Simple Summary
Recent evidence indicates that opioids can be active at a receptor that is abundantly expressed on innate immune cells as well as cancer cells: the receptor is termed toll-like receptor 4 (TLR4). TLR4 is increasingly recognised as playing key roles in tumour biology and anticancer defences. However, the issue of whether TLR4 mediates some of the effects of opioids on tumour growth and metastasis is entirely unknown. We review existing evidence, mechanisms, and functional consequences of the action of opioids at TLR4. This opens new avenues of research on the role of opioids in cancer.
Abstract
The innate immune receptor toll-like receptor 4 (TLR4) is known as a sensor for the gram-negative bacterial cell wall component lipopolysaccharide (LPS). TLR4 activation leads to a strong pro-inflammatory response in macrophages; however, it is also recognised to play a key role in cancer. Recent studies of the opioid receptor (OR)-independent actions of opioids have identified that TLR4 can respond to opioids. Opioids are reported to weakly activate TLR4, but to significantly inhibit LPS-induced TLR4 activation. The action of opioids at TLR4 is suggested to be non-stereoselective, this is because OR-inactive (+)-isomers of opioids have been shown to activate or to inhibit TLR4 signalling, although there is some controversy in the literature. While some opioids can bind to the lipopolysaccharide (LPS)-binding cleft of the Myeloid Differentiation factor 2 (MD-2) co-receptor, pharmacological characterisation of the inhibition of opioids on LPS activation of TLR4 indicates a noncompetitive mechanism. In addition to a direct interaction at the receptor, opioids affect NF-κB activation downstream of both TLR4 and opioid receptors and modulate TLR4 expression, leading to a range of in vivo outcomes. Here, we review the literature reporting the activity of opioids at TLR4, its proposed mechanism(s), and the complex functional consequences of this interaction.
1. Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors first identified in Drosophila, and to date, ten members of the TLR family are recognised in humans [1]. One of the most widely studied TLRs is TLR4, a membrane-bound receptor that has an extracellular leucine-rich repeat domain, a transmembrane domain, and a cytoplasmic toll/interleukin-1 receptor (TIR) domain [2]. Among other pattern recognition receptors, TLR4 plays a role in the innate immune system by recognising various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), and by triggering host defences to produce inflammatory cytokines, eliminating the cause of danger [3]. The activation of TLR4 by natural ligand lipopolysaccharide (LPS)—a PAMP contained in gram-negative bacteria cell membranes—requires the involvement of the following accessory proteins: lipopolysaccharide binding protein (LBP), the cluster of differentiation antigen 14 (CD14), and myeloid differentiation protein 2 (MD-2) [4] (Figure 1). Signal transduction is initiated by the binding of LPS to LBP, which traffics it to CD14. CD14 then binds to the cell and delivers LPS to the TLR4-associated MD-2. The detection of LPS by TLR4 causes the dimerisation of the TLR4 cytoplasmic TIR domain, which can then recruit different sets of adaptor proteins and initiate two downstream signalling pathways: the MyD88- and TRIF/TRAM-dependent pathways. Triggering of the MyD88-dependent signalling pathway activates the transcription factors NF-κB, IRF5, AP-1, and CREB, and the subsequent production of pro-inflammatory cytokines. The endocytosis of the TLR4 complex, however, recruits the TRIF/TRAM pathway, resulting in the activation of IRF-3 and the production of type I interferons, or, alternatively, the activation of NF-κB via RIP1 kinase interactions with TRAF6 and the production of cytokines [5,6].
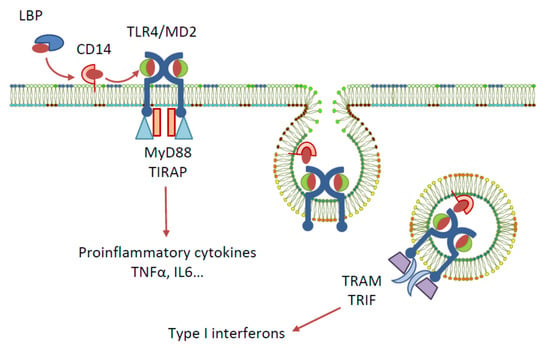
Figure 1.
Signalling pathways elicited by LPS activation of TLR4.
TLR4 is increasingly recognized as playing important roles in cancer (reviewed in [7,8]). It is abundantly expressed on tumoural immune and non-immune stromal cells and is often overexpressed by tumour cells [7,9]. TLR4 activation of cancer cells is mostly reported to promote aggressive behaviour of cancer cells, including epithelial to mesenchymal transition, migration, and invasion [10,11,12,13,14]; furthermore, TLR4 overexpression has been correlated with increased metastasis (reviewed in [8]). TLR4 activation in the tumour microenvironment further maintains a tumour-favourable inflammatory response [15] and DAMPs expressed by cancer cells can promote angiogenesis [16]. A major endogenous TLR4 agonist, that is relevant to cancer, is the DAMP high-mobility group box 1 (HMGB1), which possesses protumour characteristics through sustaining an anti-inflammatory environment and promoting invasion metastasis and angiogenesis [15]. HMGB1 that is produced by tumour cells interacts with TLR4 on platelets, causing their activation, adhesion, and release of pro-metastatic factors, resulting in metastasis in mice [17]. On the other hand, it has also been documented that TLR4 activation on immune cells is protective in the context of cancer [18] and is necessary for the efficacy of chemotherapy or radiotherapy [19]. Treatment with a synthetic TLR4 agonist increased innate and adaptive immunity and led to reduced metastasis in several rodent models [20]. An elegant study has dissected out a pathway—essential in the study of anti-cancer immunity in mice and humans—whereby HMGB1 that is secreted by dying tumour cells activates TLR4 on dendritic cells. Functional TLR4, and the adaptor MyD88, are required for dendritic cells to cross-present antigens from the dying tumour cells and activate tumour-specific T-cell immune response [19]. TLR4 therefore plays a dual role in cancer.
Contemporary literature indicates that opioids can be active at TLR4; however, whether this contributes to the action of opioids on tumour growth and metastasis is, to date, entirely unexplored. We reviewed existing evidence, mechanisms, and functional consequences of the action of opioids at TLR4.
2. Opioids Inhibit LPS-Induced Activation
Evidence of a possible in vitro connection between opioids and TLR4 originates from studies that examined the effects of LPS, the classical TLR4 agonist, on cultured primary brain cells, as well as the ability of opioids to inhibit these effects [21,22,23,24]. Das et al. reported a concentration-dependent increase in the secretion of IL-lβ, upon treating the mixed brain cell cultures of embryonic mice with either LPS or with the endogenous opioid peptide [Met 5]-enkephalin [21]. These effects were partially inhibited by naloxone (10−9 M–10−6 M) in a concentration-dependent fashion. Additionally, LPS induced morphological changes to the microglia in the brain cultures, which were blocked upon prior treatment with naloxone. Similar findings were reported by Kong et al., where the LPS-induced pro-inflammatory effects in mouse primary mixed glial cultures were significantly inhibited by naloxone, as well as by the endogenous opioid peptide dynorphin (dyn) A-(1–8) [22]. Naloxone (10−10–10−6 M) caused a concentration-dependent inhibition of LPS effects, reaching maxima of 55% and 21% inhibitions of NO and TNF-α production, respectively. The suppression of LPS effects by ultra-low dynorphin concentrations (10−16–10−12 M) was also observed, resulting in inhibitions of up to 29%, 39%, 32%, and 25% of the LPS-induced secretions of NO, TNF-α, IL-1α, and IL-6, respectively. Naloxone was later shown to prevent the effects of LPS in BV-2 microglial cells [25]. Further studies employed immune cells to evaluate the effect of opioids on LPS-induced activation. At very high concentrations (1 mM), morphine was reported to inhibit LPS-induced lymphocyte proliferation in a naloxone-insensitive fashion; however, this study did not demonstrate whether the cells were still viable at such concentrations [26]. Also at elevated concentrations (10−5–10−6 M), deltorphin-Dvariant—the δ2-specific opioid receptor agonist—was documented to suppress LPS-induced MAPK activation and the expression of TNF and MIP-2 in RAW264.7 macrophages [27]. Remifentanil (but not the structurally related compounds fentanyl, sufentanil, or alfentanil) was able to attenuate LPS-induced activation of neutrophils in vitro [28]. In this study, however, the effect of remifentanil was reversed by a κ-opioid receptor antagonist. Interestingly, the abilities of mM concentrations of morphine, tramadol, or ketobemidone to prevent the LPS-induced release of TNF-α and IL-8 by U-937 cells in vitro was facilitated, rather than countered, by naloxone [29]. In vitro studies of opioids modifying the effects of LPS are summarised in Table 1. It is noteworthy that opioids with heterogenous structural features can interfere with LPS-induced activation, which may indicate a variety of levels of action. Moreover, inhibition of the effects of LPS could not be correlated with opioid receptor subtype, since μ (e.g., ketobemidone, fentanyl), κ (e.g., salvinorin A, U50488), δ (e.g., deltorphine, TAN-67)-selective agonists, or non-selective agonists (e.g., oxycodone) all inhibited the effects of LPS. The observation that opioid agonists, antagonists, or endogenous opioids prevented the effects of LPS was verified in a number of in vivo models; however, the in vivo setting could denote an indirect effect, and this was not interpreted to mean that opioids interfere directly with TLR4 activation. For example:
- ○
- Morphine (1 mg/L) prevented waterborne LPS-induced signalling in zebrafish embryos [30], although lower concentrations (100 ng/L or 100 μg/L) had the opposite effect and exacerbated LPS-induced inflammation;
- ○
- Morphine prevented LPS-induced synovial inflammation when both were injected intra-articularly in horses [31];
- ○
- Naltrexone prevented iNOS induction in splenocytes, and NO production in the circulation of rats injected intraperitoneally with LPS [32], although the possibility that this effect might be medicated via TLR4 activity was not evoked—instead, the involvement of CNS endogenous opioids was suggested, since the peripheral opioid antagonist N-methyl naltrexone was ineffective in counteracting LPS unless injected intrathecally;
- ○
- Endomorphin-1, administered intravenously, was protective in a mouse model of lung injury and inflammation following intratracheal instillation of LPS [33];
- ○
- The μ opioid receptor antagonist β-funaltrexamine could prevent LPS-induced neuroinflammation in mice [34].
When viewed in combination, these results indicate that both opioid agonists and antagonists can prevent the effects of LPS in cultured cells or in live animals.

Table 1.
In vitro studies testing the effect of opioids on LPS-induced activation.
Table 1.
In vitro studies testing the effect of opioids on LPS-induced activation.
| Cells | Opioid Agent | Concentration | Readout of LPS-Induced TLR4 Activation | Impact of Opioid Agent on LPS Effect | Reference |
|---|---|---|---|---|---|
| Rat splenocytes | α- and β-endorphin | 3.3–330 nM | Splenocyte proliferation | No effect | [4] |
| Mouse embryonic mixed brain cell cultures | Naloxone | 10−9 M–10−6 M | Increase in production of IL-lβ Morphological changes to the microglia | Concentration-dependent inhibition | [21] |
| Mouse primary mixed glial cultures | Naloxone Dynorphin (dyn) A-(1–8) | 10−10–10−6 M 10−16–10−12 M | Production of NO, TNF-α, IL-1α, and IL-6. | Inhibition | [22] |
| Rat mesencephalic neuron–glia cultures Microglia-enriched cultures | (+) and (−)-Naloxone | 1 µM | Microglial activation (nitrite accumulation, rise in TNF-α and IL-1β levels) Inflammatory damage (reduction in high affinity dopamine uptake and decrease in number of healthy neurons) Superoxide generation | Significant reduction | [23] |
| Mouse primary cortical neuron–glia co-cultures | (+) and (−)-Naloxone | 1 µM | Production of TNF-α and NO Inflammatory damage (Morphological changes, decrease in number of healthy neurons and increased release of LDH) | Significant attenuation | [24] |
| RAW264.7 mouse macrophages | δ2-specific agonist Deltorphin Dvariant | 10−5 to 10−6 M | TNFα and MIP-2 production | Significant attenuation via suppression of p38 MAPK activation | [27] |
| Human dendritic cells | Morphine | 10−8 to 10−12 M | Dendritic cell maturation (upregulation of HLA-DR, CD86 CD80 CD83) | Augmentation | [35] |
| Whole blood | Remifentanil Fentanyl | 1–100 ng/mL 2–200 ng/mL | Cytokine release: TNFα, IL-10 IL-6 | Inhibition | [36] |
| BV2 microglial cells | Naloxone | 1 μM | Expression of HSP60, HSF-1, NF-κB, iNOS, TNF-α, IL-1β, IL-6, and nitric oxide | Significant inhibition | [37] |
| HEK-Blue™ hTLR4 cells | (+) and (−)-Naloxone (+) and (−)-Naltrexone | 10 μM 10 μM | Iincrease in SEAP expression | Non-stereoselective, noncompetitive inhibition | [38] |
| Rat microglial cells (HAPI) | (+)-Naloxone | 1 μM | Rrelease of IL-1, CD11b, and IL-6 mRNA | Significant attenuation | |
| RAW264.7 mouse macrophages | (+) and (−)-Naloxone | Cytosol to cell membrane clearance of the green fluorescent protein-tagged Akt1 | Significant attenuation | [39] | |
| HEK-Blue™ hTLR4 cells | Morphine Fentanyl Naltrexone β-FNA | 3–100 µM 1–100 µM 30–1000 µM 3–30 µM | Increase in SEAP expression | Significant inhibition | [40] |
| Bone marrow derived mast cells | Morphine | 0.1 μM–1 mM | TNF exocytosis | Inhibition | [41] |
| U-937 human monocytic cells | Morphine Fentanyl Tramadol Ketobemidone | 0.0015–1.5 mM 0.002–2.3 μM 0.0042–4.2 mM 0.00175–1.75 mM | Increase in TNF-α and IL-8 | High concentration of morphine tramadol and ketobemidone prevents TNF-α, high concentration of tramadol and ketobemidone prevents IL-8 induction | [29] |
| Human neutrophils | Remifentanil Alfentanil Sufentanil Fentanyl | 1–100 ng/mL 10–1000 ng/mL 1–100 ng/mL 1–100 ng/mL | Increase in TNF-α, IL-6 and MAPK activation | Only remifentanil attenuated the effects of LPS | [28] |
| HEK-Blue™ hTLR4 cells | (+) and (−)-Naloxone (+) and (−)-Naltrexone | 0.01–100 μM | Increase in SEAP expression | No effect | [42] |
| BV-2 microglial cells | (+)-Naltrexone | 50–400 µM | NO production | Concentration-dependent inhibition | [43] |
| Bone marrow derived macrophages | Morphine | 1 μM | Autophagy LPS-induced autophagolysosomal fusion | Facilitation Inhibition | [44] |
| Mouse primary microglial cells | Morphine | 100 nm and 10 μM | NF-κB signalling Unstimulated NF-κB signalling | Potentiation No effect | [45] |
| THP-1 human macrophage cell line | Dynorphin N-terminal fragments | 10 nM | Nuclear factor-κB translocation, IL-1β and TNF-α release | Reduction | [46] |
| Wistar rat primary microglial cultures | Biphalin | 0.1, 1, 10, 20 μM 10 μM | NO production Increase of Iba1, phosphorylation of nuclear factor-κB, iNOS, IL-1β, IL-18, COX-2, NLRP3, IL-6, IL-10, TNF-α, and TRIF | Significant attenuation (except for 20 μM Biphalin) Significant attenuation | [47] |
| BV-2 mouse microglial cells | Naltrexone and Naloxone (+)-Naltrexone and (+)-Naloxone (+)-Naltrexone and (+)-Naloxone (+)-Naltrexone and (+)-Naloxone | 1–400 μM 10–400 μM 20–100 μM 5–200 μM | NO production ROS production and phagocytosis TNF-α production IL-1β production | Non-stereoselective concentration-dependent inhibition Concentration-dependent inhibition Concentration-dependent inhibition No effect | [48] |
| Rat primary neonatal microglia and rat primary peritoneal macrophages | (+)-Naltrexone and (+)-Naloxone (+)-Naltrexone and (+)-Naloxone | 1–400 μM 100–400 μM | NO production TNF-α production | Concentration-dependent inhibition Concentration-dependent inhibition | |
| HEK-Blue™ hTLR4 cells | Morphine M3G | 1–100 µM 1–100 µM | Increase in SEAP expression | Significant inhibition | [49] |
| Mouse primary cortical astrocytes | Naloxone Nalmefene Naloxone Nalmefene | 5–300 µM 0.01–1 µM 150 µM 0.1 µM | Superoxide generation NF-κB activation and iNOS production | Concentration-dependent inhibition Significant inhibition | [50] |
| Peripheral blood mononuclear cells (PBMC) and isolated CD14+ monocytes from PBMC | Naltrexone | 1–200 μM | Production of IL-6 and TNF-α | No effect | [51] |
| THP-1 human macrophage cell line | Dynorphin 3–14 | 10 nM and 1 µM 10−17–10−11 M 10−11–10−7 M | NF-κB/p65 nuclear translocation Release of IL-1β Release of TNF-α | Significant inhibition Significant inhibition Significant augmentation | [52] |
| HEK-Blue™ hTLR4 cells | Dynorphin 3–14 | 10−8–10−5 M | Increase in SEAP expression | Concentration dependent inhibition | |
| H9C2 cardiomyocytes | Remifentanil | 2.5 μM | Cell death, LDH release, MDA production, PKCβ activation, autophagy, and decrease SOD activity | Attenuation | [53] |
| Human aortic endothelial cells | Remifentanil | 2.5 μM | Superoxide production iNOS ICAM-1 PARP1 expression, NF-κb activation | Attenuation | [54] |
| Primary hippocampal astrocytes | Oxycodone | 5–20 μg/mL | NF-κb activation and expression of TNF-α, IL-6, and IL-1β | Inhibition | [55] |
| NR8383 Macrophages | κOR agonists salvinorin A and U50488 | 10−9–10−11 M | Nitrosative stress, TNF-α, IL-1β, iNOS, and COX2 expression | Inhibition | [56] |
| RAW264.7 murine macrophages | Naltrexone | 5 μM | IL-1β, IL-6, and mcp-1 and ERK1/2 phosphorylation | Inhibition | [57] |
| Bovine endometrial cells | β-endorphin | 20–200 pg/mL | NF-κb activation and expression of TNF-α, IL-6, and IL-1β iNOS | Inhibition | [58] |
| Human pulmonary microvascular endothelial cells | Oxycodone | 23 μM | Permeability, apoptosis, TNF-α, IL-1β, and MMP9 caspase-3 expression | Inhibition | [59] |
| BV2 microglial cells | TAN-67 δ-opioid agonist | 3–10 μM | Cell death, IL-1β IL-6 and iNOS expression, MAPK singnalling | Inhibition | [60] |
| C6 glial cells | ((D-Arg2, Lys4)-Dermorphin-(1–4)-amide) DALDA | 10–20 μM | Nitrosative stress | Inhibition | [61] |
| Mouse bone marrow-derived macrophages | Butorphanol | 8 μM | Expression levels of iNOS, IL-6, TNF-a, and IL-1b in M1-polarized BMDMs | Inhibition | [62] |
3. Opioid Receptor Active and Inactive Isomers Both Inhibit LPS-Induced Activation
The non-stereoselective effect of opioids at TLR4 refers to the ability of (+) opioid isomers to interact with TLR4, while opioid receptors are selective for (−) isomers. The inhibitory effects of naloxone isomers on microglial activation, and the inflammatory damage of dopaminergic neurons induced by LPS, have been reported [23]. Treatment of rat mesencephalic neuron–glia cultures with LPS (0.1–100 ng/mL) induced microglial activation, as evidenced by nitrite accumulation and a rise in levels of the pro-inflammatory cytokines TNF-α and IL-1β, and resulted in inflammatory damage, as reflected by a reduction in high affinity dopamine uptake and a decrease in the number of healthy neurons. These effects were significantly reduced upon pre-treatment by 1 µM (−)- or (+)-naloxone, with equal potencies for both stereoisomers. Furthermore, naloxone caused a significant non-stereospecific, concentration-dependent reduction of the superoxide generation that was induced by LPS in mixed neuron–glia cultures and microglia-enriched cultures and was also proven to interfere with LPS binding to cell surface receptors. The same group reported similar findings in mouse primary cortical neuron–glia co-cultures [24], and the results were further confirmed by a preclinical study, where both (−) and (+)-naloxone inhibited the activation of microglia after LPS injection in the substantia nigra of rats [63]. However, toll-like receptors were not directly mentioned in this report, since opioid receptors only respond to (−)-opioid stereoisomers, the involvement of non-opioid receptors was proposed based on the similarities in inhibition observed for both (−) and (+)-opioid isomers in a number of studies (Table 1). More recently, (+)-naloxone was shown to prevent the effects of LPS in vivo in a model of inflammation-induced pre-term birth in mice [64].
4. Opioids Exert Agonistic and Antagonistic Effects at TLR4
The atypical, non-stereoselective effects of opioids continued to be unexplained until a link to TLR4 was proposed, with several reports on the effects of both opioid agonists and antagonists using different cell types, as well as in TLR4-specific reporter cell lines. Hutchinson et al. were the first group to report the blockade of LPS-induced effects by opioid antagonists in HEK-Blue™ hTLR4 cells and in rat microglial cells (HAPI) [38]. HEK-Blue™ hTLR4 cells are engineered human embryonic kidney 293 (HEK 293) cells. These cells are stably transfected to overexpress human TLR4, its accessory proteins MD-2 and CD14, and a nuclear factor kappa B (NF-κB)-inducible reporter gene (namely, secreted embryonic alkaline phosphatase, SEAP) [65]. LPS (0.01–100 ng/mL) increased SEAP expression in HEK-Blue™ hTLR4 cells, and this was significantly inhibited by 10 μM naloxone or naltrexone. The antagonism of TLR4 activation by naloxone and naltrexone was non-stereoselective and, unlike the competitive antagonistic effect of lipopolysaccharide from the photosynthetic bacterium Rhodobacter sphaeroides (LPS-RS), the effect of opioids was non-competitive in nature as indicated by the inhibition curves (i.e., the magnitude of the maximum attained response to LPS was reduced) [38]. Similar observations were reported in the same study, using the rat microglia cell line HAPI, where 1 μM (+)-naloxone significantly attenuated the LPS-induced activation of microglia, as measured by expression of IL-1, CD11b, and IL-6 mRNA. These results implied the blockade of TLR4 function by opioid antagonists, which could occur at the receptor level or at other downstream signalling sites.
Subsequently, RAW264.7 mouse macrophages were employed in a study to examine opioid effects on TLR4 signalling [39]. The activation of TLR4 triggers three main intracellular pathways, NF-κB, MAPK, and PI3k/Akt1. While NF-κB and MAPK are associated with the induction of pro-inflammatory responses, PI3k is involved in cell survival and motility. RAW264.7 cells expressing a green fluorescent protein (GFP)-tagged Akt1, show a diffuse green fluorescence in the cytosol under basal conditions, which is rapidly translocated to the plasma membrane upon activation. Robust and rapid cytosol-to-cell membrane clearance of the GFP-tagged Akt1 was induced by LPS; this effect was significantly attenuated upon pre-incubation with LPS-RS, (+)-naloxone, or (−)-naloxone. However, LPS-RS, (+)-naloxone, and (−)-naloxone did not prevent cytosol-to-cell membrane clearance when Akt1 was activated via a TLR4-independent pathway. These observations strongly linked the naloxone blocking effects to its interference with the TLR4 signalling pathway [39], either at the LPS binding level or through the TLR4 recruitment of adapters TRIF/TRAM.
In addition to the inhibition of LPS-induced signalling, a separate study showed that 200 µM (+)- and (−)-morphine, per se, produced a significant, prolonged GFP-Akt1 cytosolic clearance, suggesting that opioids can activate TLR4 signalling [39]. This effect was significantly inhibited by LPS-RS (200 ng/mL), (+)-naloxone (200 µM), and (−)-naloxone (200 µM). The ability of opioid agonists to activate TLR4 was confirmed using HEK-Blue™ hTLR4 cells, where (+)-morphine, (−)-morphine, and the opioid receptor-inactive metabolite morphine-3-glucuronide (M3G) induced significant activation. The effect of M3G was significantly stronger than that induced by an equimolar concentration of morphine, whereas the opioid receptor-active metabolite, morphine-6-glucuronide (M6G), did not show any activation potential. Several opioid analgesics were also examined, including (+)-methadone, (−)-methadone, levorphanol, pethidine, buprenorphine, fentanyl, and oxycodone (10–100 µM); all were found to produce significant TLR4 activation in HEK293 cells. The opioid receptor antagonists (−)-naloxone and (−)-naltrexone and their stereoisomers did not, however, display any TLR4 activation potential in HEK293 cells. In addition, (+)-naloxone inhibited morphine and M3G TLR4 signalling effects in a dose-dependent manner [39].
A subsequent study examined the effects of opioid agonists on LPS-induced TLR4 activation in HEK-Blue™ hTLR4 cells, with a view of characterising the pharmacological nature of the interaction [40]. Morphine (3 and 10 µM) and fentanyl (0.3 µM) were found to induce minor, but significant, increases in the activation of TLR4, compared with unstimulated cells. In contrast, morphine and fentanyl significantly inhibited the LPS-induced TLR4 activation in HEK-Blue™ hTLR4 cells. Treatment of cells with 100 ng/mL LPS in the presence of morphine (3–100 µM) resulted in significantly less TLR4 activation when compared with cells stimulated with LPS alone. Similarly, fentanyl (1–100 µM) significantly inhibited LPS (30 ng/mL)-induced TLR4 activation. Moreover, the opioid antagonists, naltrexone (30–1000 µM) and β-funaltrexamine (β-FNA) (3–30 µM), did not activate TLR4 on their own, but significantly inhibited LPS-induced TLR4 activation, as previously reported. Further adding to the confusion, Skolnick et al. attempted to replicate the work of other groups [38,66] that reported on the blockade of TLR4 signalling by opioid antagonists, but were unable to replicate the findings [42]. It has been proposed that the HEK-Blue™ hTLR4 assay fails to include the binding proteins that promote the binding of ligands to the TLR4 complex, which could contribute to the discrepancy [67] and highlight the poor translation of in vitro models to in vivo effects.
Studies reporting the effects of different opioid agents on TLR4 signalling are summarised in Table 1 and Table 2. These studies, while mostly presenting accumulating evidence on the effects of opioids via TLR4 signalling, reveal some discrepancies regarding the mode of action exerted by different opioid agents (agonists vs. antagonists). Several studies indicate that opioid receptor agonists and antagonists exert their effects at TLR4, by activating or blocking TLR4 activation, respectively, in a non-stereoselective manner. However, other studies report that opioid receptor agonists also have the ability to antagonise LPS-induced TLR4 activation. Studies conducted in our laboratory confirm some, but not all, of the above-mentioned results. We have observed that M3G can weakly but consistently activate TLR4 signalling, and that M3G and morphine both significantly inhibit LPS-induced activation [49]. It is worth noting that, rather than a direct interaction at TLR4, some of the literature invokes cross-talk between TLR4 and OR signalling pathways, including cross-targets such as p38 MAPK [27], PKCβ2 [53], or NF-κB [55]. This will be discussed in a later section.
Table 2.
In vitro studies testing TLR4 activation by various opioids.
5. Opioids Interact with TLR4 through its Accessory Protein (MD-2)
Many studies have utilised in silico molecular docking techniques to predict the interactions between opioids and TLR4. The structures of opioids, including morphine [39,72,73,74,75], naloxone [39,68,72,74,76], naltrexone [39,77], and methadone [39,74], as well as the morphine metabolites, M3G [39,68,72,73] and M6G [39], have all been docked into the crystal structure of TLR4 (PDB ID 3FXI) [78]. This crystal structure contains the symmetrical heterodimer of TLR4 and MD-2. Interestingly, in all the studies that included MD-2 in the docking procedure, the opioids preferentially docked into the LPS-binding pocket of the MD-2 protein [39,68,72,73,74,75,77].
As might be anticipated for the LPS-binding pocket, the majority of opioid–MD-2 interactions predicted by docking studies are hydrophobic interactions [72,77]. The docking of (−)-morphine, M3G, (+)-methadone, and (+)-naloxone within the pocket resulted in overlapping binding poses, suggesting competitive binding [68,74,75]. Interestingly, while opioid–MD-2 docking is non-stereoselective, with both the (+) and (−) stereoisomers predicted to interact favourably [39], the specific binding poses of the stereoisomers differ. (+)-Morphine, for example, is predicted to bind in a way that does not overlap with (+)-naloxone [74], unlike (−)-morphine, which docks with a similar pose to (+)-naloxone [75]. Despite the evidence for competitive binding based on overlapping docking poses, when (−)-morphine, M3G, (+)-methadone, and (+)-morphine were docked into the (+)-naloxone–MD-2 complex, altered binding poses were predicted, suggesting non-competitive binding is also possible [74,75].
The predicted binding pose, and the binding energies of the opioids, may also change depending on the state of the MD-2 into which the opioids are being docked. M3G and remifentanil were both predicted to bind more strongly in the TLR4/MD-2 dimer, compared with MD-2 alone [73,75]. M3G docked even more strongly with the TLR4/MD-2 heterodimer, and resulted in a different binding pose between the two TLR4 proteins [73]. Therefore, it was suggested that the activation of TLR4/MD-2 guides M3G down an energy gradient to its final binding site. The predicted binding energy of (−)-morphine was unaffected by the conformational state of TLR4/MD-2 [73,75]; however, docking into the TLR4/MD-2 heterodimer resulted in a similar binding pose as that of M3G [73].
These studies only examined the interactions of opioids with TLR4/MD-2 using static docking methods; however, more thorough investigations were conducted utilising molecular dynamics simulations to model the changes in opioid binding and protein conformations over time [69,72,77]. In these studies, hydrophobic interactions were emphasised as the most important factor for opioid binding within the LPS-binding pocket, causing the MD-2 structure to “clamshell” to accommodate the opioids [72,77]. Over the course of molecular dynamics simulations, morphine that docked solely into MD-2 lost all its interactions, while morphine docked with the TLR4/MD-2 heterodimer retained stability throughout the simulation [69,72]. This was due to the hydrophobic interactions of morphine with residues of the “Phe126 loop”, also known as the gating loop. This loop encloses the solvent-accessible region of the LPS-binding pocket and controls the activation of MD-2 through the conformation of the Phe126 amino acid. The residues of the “Phe126 loop” undergo large orientation shifts during molecular dynamics of MD-2 alone, explaining the instability of the predicted morphine binding. However, the presence of TLR4 facilitates electrostatic and hydrogen bonds between residues of the “Phe126 loop” and TLR4, stabilising the region and, consequently, the binding of morphine. This predicted binding did not change the conformation of Phe126, retaining the active state of MD-2 [72]. M3G was predicted to overlap with the scaffold of morphine and direct the glucuronide moiety to hydrogen bond with residues that are deeper within a solvent-inaccessible region of the pocket. This binding was stable across molecular dynamics simulations, irrelevant of the presence of TLR4, and did not alter the active state of MD-2. Meanwhile, naloxone acted similarly to morphine in molecular dynamics, requiring TLR4 to maintain stable binding; however, this binding caused Phe126 to change conformation, inactivating MD-2.
In conclusion, the in silico studies indicate that opioids interact with TLR4 primarily by binding in the LPS-binding pocket of MD-2. This binding is non-stereoselective, is governed by hydrophobic interactions, and varies depending on whether MD-2 is separate from, or part of a TLR4/MD-2 heterodimer. Different opioids are predicted to bind in multiple poses throughout the LPS-binding pocket and may or may not directly affect the activation state of MD-2 through the “Phe126 loop”. This suggests the possibility of both competitive and non-competitive binding with the protein.
6. The Effect of Opioids on TLR4 Is Non-Competitive
Pharmacological examination of the mechanism of the action of opioids on LPS-induced TLR4 activation indicates a non-competitive antagonism. This was inferred by comparison of the effect of opioids with those of the competitive antagonist LPS-RS on the full LPS concentration-response curve in HEK-Blue™ hTLR4 cells. LPS induced a concentration-dependent increase in TLR4 signalling. Cotreatment with 10 ng/mL LPS-RS did not change the LPS Emax value, but caused a parallel, rightwards shift of the curve, significantly increasing the EC50 value from 0.85 to 2.16 ng/mL. Conversely, cotreatment with either fentanyl or the opioid antagonist β-FNA reduced the Emax values and caused a non-parallel, rightwards shift of the LPS response curve to the right (increased EC50) and downwards (decreased Emax), which suggested a low capacity binding site or a non-competitive antagonism [40].
7. Opioids Affect NF-κB Activation, Downstream of Both TLR4 and Opioid Receptors
NF-κB is a major downstream signalling element in TLR4-mediated inflammatory pathways [79], and the effects of opioids on LPS-induced NF-κB activation have been evaluated. Opioid receptor gene ablation studies have shown that opioids activate or downregulate NF-κB signalling in different cell types, resulting in the modulation of immune and neuronal responses (reviewed by [80]). The modulatory effects of morphine, particularly on LPS-induced NF-κB activation, were examined in mouse and human immune cells [81]. In mouse peritoneal macrophages, pre-treatment with nanomolar morphine concentrations (50 nM) for 2 h increased LPS-induced NF-κB activation, as well as IL-6 and TNF-α secretion and mRNA levels; these effects were reversible through adding naloxone. Conversely, morphine micromolar concentrations (50 µM) inhibited LPS-induced IL-6 and TNF-α secretion and reduced NF-κB activation; however, these latter effects were not reversed upon adding naloxone. Further supporting differential mechanisms for the effects of different morphine concentrations on LPS-induced NF-κB activation, the transfection of primary microglial cells with siRNAs that target the expression of µ-opioid receptor blocked the potentiating effect of a low concentration of morphine (100 nM) on LPS-induced NF-κB activation, while only reducing the effect of high morphine concentrations (10 µM) [45]. These results indicated MOR-mediated effects for low concentrations of morphine, but MOR-independent effects for high concentrations of morphine. In contrast, while morphine alone did not induce any activation, morphine pre-treatment resulted in a concentration-dependent, naloxone-sensitive inhibitory effect on LPS-induced NF-κB nuclear translocation [82]. The underlying mechanism was suggested to be a capability of morphine to induce nitric oxide (NO) release, as the morphine inhibitory effect was entirely blocked by the NO synthase inhibitors Nω-nitro-L-arginine-methyl-ester and Nω-nitro-L-arginine.
The ability to modulate LPS-induced NF-κB activation was also reported for opioid peptides. The effects of the opioid peptides endomorphins 1 and 2 on human THP-1 cells differentiated into macrophage-like cells was evaluated [83]. Both peptides (10−8 and 10−6 M) augmented NF-κB nuclear translocation independently; furthermore, they significantly potentiated LPS (1 µg/m)-induced activation in a concentration-dependent fashion. However, neither of the two opioid peptides had an influence on the production of NF-κB targets IL-10 and IL-12, and they significantly mitigated their LPS-induced production in a concentration-dependent manner. The authors propose that endomorphins may induce the translocation of NF-κB homo- and hetero-dimers that are different from those translocated upon stimulation by LPS. Further studies have elaborated on the interplay between MOR and TLR4 activation by opioids, demonstrating that they have opposing impacts on NFκB activation [73].
Taken together, the data illustrate that the effects of opioids on LPS-induced activation vary between potentiation and inhibition when different opioid concentrations and different cell types are examined. While LPS induces NF-κB activation through TLR4, the actions of opioids seem to encompass more complexity, involving simultaneous activity at both opioid receptors and TLR4. This has been reported as a cross-talk between the two signalling pathways [84].
8. Opioids Modulate TLR4 Expression
Changes in the expression levels of TLR4 upon opioid treatment have been reported in several in vitro studies. The expression of TLR4 mRNA was up-regulated by morphine exposure (200 µM) in microglial BV-2 cells [85]. An increase in TLR4 protein expression in the same cell line was detected at a lower morphine concentration (10 μM) [86]. Treatment of lumbar dorsal spinal cord tissue with 100 µM morphine also significantly increased TLR4 protein expression; whereas, the respective 10 µM concentration had no effect on TLR4 protein levels [87]. In human CHME-5 microglia, morphine (10 µM) induced up-regulation in the expression of TLR4 protein in the presence of LPS. Methadone alone up-regulated TLR4 protein expression but had the opposite effect when combined with LPS, as did oxycodone or buprenorphine [88]. Similarly, endomorphin-1 inhibited the expression of TLR4 on peripheral blood dendritic cells that were stimulated by high glucose [89].
Similar effects have been reported for various opioids in several in vivo studies, employing various dosing regimens. Chronic intrathecal administration of morphine, (−)-methadone, or (+)-methadone to rats (15 µg, once daily for a week) induced spinal glial activation, as well as significant elevations in mRNA and in protein levels of TLR4 in the lumbar dorsal spinal cord [74,87]. Another study by the same group also reported spinal glial activation and elevation in TLR4 mRNA levels, following acute intrathecal administration of 0.75 μg M3G to rats [68]. Morphine exposure in vivo can induce long-term changes in TLR4 expression by microglial cells; rats exposed to morphine during adolescence have increased expressions of TLR4 mRNA and protein in the microglia of the nucleus accumbens during their adulthood [90]. Epithelial cells, isolated from the small intestines of mice implanted with 75 mg morphine pellets for 24 h, demonstrated up-regulated expression of TLR2, TLR4 mRNA, and protein levels [91]. In contrast, remifentanil preconditioning inhibited TLR4 expression in liver tissue in a mouse model of hepatic ischemia reperfusion injury [92] and the κ-opioid receptor agonist U50, 488H mitigated the ischemia/reperfusion-induced myocardial TLR4 expression [93]. Overall, the in vitro and in vivo literature indicate that opioid administration per se increases TLR4 expression on a variety of cell types of both central and peripheral relevance, while in pro-inflammatory conditions opioids prevent TLR4 induction.
Pro-inflammatory responses and glial activation following peripheral and spinal nerve injury are associated with up-regulation in the expression of spinal TLR4 mRNA [94,95,96]. The up-regulation of TLR4 mRNA, induced by SNAP (Spinal Neuropathic Avulsion Pain) surgery, was shown to be further amplified upon morphine administration [97]. Subcutaneous morphine administration for one week (10 mg/kg once daily in Sprague-Dawley rats after surgery) potentiated the SNAP-induced elevation of TLR4 mRNA and pro-inflammatory cytokines in the ipsilateral dorsal spinal cord tissue. Similarly, spinal TLR4 mRNA levels were increased following a week-long subcutaneous morphine treatment in mice with tibia fractures [98].
In contrast to these results, morphine treatment decreased TLR4 mRNA and protein levels in RAW 264.7 cells in vitro, as well as in peritoneal macrophages isolated from morphine-treated BALB/cJ mice; whereas, TLR4 mRNA and protein levels were increased upon naloxone treatment in both cases [99]. In addition to expression levels, it is possible that opioids modulate the subcellular localisation of TLR4. Experiments using the endocytosis inhibitor dynasore suggested that morphine treatment decreases the surface expression of TLR4 in microglia by causing increased TLR4 endocytosis [70].
9. Functional Consequences of TLR4 Activation by Opioids
Depending on the cell type affected, and the localisation (central or peripheral) of the interaction, TLR4 activation by opioids is proposed to cause neuro-inflammation [69,73], leading to opioid-induced hyperalgesia [39,100]; play a role in dependence, reward, and reinforcement [75,101]; and contribute to morphine-induced suppression of colon peristalsis [102,103].
9.1. Opioid Receptor-Independent Mechanisms Contribute to Opioids Deleterious Effects
Some of the earliest evidence suggesting a non-GPCR activity of opioids came from a series of studies that showed that the antinociceptive effect induced by morphine, in male CD-1 mice, was supressed upon intrathecal pre-treatment with the opioid-active (−)-morphine as well as the opioid-inactive (+)-morphine. The anti-analgesic effects of both morphine isomers were blocked upon pre-treatment with (+)-naloxone, further confirming the involvement of a non-opioid receptor-mediated mechanism. Moreover, (−)-morphine and (+)-morphine mitigated the antinociceptive effect of the δ-opioid receptor agonist deltorphin II, and the κ-opioid receptor agonist U50, 488H, in μ-opioid receptor knockout mice [104,105,106]. LPS was reported to reduce morphine analgesia in rats through several contributing mechanisms [107,108]. The effect of LPS was suppressed upon pre-treatment with the non-competitive NMDA receptor antagonist (MK-801), the glial metabolic inhibitor (fluorocitrate), and the opioid receptor antagonist (naloxone). In another study, the antinociceptive effect of morphine in male CD-1 mice was attenuated in a dose-dependent fashion upon pre-treatment with LPS (1.5–4 µg). However, this effect of LPS was reversed upon pre-treatment with both levo-naloxone and its opioid receptor-inactive isomer dextro-naloxone, denoting the involvement of a non-opioid mechanism [109].
Evidence suggesting the contribution of CNS glial cells to pain behaviours was first reported 25 years ago [110] and, subsequently, multiple studies emerged investigating the mechanisms mediating the development of neuropathic pain via glial activation [111,112]. Later, opioid-induced pro-inflammatory glial activation was shown to be responsible for the adverse effects of opioids, which include the opposition of analgesia, analgesic tolerance, hyperalgesia, respiratory depression, opioid dependence, and opioid reward. It was shown that drugs which suppress glial activation, and subsequent cytokine production, reinforce the antinociceptive effect of morphine and reverse neuropathic pain, tolerance, and hyperalgesia [113,114,115,116]. However, the mechanistic basis of these opioid pro-inflammatory effects was unclear, until the discovery of opioid activity at TLR4. This formed the basis for multiple studies employing a range of in vivo pharmacological and genetic manipulations to investigate the TLR4-mediated effects of opioids.
9.2. Central and Peripheral Neuropathic Pain
TLR4 is expressed in the central nervous system on microglia, astrocytes, and endothelial cells [117]. Sensory neuronal damage initiates several neuron-to-glia activation signals, one of which is through the activation of TLR4, as expressed on glial cells by endogenous “danger” signals released upon nerve injury [118,119]. The role of TLR4 in neuroimmune activation following nerve injury was demonstrated in animal models of neuropathy. A significant reduction in the expression of spinal microglial activation markers and pro-inflammatory cytokines, together with significant attenuation of behavioural hypersensitivity, were observed in TLR4 knockout and point mutant mice, and also upon intrathecal administration of TLR4 antisense oligodeoxynucleotide to rats [120].
Based on recent in vitro data that has established the TLR4-antagonistic effects of the neuronally inactive (+)-naloxone and (+)-naltrexone [38,48], their influence on neuropathic pain was tested using a model of peripheral neuropathy, via partial sciatic nerve chronic constriction injury. A significant attenuation of mechanical allodynia was observed after intrathecal administration of (+)-naloxone or (+)-naltrexone (60 μg), as well as following subcutaneous administration of (+)-naloxone (100 mg/kg). Moreover, the sustained delivery of (+)-naloxone or (−)-naloxone via intrathecal infusion (60 μg/h, 4 days) completely reversed the established neuropathic pain [38]. The TLR4-mediated effects of opioids were also explored in models of central neuropathy, where (+)-naloxone was reported to reverse mechanical allodynia resulting from spinal cord injury [121]. Moreover, the subcutaneous administration of morphine after spinal injury caused a significant elevation of mechanical allodynia, and this effect was blocked by co-administration of (+)-naloxone [97].
9.3. Analgesia, Hyperalgesia, Tolerance, and Dependence
TLR4 signalling may be involved in opposing acute opioid analgesia, and in the development of tolerance, hyperalgesia, and dependence [38]. Pharmacological blocking of TLR4 activation and its downstream signalling on the analgesic effects of morphine were evaluated. The evaluation demonstrated a significant potentiation of the magnitude and duration of morphine analgesia upon co-administration of the competitive TLR4 antagonist LPS-RS, or of a Toll-Interleukin-1 receptor domain, containing adaptor protein (TIRAP) inhibitor peptide. It was also reported that (+)-naloxone significantly increased systemic and intrathecal morphine analgesia and alleviated the effects of chronic morphine administration, including tolerance, hyperalgesia, and dependence.
In earlier studies, M3G was reported to cause pain enhancement and induce allodynia and hyperalgesia, since, however, M3G lacks activity at all opioid receptors, the mechanism involved remains unknown [122]. Based on subsequent in vitro cell studies that reported TLR4 activation by M3G, the triggering of a pro-inflammatory response by the TLR4-mediated activation of immune cells emerged as a possible mechanism underlying the pain-enhancing effects of M3G. Intrathecal administration of M3G (0.75 μg) to rats induced potent mechanical allodynia and thermal hyperalgesia, which were both blocked upon co-administering pro-inflammatory cytokine and glial inhibitors, and also upon co-administering either isomer of naloxone [68].
While most studies evaluating the non-neuronal effects of opioids focused on microglia and astrocytes, one study attempted to determine whether opioids may have a TLR4-mediated effect on CNS endothelial cells [73]. After confirming the expression of functional TLR4 by primary adult rat CNS endothelial cells, the effects of intrathecal injections of opioid-stimulated endothelial cells on in vivo behavioural responses were evaluated. Cells treated in vitro with M3G induced significant tactile allodynia in rats compared with saline-treated cells; whereas, cells stimulated with (−)-morphine, or co-treated with M3G and LPS-RS, did not have any significant effect.
Morphine tolerance resulting from chronic subcutaneous administration was shown to be associated with an increase in glial cell activity in the ventrolateral periaqueductal gray (vlPAG) [123]. Through subsequent pharmacological studies, it was demonstrated that TLR4 signalling in the vlPAG glia is involved in developing opioid tolerance. Intra-vlPAG microinjections of both LPS-RS and (+)-naloxone prevented the development of tolerance to systemic morphine, whereas intra-vlPAG microinjections of LPS or (+)-morphine were shown to induce naive tolerance [124].
9.4. Effect of Pharmacological or Genetic TLR4 Ablation on Algesia, Reinforcement, Tolerance, and Dependence
Hyperalgesia can be triggered by the continuous infusion, for several days, of morphine or oxymorphone to the opioid receptor of triple knockout mice; this suggests the involvement of an opioid receptor-independent mechanism [125]. Several genetic manipulation studies point to the involvement of TLR4 in mediating the effects of opioids. The dose–response curve for acute morphine analgesia (1–50 mg/kg) demonstrated a significantly higher analgesic effect for morphine in TLR4 knockout and MyD88 knockout Balb/c mice when compared with their wild-type control [39,69]. Co-administration of (+)-naloxone (60 mg/kg) significantly increased the analgesic effect of morphine (2.5 mg/kg) in wild-type mice, while having no influence in TLR4 knockout mice [39]. A study tested the effects of two small-molecule inhibitor compounds that reportedly targeted TLR4 and its accessory protein MD-2, causing disruption of the TLR4/MD-2 complex [69]. Both compounds potentiated the acute analgesic effects of morphine in rats; whereas, in Balb/c mice, the potentiating effect was only attained in the wild-type strain and not in the TLR4 knockout strain. The acute analgesic effect of oxycodone was also stronger in the absence of TLR4, where significantly longer hot plate latencies were recorded over a range of oxycodone doses (0.01–5 mg/kg, I.P.) in Balb/c TLR4−/− mice vs. wild-type mice [75]. Conversely, in a subsequent study by the same group, acute morphine analgesia produced by a single dose of morphine (7.5 mg/kg) in the same mouse strains was found to be similar in wild-type, TLR4 knockout, and MyD88 knockout mice [126]. It is worth mentioning that several factors may contribute to these inconsistent responses, including the behavioural test and assay protocol used, as well as the mouse strains employed. The opioid dose and its magnitude of response could also have an impact, where the differences between analgesic effects may be masked at doses approaching minimum or maximal analgesia.
The detrimental effects of opioids were also shown to diminish in TLR4 mutant and knockout mice. Repetitive injections of escalating doses of morphine to wild-type Balb/c and MyD88 knockout mice over 3 successive days resulted in significant loss of analgesia and the development of tolerance. However, analgesic tolerance was not observed in TLR4 knockout mice, implying the involvement of a MyD88-independent TLR4-mediated mechanism [126]. Intraperitoneal M3G (25 mg/kg) induced tactile hyperalgesia only in wild-type (C57BL/6) mice; whereas, hyperalgesia was significantly abolished in TLR4 mutant mice (C57BL/10ScNJ) [127]. Thermal hyperalgesia and mechanical allodynia, resulting from repeated intraperitoneal administration of codeine (21 mg/kg) or morphine (20 mg/kg) to wild-type Balb/c mice, were not observed in TLR4 null mice [128]. TLR4 ablation also protected against mechanical hyperalgesia that was induced by subcutaneous infusion of remifentanil (4 mg/kg/min, 1 h, for 3 days) [100]. The behavioural reinforcing actions of oxycodone, assessed by conditioned place preference tests, were significantly reduced in TLR4 knockout and MyD88 knockout Balb/c mice compared with the wild-type control [75], suggesting the involvement of TLR4 signalling in opioid reinforcement and reward effects.
Interestingly, studies have also presented evidence that supports the absence of a role for TLR4 signalling in some deleterious opioid actions. No difference was found between the extent of tactile hyperalgesia induced by morphine in TLR4 mutant mice (C3H/HeJ) and that observed in their wild-type controls (C3H/HeOuJ) [129]. Chronic morphine administration (60 mg/kg, once daily for 5 days) was also reported to induce analgesic tolerance to the same extent in TLR4 mutant mice (C3H/HeJ) when compared to the wild-type (C3H/HeN), as well as in TLR4 knockout C57BL/6 mice compared to wild-type. Moreover, the mRNA expression of the microglial activation marker CD11b was significantly increased after chronic morphine administration in knockout and wild-type mice, suggesting that TLR4 is not involved in opioid–microglial activation [130]. In another study, two mouse strains were employed to test opioid effects: TLR4 mutant (C3H/HeJ) vs. wild-type (C3H/HeOuJ), and TLR4 null (B10ScNJ) vs. wild-type (B10ScSNJ). All morphine effects including antinociception, hyperalgesia, tolerance, and physical dependence were unchanged in TLR4 mutant and TLR4-null mice [131]. The discrepancies in the effect of TLR4 ablation on morphine pharmacodynamics have been proposed to be due to confounders in studies, e.g., higher morphine-induced analgesia in TLR4-knockout animals compared with the control strain, the effect of opioid binding to MD2 in absence of TLR4, and the concentration of opioids to which receptors are exposed in vivo [130,131].
9.5. GIT and Colon Motility
It is well recognised that one of the major undesirable effects of opioid analgesics is their gastrointestinal side effects, which are perceived as a key limitation to their therapeutic utility [132,133]. Toll-like receptors, including TLR4, are widely expressed in the GIT and have been linked to the development of GIT immune responses and to several gastrointestinal pathologies [134]. Owing to the observations of opioid interactions with TLR4 in the nervous system, the role of toll-like receptors in mediating opioid GIT effects have been investigated. One of the mechanisms implicated in the disruption of intestinal tight junctions is through the activation of toll-like receptors [135,136]. Accordingly, TLR involvement in mediating the effects of morphine on the intestinal barrier function was investigated. Morphine-induced gut bacterial translocation to the mesenteric lymph node and liver was completely abolished in μ-opioid receptor knockout mice and was significantly mitigated in TLR4−/−, TLR2−/−, and TLR2/4−/− double knockout mice [91]. Morphine was shown to disrupt the tight junction protein organisation between intestinal epithelial cells. This effect was significantly attenuated in TLR4−/− mice and completely abolished in mice lacking μ-opioid receptor, and in TLR2−/− and TLR2/4−/− double knockout mice, which implies that disruption of intestinal barrier function by morphine is partially mediated by TLR4.
Opioids are well-known for their inhibitory effects on the gastrointestinal motility, and opioid-induced constipation is a serious limitation of opioid therapy [137,138]. TLR4 activation by opioids is proposed to contribute to the morphine-induced suppression of colon peristalsis [102,103]. Pre-treatment with the TLR4 antagonist TAK-242 significantly alleviated the morphine-induced inhibition of colon peristalsis and propulsion velocity in the isolated guinea pig colon in vitro and in mice in vivo [102]. In a more recent study, morphine’s inhibitory effects on the gastrointestinal transit in wild-type BALB/c mice were shown to be significantly attenuated in TLR4−/−, TLR2/4−/− and MyD88−/− knockout mice, where a subcutaneous injection of morphine (10 mg/kg) was shown to retard the movement of ingested content along the GIT in wild-type mice. However, this differential effect was not replicated in vitro, where no differences were observed between the responses to morphine for wild-type and TLR2/4−/− isolated colon preparations, suggesting the involvement of a pathway extrinsic to the colon [103]. Taken cumulatively, these data suggest that morphine may exert its effects on the GIT through acting directly on toll-like receptors or through a mechanism involving cross-talk between μ-opioid and toll-like receptor signalling.
10. Whether TLR4 Mediate the Effects of Opioids on Tumour Growth and Metastasis Is Unexplored
It is quite remarkable that, despite a well-documented link between TLR4 and cancer (reviewed in [7,139,140]), and reasonably convincing evidence that opioids are active at TLR4, the possibility that TLR4 may mediate the effects of opioids on tumour growth and metastasis has not, to date, been explored.
From a clinical perspective, opioids will remain the mainstay analgesics in patients with cancer, although they have been scrutinized for negatively affecting tumour biology. Opium has been implicated in cancer development [141] most likely due to mutagenic compounds induced by pyrolysis. Chronic use of prescription opioids has been associated with a higher morbidity and overall mortality; however, evidence for increased carcinogenesis in these patients is lacking [142]. With respect to the perioperative use of opioids in cancer surgery, the most recent meta-analysis by Zheng et al. in a mixed cancer population indicated that neither overall survival nor progression-free survival was affected by the intraoperative use of opioids [143]. For those patients with advanced cancer, higher opioid doses are usually required to effectively treat pain.
Pain itself is a major contributor impairing host resistance and promoting tumour progression; thus, there has been advocation for adequate pain treatment to be tumour-protective [144]. Differentiation between the suggested “tumour-protective” action of opioids—due to their effective analgesic properties—and their hypothesized immune-related “tumour-promoting” effects, regarding clinical relevance in oncological patients, is challenging. It is thus not surprising that, in a recently published large scaled trial including 2132 patients with breast cancer, the use of regional anaesthesia—which decreases opioid administration—did no better than perioperative use of opioids, with regard to local or metastatic breast cancer recurrence [145].
Recognition that some opioids weakly activate TLR4, but significantly prevent TLR4 activation induced by agonists, needs to be added to the list of factors that may contribute to the complexity and existing discrepancy in the field of opioid influence on cancer. We have demonstrated that opioid activity of perioperative plasma samples correlates with inhibition of TLR4 activation [65]. Interestingly, using linear mixed models, this study also found that the ability of plasma samples to activate TLR4 has a significant effect that explains pain scores [65]. Therefore, the interrelation between pain, TLR4 activation, and opioids is anticipated to be an important avenue of research for basic scientists and clinicians.
11. Conclusions
The discovery of opioid activity at TLR4 has allowed us to explain a number of OR-independent effects for this class of drugs and forced a number of paradigms to evolve in opioid pharmacology. The discovery also suggests the possibility that a number of undesirable effects of opioids may be mitigated by the development of pharmacotherapies targeting TLR4, to enhance the safety and efficacy of opioids. Furthermore, research in the near future will likely attempt to identify whether opioids interfere with the activation of TLR4 by the endogenous molecular patterns (DAMPs) that are relevant to a number of pathologies in which patients are likely to be administered opioids.
Author Contributions
Writing—original draft preparation, M.-O.P., M.M.G. and J.A.M.; writing—review and editing, I.S., B.P.R., P.N.S. and M.W.H.; visualization, M.-O.P.; supervision, M.-O.P.; clinical validation, M.W.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
M.M.G., I.S., J.A.M., B.P.R., P.N.S. and M.-O.P. declare no conflict of interest. Markus W. Hollmann has received research funding from CSL Behring, ZonMw, the Society of Cardiovasular Anesthesiologists (SCA) and the European Association of Cardiothoracic Anaesthesiology (EACTA), and has received compensation from Eurocept Pharmaceuticals, BV, and IDD for services as a consultant. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- García Bueno, B.; Caso, J.R.; Madrigal, J.L.M.; Leza, J.C. Innate immune receptor Toll-like receptor 4 signalling in neuropsychiatric diseases. Neurosci. Biobehav. Rev. 2016, 64, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Chauhan, A.; Jain, N. TLR4 Polymorphisms and Expression in Solid Cancers. Mol. Diagn. Ther. 2018, 22, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef]
- Li, J.; Yang, F.; Wei, F.; Ren, X. The role of toll-like receptor 4 in tumor microenvironment. Oncotarget 2017, 8, 66656–66667. [Google Scholar] [CrossRef]
- Mai, C.W.; Kang, Y.B.; Pichika, M.R. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: Its expression and effects in the ten most common cancers. Onco Targets 2013, 6, 1573–1587. [Google Scholar] [CrossRef]
- Khademalhosseini, M.; Arababadi, M.K. Toll-like receptor 4 and breast cancer: An updated systematic review. Breast Cancer 2019, 26, 265–271. [Google Scholar] [CrossRef]
- Sato, Y.; Goto, Y.; Narita, N.; Hoon, D.S. Cancer Cells Expressing Toll-like Receptors and the Tumor Microenvironment. Cancer Microenviron. 2009, 2 (Suppl. 1), 205–214. [Google Scholar] [CrossRef]
- Molteni, M.; Marabella, D.; Orlandi, C.; Rossetti, C. Melanoma cell lines are responsive in vitro to lipopolysaccharide and express TLR-4. Cancer Lett. 2006, 235, 75–83. [Google Scholar] [CrossRef]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N.; et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef]
- Liao, S.J.; Zhou, Y.H.; Yuan, Y.; Li, D.; Wu, F.H.; Wang, Q.; Zhu, J.H.; Yan, B.; Wei, J.J.; Zhang, G.M.; et al. Triggering of Toll-like receptor 4 on metastatic breast cancer cells promotes alphavbeta3-mediated adhesion and invasive migration. Breast Cancer Res Treat 2012, 133, 853–863. [Google Scholar] [CrossRef]
- Chung, Y.H.; Kim, D. Enhanced TLR4 Expression on Colon Cancer Cells After Chemotherapy Promotes Cell Survival and Epithelial–Mesenchymal Transition through Phosphorylation of GSK3β. Anticancer Res. 2016, 36, 3383–3394. [Google Scholar]
- Sun, Y.; Wu, C.; Ma, J.; Yang, Y.; Man, X.; Wu, H.; Li, S. Toll-like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp. Cell Res. 2016, 347, 274–282. [Google Scholar] [CrossRef]
- Kang, R.; Zhang, Q.; Zeh, H.J.; Lotze, M.T.; Tang, D. HMGB1 in Cancer: Good, Bad, or Both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef]
- Schlueter, C.; Weber, H.; Meyer, B.; Rogalla, P.; Röser, K.; Hauke, S.; Bullerdiek, J. Angiogenetic Signaling through Hypoxia: HMGB1: An Angiogenetic Switch Molecule. Am. J. Pathol. 2005, 166, 1259–1263. [Google Scholar] [CrossRef]
- Yu, L.-X.; Yan, L.; Yang, W.; Wu, F.-Q.; Ling, Y.; Chen, S.-Z.; Tang, L.; Tan, Y.-X.; Cao, D.; Wu, M.-C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef]
- Afsharimoghaddam, A.; Soleimani, M.; Lashay, A.; Dehghani, M.; Sepehri, Z. Controversial roles played by toll like receptor 4 in urinary bladder cancer; A systematic review. Life Sci. 2016, 158, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050. [Google Scholar] [CrossRef] [PubMed]
- Matzner, P.; Sorski, L.; Shaashua, L.; Elbaz, E.; Lavon, H.; Melamed, R.; Rosenne, E.; Gotlieb, N.; Benbenishty, A.; Reed, S.G.; et al. Perioperative treatment with the new synthetic TLR-4 agonist GLA-SE reduces cancer metastasis without adverse effects. Int. J. Cancer 2015, 138, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Das, K.P.; McMillian, M.K.; Bing, G.; Hong, J.S. Modulatory effects of [Met5]-enkephalin on interleukin-1 beta secretion from microglia in mixed brain cell cultures. J. Neuroimmunol. 1995, 62, 9–17. [Google Scholar] [CrossRef]
- Kong, L.Y.; McMillian, M.K.; Hudson, P.M.; Jin, L.; Hong, J.S. Inhibition of lipopolysaccharide-induced nitric oxide and cytokine production by ultralow concentrations of dynorphins in mixed glia cultures. J. Pharmacol. Exp. Ther. 1997, 280, 61–66. [Google Scholar] [PubMed]
- Liu, B.; Du, L.; Hong, J.S. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000, 293, 607–617. [Google Scholar] [PubMed]
- Liu, B.; Du, L.; Kong, L.Y.; Hudson, P.M.; Wilson, B.C.; Chang, R.C.; Abel, H.H.; Hong, J.S. Reduction by naloxone of lipopolysaccharide-induced neurotoxicity in mouse cortical neuron-glia co-cultures. Neuroscience 2000, 97, 749–756. [Google Scholar] [CrossRef]
- Cheng, W.; Li, Y.; Hou, X.; Zhang, N.; Ma, J.; Ding, F.; Li, F.; Miao, Z.; Zhang, Y.; Qi, Q. HSP60 is involved in the neuroprotective effects of naloxone. Mol. Med. Rep. 2014, 10, 2172–2176. [Google Scholar] [CrossRef]
- Bian, T.-H.; Wang, X.-F.; Li, X.-Y. Effects of morphine and naloxone on proliferation of lymphocytes in vitro. Acta Pharmacol. Sin. 1995, 16, 315–318. [Google Scholar]
- Husted, T.L.; Govindaswami, M.; Oeltgen, P.R.; Rudich, S.M.; Lentsch, A.B. A δ2-opioid agonist inhibits p38 MAPK and suppresses activation of murine macrophages. J. Surg. Res. 2005, 128, 45–49. [Google Scholar] [CrossRef]
- Hyejin, J.; Mei, L.; Seongheon, L.; Cheolwon, J.; Seokjai, K.; Hongbeom, B.; Minsun, K.; Sungsu, C.; Sanghyun, K. Remifentanil attenuates human neutrophils activation induced by lipopolysaccharide. Immunopharmacol. Immunotoxicol. 2013, 35, 264–271. [Google Scholar] [CrossRef]
- Bastami, S.; Norling, C.; Trinks, C.; Holmlund, B.; Walz, T.M.; Ahlner, J.; Uppugunduri, S. Inhibitory effect of opiates on LPS mediated release of TNF and IL-8. Acta Oncol. 2013, 52, 1022–1033. [Google Scholar] [CrossRef]
- Mottaz, H.; Schönenberger, R.; Fischer, S.; Eggen, R.I.L.; Schirmer, K.; Groh, K.J. Dose-dependent effects of morphine on lipopolysaccharide (LPS)-induced inflammation, and involvement of multixenobiotic resistance (MXR) transporters in LPS efflux in teleost fish. Environ. Pollut. 2017, 221, 105–115. [Google Scholar] [CrossRef]
- van Loon, J.P.; de Grauw, J.C.; van Dierendonck, M.; L’Ami, J.J.; Back, W.; van Weeren, P.R. Intra-articular opioid analgesia is effective in reducing pain and inflammation in an equine LPS induced synovitis model. Equine Vet. J. 2010, 42, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Lysle, D.T.; How, T. Endogenous opioids regulate the expression of inducible nitric oxide synthase by splenocytes. J. Pharmacol. Exp. Ther. 1999, 288, 502–508. [Google Scholar] [PubMed]
- Ji, S.; Wang, L. μ-Opioid receptor signalling via PI3K/Akt pathway ameliorates lipopolysaccharide-induced acute respiratory distress syndrome. Exp. Physiol. 2019, 104, 1555–1561. [Google Scholar] [CrossRef]
- Davis, R.L.; Stevens, C.W.; Thomas Curtis, J. The opioid antagonist, β-funaltrexamine, inhibits lipopolysaccharide-induced neuroinflammation and reduces sickness behavior in mice. Physiol. Behav. 2017, 173, 52–60. [Google Scholar] [CrossRef]
- Messmer, D.; Hatsukari, I.; Hitosugi, N.; Schmidt-Wolf, I.G.H.; Singhal, P.C. Morphine reciprocally regulates IL-10 and IL-12 production by monocyte-derived human dendritic cells and enhances T cell activation. Mol. Med. 2006, 12, 284–290. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Zhan, J. Effects of remifentanyl and fentanyl on LPS-induced cytokine release in human whole blood in vitro. Mol. Biol. Rep. 2009, 36, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Zhang, Y.; Brown, K.; Coats, B.D.; Shridhar, M.; Sholar, P.W.; Patel, S.J.; Crysdale, N.Y.; Harrison, J.A.; Maier, S.F.; et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: Involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 2008, 28, 20–29. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Zhang, Y.; Shridhar, M.; Evans, J.H.; Buchanan, M.M.; Zhao, T.X.; Slivka, P.F.; Coats, B.D.; Rezvani, N.; Wieseler, J.; et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav. Immun. 2010, 24, 83–95. [Google Scholar] [CrossRef]
- Stevens, C.W.; Aravind, S.; Das, S.; Davis, R.L. Pharmacological characterization of LPS and opioid interactions at the toll-like receptor 4. Br. J. Pharmacol. 2013, 168, 1421–1429. [Google Scholar] [CrossRef]
- Madera-Salcedo, I.K.; Cruz, S.L.; Gonzalez-Espinosa, C. Morphine prevents lipopolysaccharide-induced TNF secretion in mast cells blocking IκB kinase activation and SNAP-23 phosphorylation: Correlation with the formation of a β-arrestin/TRAF6 complex. J. Immunol. 2013, 191, 3400–3409. [Google Scholar] [CrossRef]
- Skolnick, P.; Davis, H.; Arnelle, D.; Deaver, D. Translational potential of naloxone and naltrexone as TLR4 antagonists. Trends Pharmacol. Sci. 2014, 35, 431–432. [Google Scholar] [CrossRef]
- Selfridge, B.R.; Wang, X.; Zhang, Y.; Yin, H.; Grace, P.M.; Watkins, L.R.; Jacobson, A.E.; Rice, K.C. Structure–Activity Relationships of (+)-Naltrexone-Inspired Toll-like Receptor 4 (TLR4) Antagonists. J. Med. Chem. 2015, 58, 5038–5052. [Google Scholar] [CrossRef]
- Wan, J.; Ma, J.; Anand, V.; Ramakrishnan, S.; Roy, S. Morphine potentiates LPS-induced autophagy initiation but inhibits autophagosomal maturation through distinct TLR 4-dependent and independent pathways. Acta Physiol. 2015, 214, 189–199. [Google Scholar] [CrossRef]
- Gessi, S.; Borea, P.A.; Bencivenni, S.; Fazzi, D.; Varani, K.; Merighi, S. The activation of μ-opioid receptor potentiates LPS-induced NF-kB promoting an inflammatory phenotype in microglia. FEBS Lett. 2016, 590, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Fazalul Rahiman, S.S.; Morgan, M.; Gray, P.; Shaw, P.N.; Cabot, P.J. Dynorphin 1-17 and Its N-Terminal Biotransformation Fragments Modulate Lipopolysaccharide-Stimulated Nuclear Factor-kappa B Nuclear Translocation, Interleukin-1beta and Tumor Necrosis Factor-alpha in Differentiated THP-1 Cells. PLoS ONE 2016, 11, e0153005. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Piotrowska, A.; Makuch, W.; Mika, J. Biphalin, a Dimeric Enkephalin, Alleviates LPS-Induced Activation in Rat Primary Microglial Cultures in Opioid Receptor-Dependent and Receptor-Independent Manners. Neural Plast. 2017, 2017, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Peng, Y.; Hutchinson, M.R.; Rice, K.C.; Yin, H.; Watkins, L.R. Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br. J. Pharmacol. 2016, 173, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Gomes, F.P.; Deora, V.; Gregory, K.; Vithanage, T.; Nassar, Z.D.; Cabot, P.J.; Sturgess, D.; Shaw, P.N.; Parat, M.-O. Activation of μ-opioid receptor and Toll-like receptor 4 by plasma from morphine-treated mice. Brain Behav. Immun. 2017, 61, 244–258. [Google Scholar] [CrossRef]
- Montesinos, J.; Gil, A.; Guerri, C. Nalmefene Prevents Alcohol-Induced Neuroinflammation and Alcohol Drinking Preference in Adolescent Female Mice: Role of TLR4. Alcohol. Clin. Exp. Res. 2017, 41, 1257–1270. [Google Scholar] [CrossRef]
- Cant, R.; Dalgleish, A.G.; Allen, R.L. Naltrexone Inhibits IL-6 and TNFα Production in Human Immune Cell Subsets following Stimulation with Ligands for Intracellular Toll-Like Receptors. Front. Immunol. 2017, 8, 809. [Google Scholar] [CrossRef]
- Rahiman, S.S.F.; Morgan, M.; Gray, P.; Shaw, P.N.; Cabot, P.J. Inhibitory effects of dynorphin 3-14 on the lipopolysaccharide-induced toll-like receptor 4 signalling pathway. Peptides 2017, 90, 48–54. [Google Scholar] [CrossRef]
- Lei, S.; Zhang, Y.; Su, W.; Zhou, L.; Xu, J.; Xia, Z.-y. Remifentanil attenuates lipopolysaccharide-induced oxidative injury by downregulating PKCβ2 activation and inhibiting autophagy in H9C2 cardiomyocytes. Life Sci. 2018, 213, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.N.; Ma, Y.; Wei, X.Y.; Liu, K.Y.; Wang, H.; Han, H.; Cui, Y.; Zhang, M.X.; Qin, W.D. Remifentanil Protects against Lipopolysaccharide-Induced Inflammation through PARP-1/NF-κB Signaling Pathway. Mediat. Inflamm. 2019, 2019, 3013716. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fan, L.; Kong, C.; Miao, F.; Wu, Y.; Wang, T. Oxycodone suppresses the lipopolysaccharide-induced neuroinflammation by downregulating nuclear factor-κB in hippocampal astrocytes of Sprague–Dawley rats. NeuroReport 2020, 31, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhong, Y.; Xiao, J.; Ji, J.; Xi, J.; Wei, X.; Liu, R. Kappa Opioid Receptor on Pulmonary Macrophages and Immune Function. Transl. Perioper. Pain Med. 2020, 7, 225–233. [Google Scholar] [CrossRef][Green Version]
- Choubey, A.; Dehury, B.; Kumar, S.; Medhi, B.; Mondal, P. Naltrexone a potential therapeutic candidate for COVID-19. J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef]
- Cui, L.; Cai, H.; Sun, F.; Wang, Y.; Qu, Y.; Dong, J.; Wang, H.; Li, J.; Qian, C.; Li, J. Beta-endorphin inhibits the inflammatory response of bovine endometrial cells through δ opioid receptor in vitro. Dev. Comp. Immunol. 2021, 121, 104074. [Google Scholar] [CrossRef]
- Li, X.; Li, R.; Fang, Q.; Jamal, M.; Wang, C.; Wang, Y.; Zhang, Z.; Wu, X.; Song, X. Oxycodone attenuates vascular leak and lung inflammation in a clinically relevant two-hit rat model of acute lung injury. Cytokine 2021, 138, 155346. [Google Scholar] [CrossRef]
- Cheng, M.; Geng, Y.; Chen, Y.; Zhang, Y.; Guo, R.; Xu, H.; Liang, J.; Xie, J.; Zhang, Z.; Tian, X. δ-Opioid receptor activation ameliorates lipopolysaccharide-induced inflammation and apoptosis by inhibiting the MAPK/caspase-3 pathway in BV2 microglial cells. Exp. Brain Res. 2021, 239, 401–412. [Google Scholar] [CrossRef]
- Vaidya, S.; Shantanu, P.A.; Tiwari, V. Attenuation of ongoing neuropathic pain by peripheral acting opioid involves activation of central dopaminergic neurocircuitry. Neurosci. Lett. 2021, 754, 135751. [Google Scholar] [CrossRef]
- Luan, G.; Pan, F.; Bu, L.; Wu, K.; Wang, A.; Xu, X. Butorphanol Promotes Macrophage Phenotypic Transition to Inhibit Inflammatory Lung Injury via κ Receptors. Front. Immunol. 2021, 12, 2660. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, J.-W.; Wilson, B.C.; Du, L.; Yang, S.-N.; Wang, J.-Y.; Wu, G.-C.; Cao, X.-D.; Hong, J.-S. Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J. Pharmacol. Exp. Ther. 2000, 295, 125–132. [Google Scholar]
- Chin, P.Y.; Dorian, C.L.; Hutchinson, M.R.; Olson, D.M.; Rice, K.C.; Moldenhauer, L.M.; Robertson, S.A. Novel Toll-like receptor-4 antagonist (+)-naloxone protects mice from inflammation-induced preterm birth. Sci. Rep. 2016, 6, 36112. [Google Scholar] [CrossRef]
- Xie, N.; Matigian, N.; Vithanage, T.; Gregory, K.; Nassar, Z.D.; Cabot, P.J.; Shaw, P.N.; Kirkpatrick, C.M.J.; Cao, K.-A.L.; Sturgess, D.; et al. Effect of Perioperative Opioids on Cancer-Relevant Circulating Parameters: Mu Opioid Receptor and Toll-Like Receptor 4 Activation Potential, and Proteolytic Profile. Clin. Cancer Res. 2018, 24, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.R.; Hutchinson, M.R.; Rice, K.C.; Maier, S.F. The “Toll” of opioid-induced glial activation: Improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol. Sci. 2009, 30, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.R.; Wang, X.; Mustafa, S.; Hutchinson, M.R. In vivo veritas: (+)-Naltrexone’s actions define translational importance. Trends Pharmacol. Sci. 2014, 35, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.S.; Hutchinson, M.R.; Rezvani, N.; Loram, L.C.; Zhang, Y.; Maier, S.F.; Rice, K.C.; Watkins, L.R. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience 2010, 165, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loram, L.C.; Ramos, K.; de Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R.; et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef]
- Liang, Y.; Chu, H.; Jiang, Y.; Yuan, L. Morphine enhances IL-1β release through toll-like receptor 4-mediated endocytic pathway in microglia. Purinergic. Signal. 2016, 12, 637–645. [Google Scholar] [CrossRef]
- Iqbal, S.; Parker, L.M.; Everest-Dass, A.V.; Moh, E.S.X.; Sayyadi, N.; Hutchinson, M.R.; Packer, N.H. Lipopolysaccharide and Morphine-3-Glucuronide-Induced Immune Signalling Increases the Expression of Polysialic Acid in PC12 Cells. Mol. Neurobiol. 2020, 57, 964–975. [Google Scholar] [CrossRef]
- Shah, M.; Anwar, M.A.; Yesudhas, D.; Krishnan, J.; Choi, S. A structural insight into the negative effects of opioids in analgesia by modulating the TLR4 signaling: An in silico approach. Sci. Rep. 2016, 6, 39271. [Google Scholar] [CrossRef]
- Grace, P.M.; Ramos, K.M.; Rodgers, K.M.; Wang, X.; Hutchinson, M.R.; Lewis, M.T.; Morgan, K.N.; Kroll, J.L.; Taylor, F.R.; Strand, K.A.; et al. Activation of adult rat CNS endothelial cells by opioid-induced toll-like receptor 4 (TLR4) signaling induces proinflammatory, biochemical, morphological, and behavioral sequelae. Neuroscience 2014, 280, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.R.; Lewis, S.S.; Coats, B.D.; Rezvani, N.; Zhang, Y.; Wieseler, J.L.; Somogyi, A.A.; Yin, H.; Maier, S.F.; Rice, K.C.; et al. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience 2010, 167, 880–893. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Northcutt, A.L.; Hiranita, T.; Wang, X.; Lewis, S.S.; Thomas, J.; van Steeg, K.; Kopajtic, T.A.; Loram, L.C.; Sfregola, C.; et al. Opioid Activation of Toll-Like Receptor 4 Contributes to Drug Reinforcement. J. Neurosci. 2012, 32, 11187–11200. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.S.; Hutchinson, M.R.; Zhang, Y.; Hund, D.K.; Maier, S.F.; Rice, K.C.; Watkins, L.R. Glucuronic acid and the ethanol metabolite ethyl-glucuronide cause toll-like receptor 4 activation and enhanced pain. Brain Behav. Immun. 2013, 30, 24–32. [Google Scholar] [CrossRef]
- Zhang, X.; Cui, F.; Chen, H.; Zhang, T.; Yang, K.; Wang, Y.; Jiang, Z.; Rice, K.C.; Watkins, L.R.; Hutchinson, M.R.; et al. Dissecting the Innate Immune Recognition of Opioid Inactive Isomer (+)-Naltrexone Derived Toll-like Receptor 4 (TLR4) Antagonists. J. Chem. Inf. Modeling 2018, 58, 816–825. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Chen, Y.L.; Law, P.-Y.; Loh, H.H. Nuclear factor kappaB signaling in opioid functions and receptor gene expression. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2006, 1, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Cain, K.J.; Chapin, R.B.; Charboneau, R.G.; Barke, R.A. Morphine Modulates NFκB Activation in Macrophages. Biochem. Biophys. Res. Commun. 1998, 245, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Welters, I.D.; Menzebach, A.; Goumon, Y.; Cadet, P.; Menges, T.; Hughes, T.K.; Hempelmann, G.; Stefano, G.B. Morphine inhibits NF-kappaB nuclear binding in human neutrophils and monocytes by a nitric oxide-dependent mechanism. Anesthesiology 2000, 92, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Ohura, K. Endomorphins 1 and 2 inhibit IL-10 and IL-12 production and innate immune functions, and potentiate NF-kappaB DNA binding in THP-1 differentiated to macrophage-like cells. Scand. J. Immunol. 2002, 56, 260–269. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, M.; Chen, C.; Liu, L.; Wei, X.; Zeng, S. Toll-Like Receptor 4 (TLR4)/Opioid Receptor Pathway Crosstalk and Impact on Opioid Analgesia, Immune Function, and Gastrointestinal Motility. Front. Immunol. 2020, 11, 1455. [Google Scholar] [CrossRef]
- Pan, Y.; Sun, X.; Jiang, L.; Hu, L.; Kong, H.; Han, Y.; Qian, C.; Song, C.; Qian, Y.; Liu, W. Metformin reduces morphine tolerance by inhibiting microglial-mediated neuroinflammation. J. Neuroinflamm. 2016, 13, 294. [Google Scholar] [CrossRef]
- Chen, J.; Wang, G.; Sun, T.; Ma, C.; Huo, X.; Kong, Y. Involvement of TCF7L2 in generation of morphine-induced antinociceptive tolerance and hyperalgesia by modulating TLR4/NF-κB/NLRP3 in microglia. Toxicol. Appl. Pharmacol. 2021, 416, 115458. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Coats, B.D.; Lewis, S.S.; Zhang, Y.; Sprunger, D.B.; Rezvani, N.; Baker, E.M.; Jekich, B.M.; Wieseler, J.L.; Somogyi, A.A.; et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav. Immun. 2008, 22, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Summer, D.; Randall, D.; Leandra, F.; Craig, S. Opioid-Induced Alterations of TLR4 Protein Expression in a Human Microglial Cell Line. FASEB J. 2015, 29, 770–774. [Google Scholar]
- Liu, C.M.; Yang, T.H.; Huang, M.; Zhou, C.; Li, Y.H.; Li, Z.H. Effect of Endomorphin-1 on Maturation and Expression of TLR4 in Peripheral Blood Dendritic Cells Induced by High Glucose. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2018, 26, 886–893. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Bilbo, S.D. Adolescent Morphine Exposure Affects Long-Term Microglial Function and Later-Life Relapse Liability in a Model of Addiction. J. Neurosci. 2013, 33, 961–971. [Google Scholar] [CrossRef]
- Meng, J.; Yu, H.; Ma, J.; Wang, J.; Banerjee, S.; Charboneau, R.; Barke, R.A.; Roy, S. Morphine Induces Bacterial Translocation in Mice by Compromising Intestinal Barrier Function in a TLR-Dependent Manner. PLoS ONE 2013, 8, e54040. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, C.; Cui, C.; Jiao, Y.; Li, P.; Zhu, L.; Yu, W.; Xia, Q.; Wen, D.; Yang, L. Indispensable role of β-arrestin2 in the protection of remifentanil preconditioning against hepatic ischemic reperfusion injury. Sci. Rep. 2019, 9, 2087. [Google Scholar] [CrossRef]
- Lin, J.; Wang, H.; Li, J.; Wang, Q.; Zhang, S.; Feng, N.; Fan, R.; Pei, J. κ-Opioid receptor stimulation modulates TLR4/NF-κB signaling in the rat heart subjected to ischemia–reperfusion. Cytokine 2013, 61, 842–848. [Google Scholar] [CrossRef]
- Tanga, F.Y.; Raghavendra, V.; DeLeo, J.A. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem. Int. 2004, 45, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, V.; Tanga, F.Y.; DeLeo, J.A. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur. J. Neurosci. 2004, 20, 467–473. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Lai, W.; Rivest, S.; Hart, R.P.; Satoskar, A.R.; Popovich, P.G. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J. Neurochem. 2007, 102, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.; Grace, P.M.; Wieseler, J.; Favret, J.; Springer, K.; Skarda, B.; Ayala, M.; Hutchinson, M.R.; Falci, S.; Rice, K.C.; et al. Morphine amplifies mechanical allodynia via TLR4 in a rat model of spinal cord injury. Brain Behav. Immun. 2016, 58, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-Y.; Li, W.-W.; Nwaneshiudu, C.; Irvine, K.-A.; Clark, J.D. Pharmacological Characters of Oliceridine, a μ-Opioid Receptor G-Protein-Biased Ligand in Mice. Anesth. Analg. 2018, 129, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Franchi, S.; Moretti, S.; Castelli, M.; Lattuada, D.; Scavullo, C.; Panerai, A.E.; Sacerdote, P. Mu opioid receptor activation modulates Toll like receptor 4 in murine macrophages. Brain Behav. Immun. 2012, 26, 480–488. [Google Scholar] [CrossRef]
- Aguado, D.; Bustamante, R.; Gómez de Segura, I.A. Toll-like receptor 4 deficient mice do not develop remifentanil-induced mechanical hyperalgesia. Eur. J. Anaesthesiol. 2018, 35, 1. [Google Scholar] [CrossRef]
- Jacobsen, J.H.; Watkins, L.R.; Hutchinson, M.R. Discovery of a novel site of opioid action at the innate immune pattern-recognition receptor TLR4 and its role in addiction. Int. Rev. Neurobiol. 2014, 118, 129–163. [Google Scholar] [CrossRef] [PubMed]
- Farzi, A.; Halicka, J.; Mayerhofer, R.; Fröhlich, E.E.; Tatzl, E.; Holzer, P. Toll-like receptor 4 contributes to the inhibitory effect of morphine on colonic motility in vitro and in vivo. Sci. Rep. 2015, 5, 9499. [Google Scholar] [CrossRef] [PubMed]
- Beckett, E.A.H.; Staikopoulos, V.; Hutchinson, M.R. Differential effect of morphine on gastrointestinal transit, colonic contractions and nerve-evoked relaxations in Toll-Like Receptor deficient mice. Sci. Rep. 2018, 8, 5923. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.E.; Thompson, J.; Sun, H.-S.; Leitermann, R.J.; Fujimoto, J.M.; Tseng, L.F. Nonopioidergic Mechanism Mediating Morphine-Induced Antianalgesia in the Mouse Spinal Cord. J. Pharmacol. Exp. Ther. 2004, 310, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.E.; Thompson, J.; Sun, H.-S.; Terashvili, M.; Tseng, L.F. Antianalgesia: Stereoselective Action of dextro-Morphine over levo-Morphine on Glia in the Mouse Spinal Cord. J. Pharmacol. Exp. Ther. 2005, 314, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-e.; Sun, H.-S.; Terashivili, M.; Schwasinger, E.; Sora, I.; Scott Hall, F.; Uhl, G.R.; Tseng, L.F. dextro- and levo-morphine attenuate opioid δ and κ receptor agonist produced analgesia in μ-opioid receptor knockout mice. Eur. J. Pharmacol. 2006, 531, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Johnston, I.N.; Westbrook, R.F. Acute and conditioned sickness reduces morphine analgesia. Behav. Brain Res. 2003, 142, 89–97. [Google Scholar] [CrossRef]
- Johnston, I.N.; Westbrook, R.F. Inhibition of morphine analgesia by LPS: Role of opioid and NMDA receptors and spinal glia. Behav. Brain Res. 2005, 156, 75–83. [Google Scholar] [CrossRef]
- Wu, H.-E.; Sun, H.-S.; Cheng, C.W.; Terashvili, M.; Tseng, L.F. dextro-Naloxone or levo-naloxone reverses the attenuation of morphine antinociception induced by lipopolysaccharide in the mouse spinal cord via a non-opioid mechanism. Eur. J. Neurosci. 2006, 24, 2575–2580. [Google Scholar] [CrossRef]
- Meller, S.T.; Dykstra, C.; Grzybycki, D.; Murphy, S.; Gebhart, G.F. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 1994, 33, 1471–1478. [Google Scholar] [CrossRef]
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 716, 106–119. [Google Scholar] [CrossRef]
- Jha, M.K.; Jeon, S.; Suk, K. Glia as a Link between Neuroinflammation and Neuropathic Pain. Immune Netw. 2012, 12, 41–47. [Google Scholar] [CrossRef]
- Song, P.; Zhao, Z.-Q. The involvement of glial cells in the development of morphine tolerance. Neurosci. Res. 2001, 39, 281–286. [Google Scholar] [CrossRef]
- Raghavendra, V.; Rutkowski, M.D.; DeLeo, J.A. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 9980–9989. [Google Scholar] [CrossRef]
- Raghavendra, V.; Tanga, F.; Rutkowski, M.D.; DeLeo, J.A. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: Mechanistic implications of spinal glia and proinflammatory cytokines. Pain 2003, 104, 655–664. [Google Scholar] [CrossRef]
- Raghavendra, V.; Tanga, F.Y.; DeLeo, J.A. Attenuation of Morphine Tolerance, Withdrawal-Induced Hyperalgesia, and Associated Spinal Inflammatory Immune Responses by Propentofylline in Rats. Neuropsychopharmacology 2004, 29, 327–334. [Google Scholar] [CrossRef]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef]
- Liu, T.; Gao, Y.-J.; Ji, R.-R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012, 28, 131–144. [Google Scholar] [CrossRef]
- Kofler, J.; Wiley, C.A. Microglia: Key innate immune cells of the brain. Toxicol. Pathol. 2011, 39, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Tanga, F.Y.; Nutile-McMenemy, N.; DeLeo, J.A. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. USA 2005, 102, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.; Wieseler, J.; Favret, J.; Johnson, K.W.; Rice, K.C.; Maier, S.F.; Falci, S.; Watkins, L.R. Systemic Administration of Propentofylline, Ibudilast, and (+)-Naltrexone Each Reverses Mechanical Allodynia in a Novel Rat Model of Central Neuropathic Pain. J. Pain 2014, 15, 407–421. [Google Scholar] [CrossRef]
- Sakurada, T.; Komatsu, T.; Sakurada, S. Mechanisms of Nociception Evoked by Intrathecal High-dose Morphine. NeuroToxicology 2005, 26, 801–809. [Google Scholar] [CrossRef]
- Eidson, L.N.; Murphy, A.Z. Persistent peripheral inflammation attenuates morphine-induced periaqueductal gray glial cell activation and analgesic tolerance in the male rat. J. Pain Off. J. Am. Pain Soc. 2013, 14, 393–404. [Google Scholar] [CrossRef]
- Eidson, L.N.; Murphy, A.Z. Blockade of Toll-Like Receptor 4 Attenuates Morphine Tolerance and Facilitates the Pain Relieving Properties of Morphine. J. Neurosci. 2013, 33, 15952–15963. [Google Scholar] [CrossRef]
- Juni, A.; Klein, G.; Pintar, J.E.; Kest, B. Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 2007, 147, 439–444. [Google Scholar] [CrossRef]
- Liu, L.; Coller, J.K.; Watkins, L.R.; Somogyi, A.A.; Hutchinson, M.R. Naloxone-precipitated morphine withdrawal behavior and brain IL-1β expression: Comparison of different mouse strains. Brain Behav. Immun. 2011, 25, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Due, M.R.; Piekarz, A.D.; Wilson, N.; Feldman, P.; Ripsch, M.S.; Chavez, S.; Yin, H.; Khanna, R.; White, F.A. Neuroexcitatory effects of morphine-3-glucuronide are dependent on Toll-like receptor 4 signaling. J. Neuroinflamm. 2012, 9, 725. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Rolan, P.E.; Johnson, M.E.; Bobrovskaya, L.; Williams, D.B.; Johnson, K.; Tuke, J.; Hutchinson, M.R. Codeine-induced hyperalgesia and allodynia: Investigating the role of glial activation. Transl. Psychiatry 2014, 4, e482. [Google Scholar] [CrossRef]
- Ferrini, F.; Trang, T.; Mattioli, T.-A.M.; Laffray, S.; Del’Guidice, T.; Lorenzo, L.-E.; Castonguay, A.; Doyon, N.; Zhang, W.; Godin, A.G.; et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl−homeostasis. Nat. Neurosci. 2013, 16, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, H.; Koyama, T.; Kakuyama, M.; Fukuda, K. Microglial activation involved in morphine tolerance is not mediated by toll-like receptor 4. J. Anesth. 2013, 27, 93–97. [Google Scholar] [CrossRef]
- Mattioli, T.A.; Leduc-Pessah, H.; Skelhorne-Gross, G.; Nicol, C.J.B.; Milne, B.; Trang, T.; Cahill, C.M. Toll-Like Receptor 4 Mutant and Null Mice Retain Morphine-Induced Tolerance, Hyperalgesia, and Physical Dependence. PLoS ONE 2014, 9, e97361. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, H.U.; Cremonini, F.; Park, M.I.; Camilleri, M. Opioids and the gut: Pharmacology and current clinical experience. Neurogastroenterol. Motil. 2004, 16, 383–394. [Google Scholar] [CrossRef]
- Nee, J.; Rangan, V.; Lembo, A. Reduction in pain: Is it worth the gain? The effect of opioids on the GI tract. Neurogastroenterol. Motil. 2018, 30, e13367. [Google Scholar] [CrossRef] [PubMed]
- Testro, A.G.; Visvanathan, K. Toll-like receptors and their role in gastrointestinal disease. J. Gastroenterol. Hepatol. 2009, 24, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Opioid-Induced Constipation: Challenges and Therapeutic Opportunities. Am. J. Gastroenterol. 2011, 106, 835–842. [Google Scholar] [CrossRef]
- Baldini, A.; Von Korff, M.; Lin, E.H.B. A Review of Potential Adverse Effects of Long-Term Opioid Therapy: A Practitioner’s Guide. Prim. Care Companion CNS Disord. 2012, 14, PCC.11m01326. [Google Scholar] [CrossRef]
- Kashani, B.; Zandi, Z.; Pourbagheri-Sigaroodi, A.; Bashash, D.; Ghaffari, S.H. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J. Cell Physiol. 2021, 236, 4121–4137. [Google Scholar] [CrossRef]
- Awasthi, S. Toll-Like Receptor-4 Modulation for Cancer Immunotherapy. Front. Immunol. 2014, 5, 328. [Google Scholar] [CrossRef]
- Sheikh, M.; Shakeri, R.; Poustchi, H.; Pourshams, A.; Etemadi, A.; Islami, F.; Khoshnia, M.; Gharavi, A.; Roshandel, G.; Khademi, H.; et al. Opium use and subsequent incidence of cancer: Results from the Golestan Cohort Study. Lancet Glob. Health 2020, 8, e649–e660. [Google Scholar] [CrossRef]
- Ramirez, M.F.; Gorur, A.; Cata, J.P. Opioids and cancer prognosis: A summary of the clinical evidence. Neurosci. Lett. 2021, 746, 135661. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; He, J.; Wang, W.; Zhou, H.; Cai, S.; Zhu, L.; Qian, X.; Wang, J.; Lu, Z.; Huang, C. The impact of pain and opioids use on survival in cancer patients: Results from a population-based cohort study and a meta-analysis. Medicine 2020, 99, e19306. [Google Scholar] [CrossRef]
- Sekandarzad, M.W.; Doornebal, C.; Hollmann, M.W. Opiophobia in Cancer Biology- Justified?—The Role of Perioperative Use of Opioids in Cancer Recurrence. Curr. Pharm. Des. 2019, 25, 3020–3027. [Google Scholar] [CrossRef]
- Sessler, D.I.; Pei, L.; Huang, Y.; Fleischmann, E.; Marhofer, P.; Kurz, A.; Mayers, D.B.; Meyer-Treschan, T.A.; Grady, M.; Tan, E.Y.; et al. Recurrence of breast cancer after regional or general anaesthesia: A randomised controlled trial. Lancet 2019, 394, 1807–1815. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).