarlane, M.R.; Boet, R.; Baguley, B.C.; et al. Association of Mutant TP53 with Alternative Lengthening of Telomeres and Favorable Prognosis in Glioma. Cancer Res. 2006, 66, 6473–6476. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.-Y.; Ma, J.; Wang, J.-F.; Li, S.-W.; Jiang, T.; Dai, J.-P. Prognostic value of MGMT promoter methylation and TP53 mutation in glioblastomas depends on IDH1 mutation. Asian Pac. J. Cancer Prev. 2015, 15, 10893–10898. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K.; Liebner, D.; Chen, J.L. TP53 mutational status is predictive of pazopanib response in advanced sarcomas. Ann. Oncol. 2016, 27, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Li, V.D.; Li, K.H.; Li, J.T. TP53 mutations as potential prognostic markers for specific cancers: Analysis of data from The Cancer Genome Atlas and the International Agency for Research on Cancer TP53 Database. J. Cancer Res. Clin. Oncol. 2018, 145, 625–636. [Google Scholar] [CrossRef]
- Freier, C.P.; Stiasny, A.; Kuhn, C.; Mayr, D.; Alexiou, C.; Janko, C.; Wiest, I.; Jeschke, U.; Kost, B. Immunohistochemical Evaluation of the Role of p53 Mutation in Cervical Cancer: Ser-20 p53-Mutant Correlates with Better Prognosis. Anticancer. Res. 2016, 36, 3131–3137. [Google Scholar] [PubMed]
- Groenendijk, F.H.; Taal, W.; Dubbink, H.J.; Haarloo, C.R.; Kouwenhoven, M.C.; Bent, M.J.V.D.; Kros, J.M.; Dinjens, W.N.M. MGMT promoter hypermethylation is a frequent, early, and consistent event in astrocytoma progression, and not correlated with TP53 mutation. J. Neuro-Oncol. 2011, 101, 405–417. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ono, Y.; Tamiya, T.; Ichikawa, T.; Matsumoto, K.; Furuta, T.; Ohmoto, T.; Akiyama, K.; Seki, S.; Ueki, K.; Louis, D.N. Accumulation of wild-type p53 in astrocytomas is associated with increased p21 expression. Acta Neuropathol. 1997, 94, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Mellai, M.; Piazzi, A.; Caldera, V.; Monzeglio, O.; Cassoni, P.; Valente, G.; Schiffer, D. IDH1 and IDH2 mutations, immunohistochemistry and associations in a series of brain tumors. J. Neuro-Oncol. 2011, 105, 345–357. [Google Scholar] [CrossRef]
- Peraud, A.; Kreth, F.W.; Wiestler, O.D.; Kleihues, P.; Reulen, H.-J. Prognostic impact of TP53 mutations and P53 protein overexpression in supratentorial WHO grade II astrocytomas and oligoastrocytomas. Clin. Cancer Res. 2002, 8, 1117–1124. [Google Scholar] [PubMed]
- Sarma, P.P.; Dutta, D.; Mirza, Z.; Saikia, K.K.; Baishya, B.K. Point mutations in the DNA binding domain of p53 contribute to glioma progression and poor prognosis. Mol. Biol. 2017, 51, 334–341. [Google Scholar] [CrossRef]
- Hartmann, C.; Hentschel, B.; Tatagiba, M.; Schramm, J.; Schnell, O.; Seidel, C.; Stein, R.; Reifenberger, G.; Pietsch, T.; von Deimling, A.; et al. Molecular Markers in Low-Grade Gliomas: Predictive or Prognostic? Clin. Cancer Res. 2011, 17, 4588–4599. [Google Scholar] [CrossRef] [PubMed]
- Guichet, P.-O.; Masliantsev, K.; Tachon, G.; Petropoulos, C.; Godet, J.; Larrieu-Ciron, D.; Milin, S.; Wager, M.; Karayan-Tapon, L. Fatal correlation between YAP1 expression and glioma aggressiveness: Clinical and molecular evidence. J. Pathol. 2018, 246, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Katayama, Y.; Komine, C.; Yoshino, A.; Ogino, A.; Ohta, T.; Fukushima, T. O6-methylguanine-DNA methyltransferase methylation and TP53 mutation in malignant astrocytomas and their relationships with clinical course. Int. J. Cancer 2004, 113, 581–587. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at diagnosis, median ± SD (years) | 36 ± 10 | |
| Median follow-up (years, 95% CI) | 9.0 (7.9–10.1) | |
| Gender, n (%) | F | 53 (51.9%) |
| M | 49 (48.0%) | |
| Diagnosis, n (%) | Astrocytoma grade II | 32 (31.1%) |
| Astrocytoma grade III | 19 (18.4%) | |
| Oligodendroglioma grade II | 26 (26.2%) | |
| Oligodendroglioma grade III | 25 (24.3%) | |
| IDH1 mutation, n (%) | Wild-type | 17 (17%) |
| Mutant | 85 (83%) | |
| Anatomical Location of Tumour, n (%) | Frontal | 45 (44.1%) |
| Insular | 6 (5.9%) | |
| Motor Cortex | 1 (1.0%) | |
| Parietal | 13 (12.7%) | |
| Temporal | 22 (21.6%) | |
| Unknown | 15 (14.7%) | |
| Treatment, n (%) | Surgery and chemoradiation | 28 (27.5%) |
| Surgery and chemotherapy | 15 (14.7%) | |
| Surgery and radiotherapy | 11 (10.8%) | |
| Surgery only | 27 (26.5%) | |
| Unknown adjuvant therapy | 21 (20.5%) |
| Mutation Syntax | Amino Acid Change | Frequency | Type of Mutation | Ref SNP ID | Outcome |
|---|---|---|---|---|---|
| c.215C>G | p.P72R | 24 | Missense | rs1042522 | Neutral |
| c.321C>G | p.Y107 * | 1 | Nonsense | rs770776262 | Neutral |
| c.377A>G | p.Y126C | 2 | Missense | rs1555526335 | Pathogenic |
| c.469_470delGT | 1 | Frameshift Deletion | Novel mutation | Pathogenic | |
| c.473G>A | p.R158H | 1 | Missense | rs587782144 | Pathogenic |
| c.476C>A | p.A159D | 1 | Missense | unavailable | Pathogenic |
| c.480G>A | p.M160I | 1 | Missense | rs193920817 | Pathogenic |
| c.488A>G | p.Y163C | 1 | Missense | rs148924904 | Pathogenic |
| c.517G>C | p.V173L | 1 | Missense | rs876660754 | Pathogenic |
| c.524G>C | p.R175P | 2 | Missense | rs28934578 | Pathogenic |
| c.526T>G | p.C176G | 1 | Missense | rs967461896 | Pathogenic |
| c.537T>A | p.H179Q | 1 | Missense | rs876660821 | Neutral |
| c.581T>G | p.L194R | 1 | Missense | rs1057519998 | Pathogenic |
| c.639A>G | p.R213R | 3 | Silent | rs1800372 | Neutral |
| c.643A>G | p.S215G | 1 | Missense | rs886039484 | Pathogenic |
| c.659A>G | p.Y220C | 2 | Missense | rs121912666 | Pathogenic |
| c.700T>C | p.Y234H | 1 | Missense | rs864622237 | Pathogenic |
| c.711G>A | p.M237I | 1 | Missense | rs587782664 | Pathogenic |
| c.734G>A | p.G245D | 1 | Missense | rs121912656 | Pathogenic |
| c.742C>T | p.R248W | 1 | Missense | rs121912651 | Pathogenic |
| c.743G>A | p.R248Q | 1 | Missense | rs11540652 | Pathogenic |
| c.797G>A | p.G266E | 1 | Missense | rs193920774 | Pathogenic |
| c.817C>T | p.R273C | 15 | Missense | rs121913343 | Pathogenic |
| c.818G>A | p.R273H | 3 | Missense | rs28934576 | Pathogenic |
| c.823T>G | p.C275G | 1 | Missense | rs1029688274 | Pathogenic |
| c.861G>A | p.E287E | 1 | Silent | rs748891343 | Neutral |
| TP53 Mutation Status | p-Value | |||
|---|---|---|---|---|
| Wild-Type | Mutant | |||
| n (%) | n (%) | |||
| Age at diagnosis, mean ± SD 1, (years) | 38 ± 11 | 34 ± 9 | 0.046 | |
| Gender | F | 32 (52%) | 20 (50%) | 0.694 |
| M | 29 (48%) | 20 (50%) | ||
| Diagnosis | Astrocytoma II | 8 (13%) | 24 (60%) | <0.001 |
| Astrocytoma III | 12 (19%) | 7 (18%) | ||
| Oligodendroglioma II | 21 (34%) | 5 (12%) | ||
| Oligodendroglioma III | 20 (34%) | 4 (10%) | ||
| IDH1 mutation status | Wild-type | 14 (23%) | 2 (5%) | 0.009 |
| Mutant | 47 (77%) | 37 (95%) | ||
| 1p/19q co-deletion status | Co-deleted | 22 (36%) | 0 (0%) | <0.001 |
| Non-co-deleted | 9 (15%) | 18 (45%) | ||
| Unknown | 30 (49%) | 22 (55%) | ||
| p53 expression level 2 | Unknown | 22 (36%) | 17 (43%) | <0.001 |
| Low | 25 (41%) | 1 (1%) | ||
| High | 14 (23%) | 22 (55%) | ||
| Treatment | Surgery and chemoradiation | 16 (26%) | 12 (30%) | 0.016 |
| Surgery and chemotherapy | 10 (16%) | 5 (13%) | ||
| Surgery and radiotherapy | 8 (13%) | 3 (7%) | ||
| Surgery only Unknown | 21 (35%) | 6 (15%) | ||
| 6 (10%) | 14 (35%) | |||
| Overall Survival | ||||
|---|---|---|---|---|
| Univariate Analysis | Multivariate Analysis | |||
| TP53 Mutation | Hazard Ratio (95% CI) | p-Value | Hazard Ratio (95% CI) | p-Value |
| TP53 combined vs. WT | 0.331 (0.138–0.796) | 0.014 | 0.392 (0.132–1.163) | 0.091 |
| TP53 codon 273 vs. other | 0.466 (0.172–1.262) | 0.133 | 0.175 (0.04–0.759) | 0.02 |
| TP53 codon 273 vs. WT | 0.232 (0.076–0.711) | 0.011 | 0.169 (0.036–0.766) | 0.021 |
| Other vs. WT | 0.500 (0.150–1.590) | 0.241 | 1.042 (0.278–3.906) | 0.951 |
| Dataset | TP53 Mutation | Hazard Ratio (95% CI) | p-Value |
|---|---|---|---|
| TCGA OS | TP53 combined vs. WT | 0.199 (0.113–0.350) | <0.0001 |
| Codon 273 vs. WT | 0.182 (0.0069–0.479) | <0.001 | |
| Other vs. WT | 0.210 (0.116–0.379) | <0.0001 | |
| TCGA PFS | TP53 combined vs. WT | 0.336 (0.218–0.517) | <0.0002 |
| Codon 273 vs. WT | 0.260 (0.125–0.539) | <0.0001 | |
| Other vs. WT | 0.360 (0.229–0.567) | <0.0001 | |
| MSKCC OS | Codon 273 vs. WT | 0.124 (0.053–0.289) | <0.0001 |
| Other vs. WT | 0.231 (0.136–0.392) | <0.0001 | |
| MSKCC PFS | Codon 273 vs. WT | 0.154 (0.83–0.286) | <0.0001 |
| Other vs. WT | 0.187 (0.117–0.297) | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noor, H.; Briggs, N.E.; McDonald, K.L.; Holst, J.; Vittorio, O. TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers 2021, 13, 5362. https://doi.org/10.3390/cancers13215362
Noor H, Briggs NE, McDonald KL, Holst J, Vittorio O. TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers. 2021; 13(21):5362. https://doi.org/10.3390/cancers13215362
Chicago/Turabian StyleNoor, Humaira, Nancy E. Briggs, Kerrie L. McDonald, Jeff Holst, and Orazio Vittorio. 2021. "TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy" Cancers 13, no. 21: 5362. https://doi.org/10.3390/cancers13215362
APA StyleNoor, H., Briggs, N. E., McDonald, K. L., Holst, J., & Vittorio, O. (2021). TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers, 13(21), 5362. https://doi.org/10.3390/cancers13215362