
HMGA2 Supports Cancer Hallmarks in Triple-Negative Breast Cancer

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis of Human Patient Samples
2.2. Cell Culture and Lentiviral Transductions
2.3. qRT-PCR Gene Expression
2.4. Western Blot
2.5. Immunohistochemical Staining
2.6. ELISA Assay
2.7. Crystal Violet Assay
2.8. EDU Assay
2.9. Cell Cycle FACS Assay
2.10. Migration Assay
2.11. Invasion Assay
2.12. Colony Formation Assay
2.13. Spheroid Assay
2.14. CD44 and CD24 FACS Assay
2.15. Xenograft Model
2.16. Statistical Analysis
3. Results
3.1. HMGA2 Is Overexpressed in Triple-Negative Breast Cancer
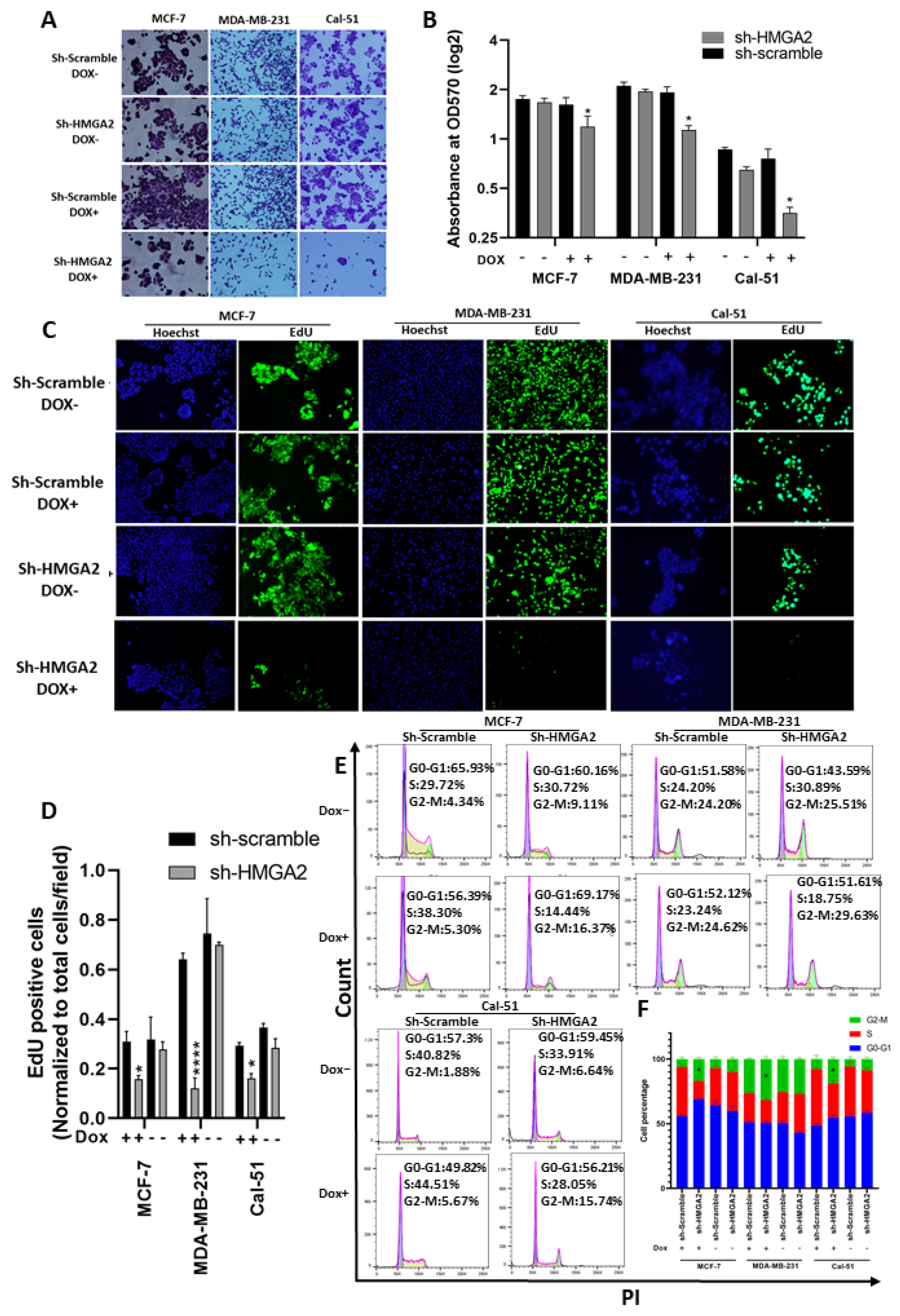
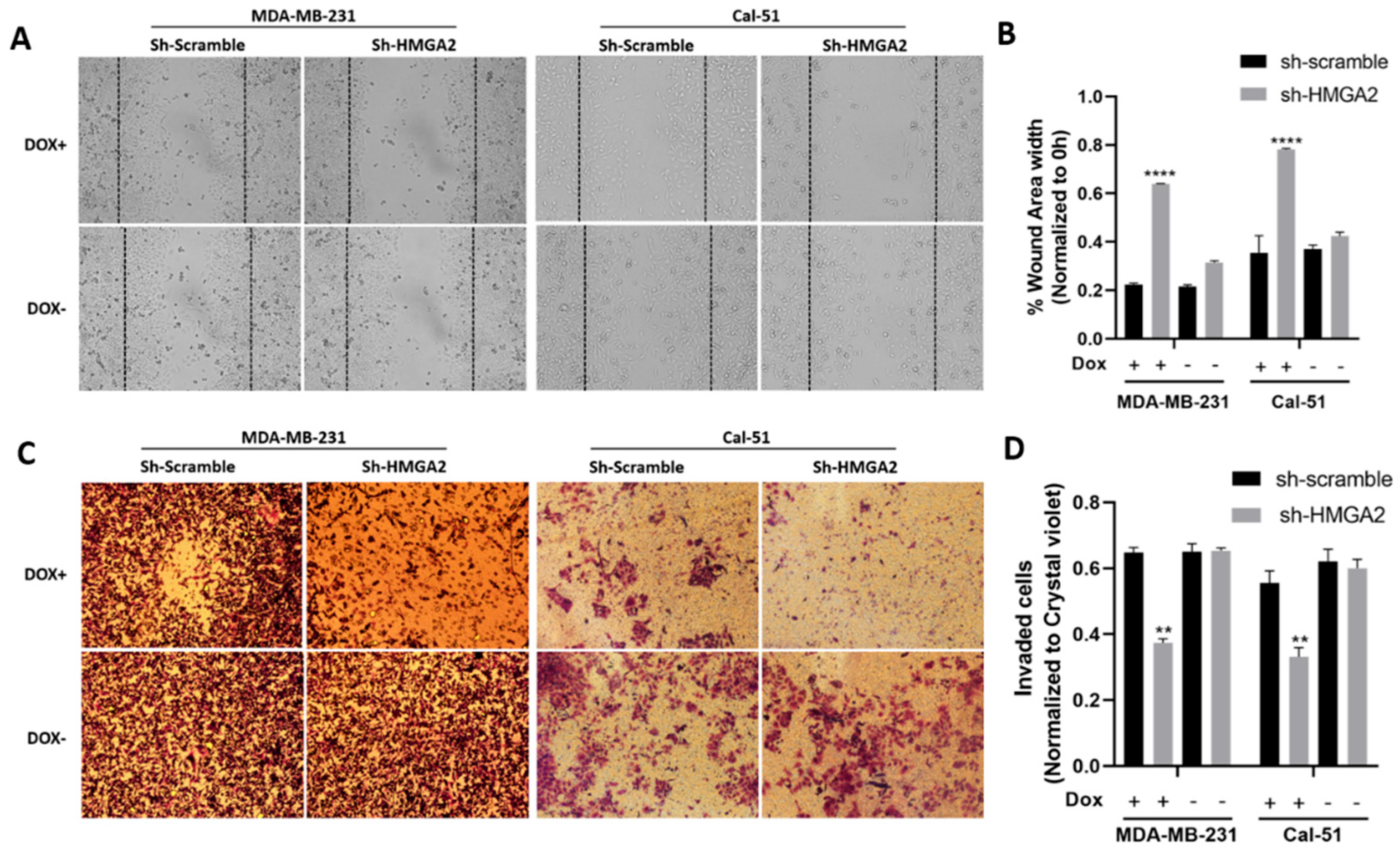
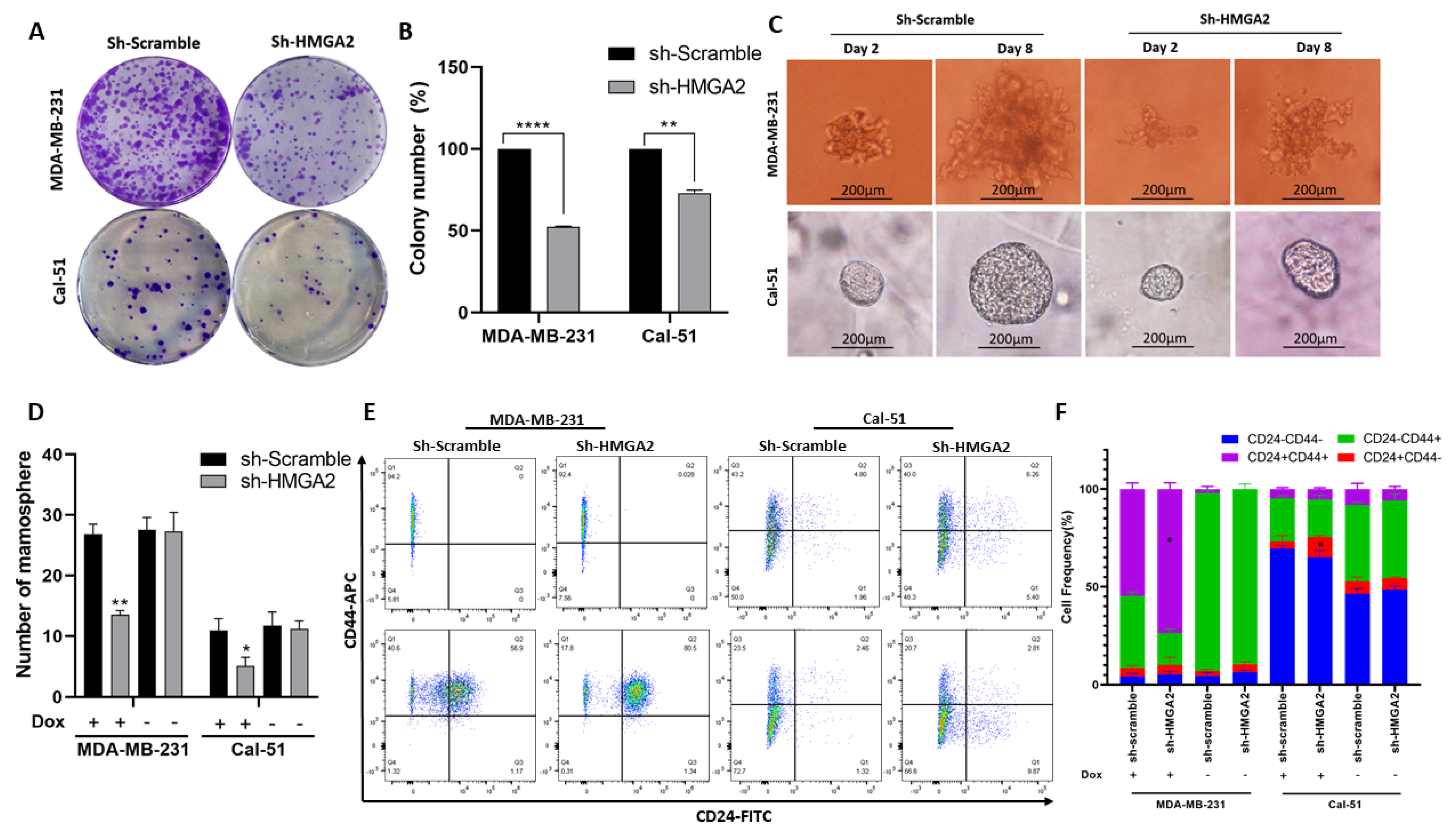
3.2. HMGA2 Supports Hallmark Phenotypes of TNBC Cells
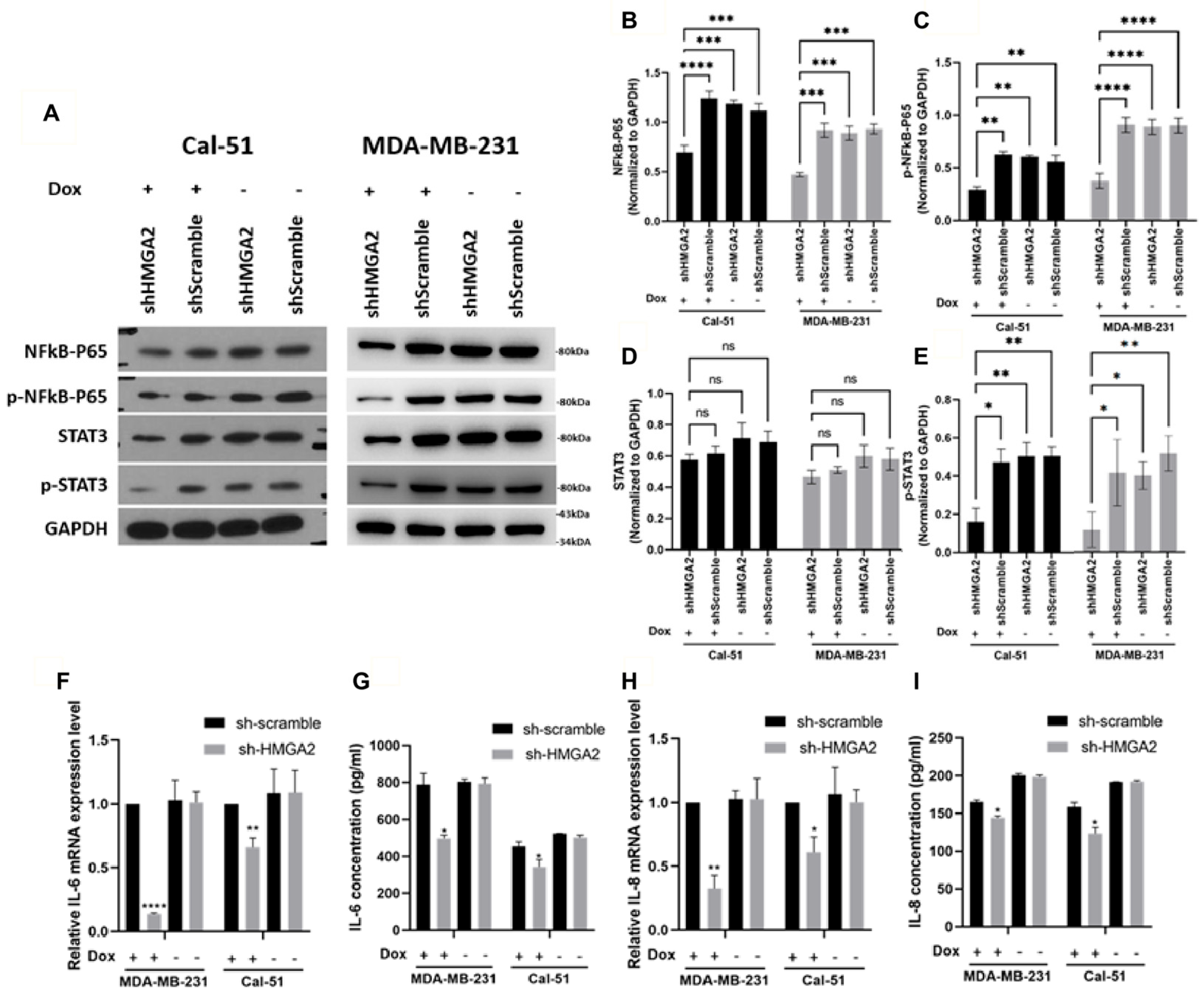
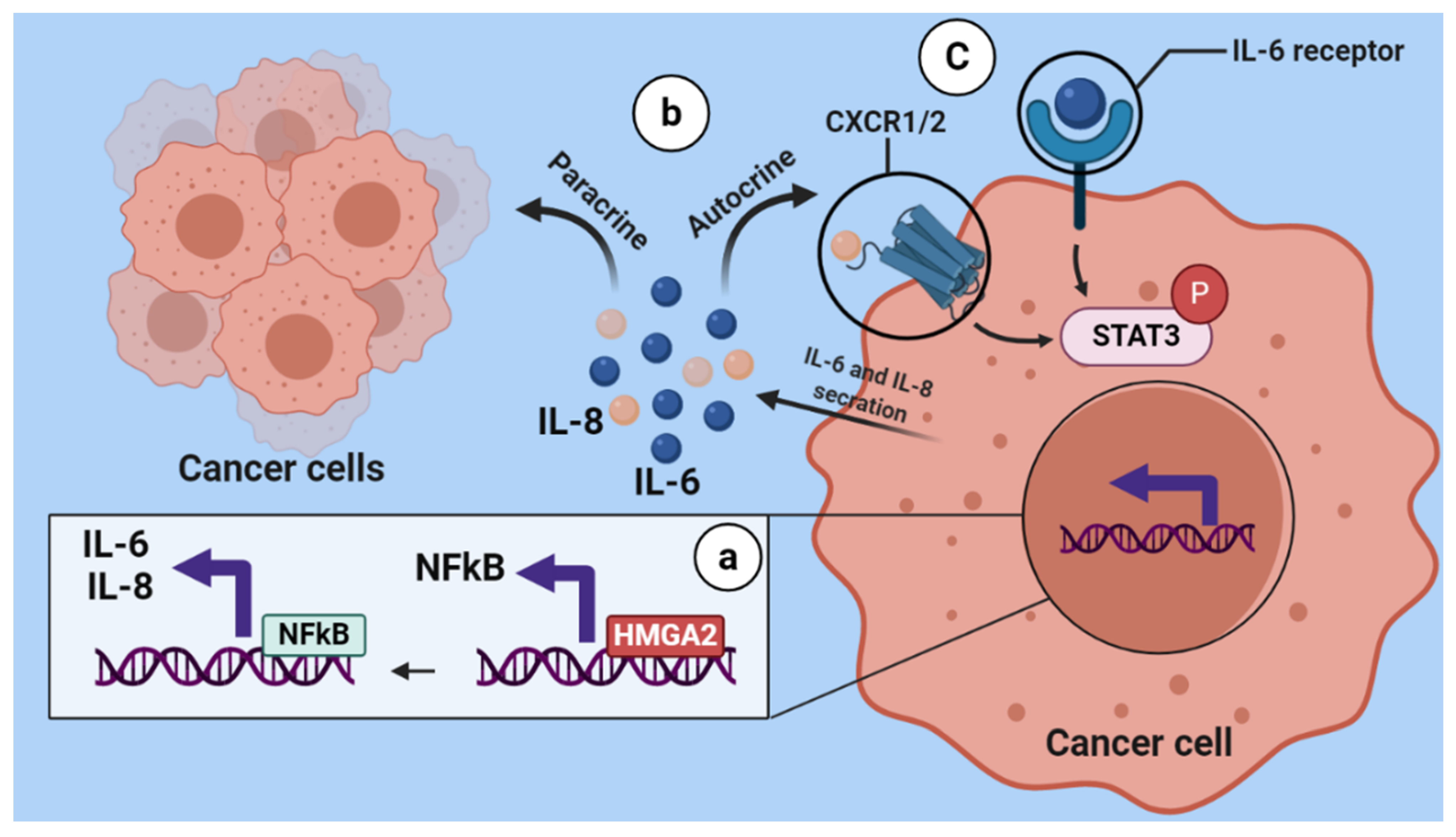
3.3. HMGA2 Silencing Affects NF-kβ/IL-6/STAT3 Signaling
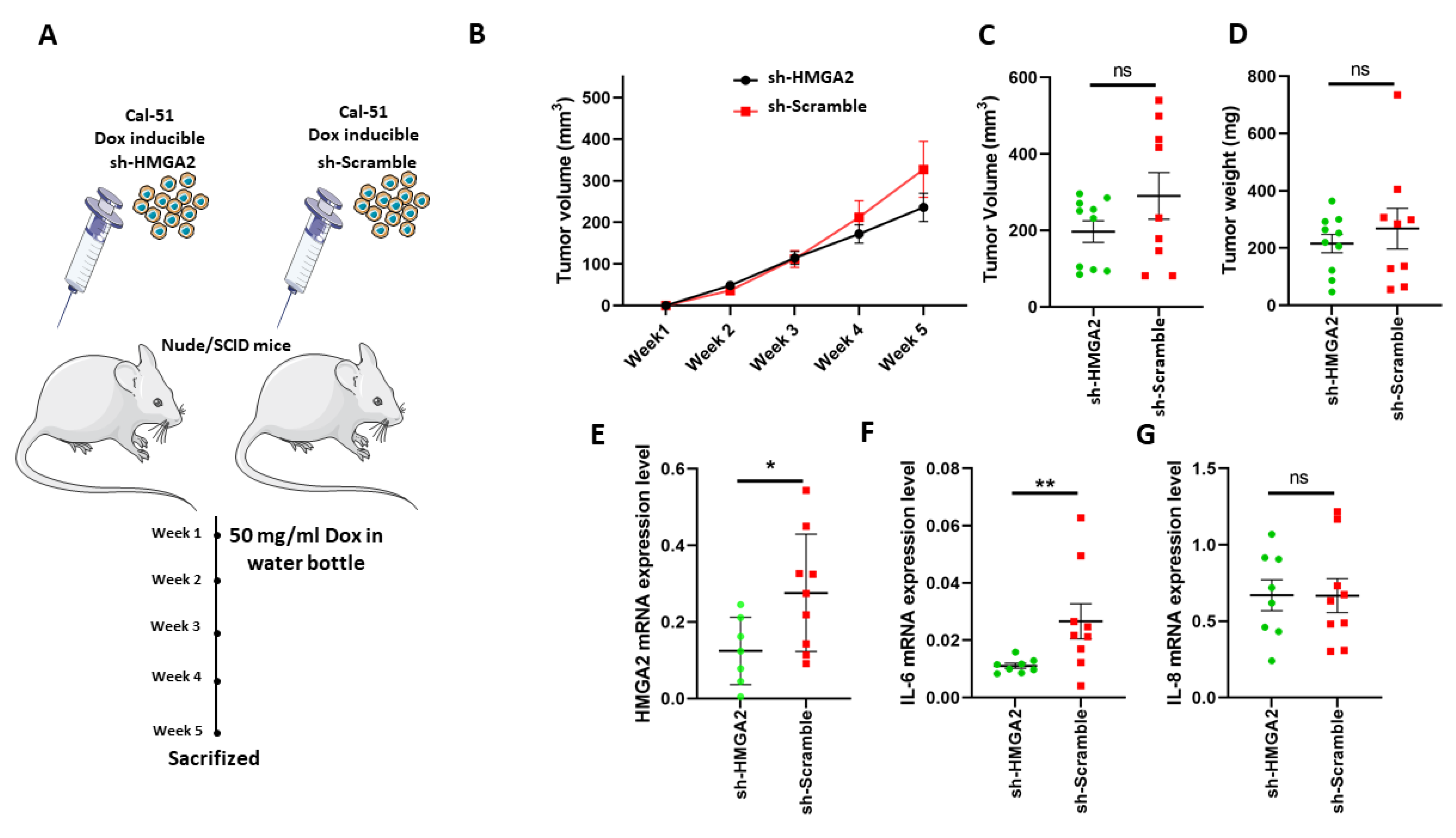
3.4. HMGA2 Silencing Decreased IL-6 Levels in TNBC Xenograft Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Voduc, K.D.; Cheang, M.C.; Tyldesley, S.; Gelmon, K.; Nielsen, T.O.; Kennecke, H. Breast cancer subtypes and the risk of local and regional relapse. J. Clin. Oncol. 2010, 28, 1684–1691. [Google Scholar] [CrossRef]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef]
- Anders, C.; Carey, L.A. Understanding and treating triple-negative breast cancer. Oncology 2008, 22, 1233. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. HMGI-C suppressing induces P53/caspase9 axis to regulate apoptosis in breast adenocarcinoma cells. Cell Cycle 2016, 15, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Vignali, R.; Marracci, S. HMGA Genes and Proteins in Development and Evolution. Int. J. Mol. Sci. 2020, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Ditzel, H.J.; Duijf, P.H.; Khaze, V.; Gjerstorff, M.F.; Baradaran, B. HMGA2 as a Critical Regulator in Cancer Development. Genes 2021, 12, 269. [Google Scholar] [CrossRef] [PubMed]
- Esmailzadeh, S.; Mansoori, B.; Mohammadi, A.; Shanehbandi, D.; Baradaran, B. siRNA-Mediated Silencing of HMGA2 Induces Apoptosis and Cell Cycle Arrest in Human Colorectal Carcinoma. J. Gastrointest. Cancer 2017, 48, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, J.; Cheng, Q.; Xu, P.; Wang, J.; Zhang, Z.; Fan, W.; Wang, P.; Yu, M. Prognostic value of HMGA2 in human cancers: A meta-analysis based on literatures and TCGA datasets. Front. Physiol. 2018, 9, 776. [Google Scholar] [CrossRef]
- Mansoori, B.; Duijf, P.H.G.; Mohammadi, A.; Najafi, S.; Roshani, E.; Shanehbandi, D.; Hajiasgharzadeh, K.; Shirjang, S.; Ditzel, H.J.; Kazemi, T.; et al. Overexpression of HMGA2 in breast cancer promotes cell proliferation, migration, invasion and stemness. Expert Opin. Ther. Targets 2020, 24, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.-H.; Moustakas, A. Transforming growth factor-β employs HMGA2 to elicit epithelial–mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Parisi, S.; Piscitelli, S.; Passaro, F.; Russo, T. HMGA Proteins in Stemness and Differentiation of Embryonic and Adult Stem Cells. Int. J. Mol. Sci. 2020, 21, 362. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Asadzadeh, Z.; Shirjang, S.; Minouei, M.; Abedi Gaballu, F.; Shajari, N.; Kazemi, T.; Gjerstorff, M.F.; Duijf, P.H.G.; et al. HMGA2 and Bach-1 cooperate to promote breast cancer cell malignancy. J. Cell. Physiol. 2019, 234, 17714–17726. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma'ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Subramanian, A.; Kuehn, H.; Gould, J.; Tamayo, P.; Mesirov, J.P. GSEA-P: A desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007, 23, 3251–3253. [Google Scholar] [CrossRef]
- Golovine, K.; Uzzo, R.G.; Makhov, P.; Crispen, P.L.; Kunkle, D.; Kolenko, V.M. Depletion of intracellular zinc increases expression of tumorigenic cytokines VEGF, IL-6 and IL-8 in prostate cancer cells via NF-κB-dependent pathway. Prostate 2008, 68, 1443–1449. [Google Scholar] [CrossRef]
- McFarland, B.C.; Gray, G.K.; Nozell, S.E.; Hong, S.W.; Benveniste, E.N. Activation of the NF-kappaB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 2013, 11, 494–505. [Google Scholar] [CrossRef]
- McFarland, B.C.; Hong, S.W.; Rajbhandari, R.; Twitty, G.B., Jr.; Gray, G.K.; Yu, H.; Benveniste, E.N.; Nozell, S.E. NF-kappaB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS ONE 2013, 8, e78728. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Naghizadeh, S.; Mansoori, B.; Mohammadi, A.; Kafil, H.S.; Mousavi, Z.; Sakhinia, E.; Baradaran, B. Effects of HMGA2 gene downregulation by siRNA on lung carcinoma cell migration in A549 cell lines. J. Cell. Biochem. 2019, 120, 5024–5032. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.; Bakhidze, E.; Noske, A.; Sers, C.; Aigner, A.; Schäfer, R.; Tchernitsa, O. HMGA2 gene is a promising target for ovarian cancer silencing therapy. Int. J. Cancer 2008, 123, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Krishnakumar, S.; Deepa, P.R.; Deepa, M.; Khetan, V.; Reddy, M.A. Molecular deregulation induced by silencing of the high mobility group protein A2 gene in retinoblastoma cells. Mol. Vis. 2012, 18, 2420–2437. [Google Scholar]
- Mansoori, B.; Mohammadi, A.; Naghizadeh, S.; Gjerstorff, M.; Shanehbandi, D.; Shirjang, S.; Najafi, S.; Holmskov, U.; Khaze, V.; Duijf, P.H.G.; et al. miR-330 suppresses EMT and induces apoptosis by downregulating HMGA2 in human colorectal cancer. J. Cell. Physiol. 2020, 235, 920–931. [Google Scholar] [CrossRef]
- Mansoori, B.; Duijf, P.H.; Mohammadi, A.; Safarzadeh, E.; Ditzel, H.J.; Gjerstorff, M.F.; Cho, W.C.-S.; Baradaran, B. MiR-142-3p targets HMGA2 and suppresses breast cancer malignancy. Life Sci. 2021, 276, 119431. [Google Scholar] [CrossRef]
- Sun, J.; Sun, B.; Zhu, D.; Zhao, X.; Zhang, Y.; Dong, X.; Che, N.; Li, J.; Liu, F.; Zhao, N.; et al. HMGA2 regulates CD44 expression to promote gastric cancer cell motility and sphere formation. Am. J. Cancer Res. 2017, 7, 260–274. [Google Scholar]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. Onco Targets 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Nakshatri, H.; Goulet, R.J., Jr. NF-κB and breast cancer. Curr. Probl. Cancer 2002, 26, 282–309. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Brasier, A.R. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef]
- Dethlefsen, C.; Højfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef]
- Todorović-Raković, N.; Milovanović, J. Interleukin-8 in breast cancer progression. J. Interferon Cytokine Res. 2013, 33, 563–570. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Shibayama, O.; Yoshiuchi, K.; Inagaki, M.; Matsuoka, Y.; Yoshikawa, E.; Sugawara, Y.; Akechi, T.; Wada, N.; Imoto, S.; Murakami, K.; et al. Association between adjuvant regional radiotherapy and cognitive function in breast cancer patients treated with conservation therapy. Cancer Med. 2014, 3, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Umanzor, F.G.; Shah, V.; Meirson, T.; Russo, G.; Starich, B.; Tyle, P.; Lee, J.S.H.; Khatau, S.; Gil-Henn, H.; et al. Tumor cell density regulates matrix metalloproteinases for enhanced migration. Oncotarget 2018, 9, 32556–32569. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansoori, B.; Terp, M.G.; Mohammadi, A.; Pedersen, C.B.; Ditzel, H.J.; Baradaran, B.; Gjerstorff, M.F. HMGA2 Supports Cancer Hallmarks in Triple-Negative Breast Cancer. Cancers 2021, 13, 5197. https://doi.org/10.3390/cancers13205197
Mansoori B, Terp MG, Mohammadi A, Pedersen CB, Ditzel HJ, Baradaran B, Gjerstorff MF. HMGA2 Supports Cancer Hallmarks in Triple-Negative Breast Cancer. Cancers. 2021; 13(20):5197. https://doi.org/10.3390/cancers13205197
Chicago/Turabian StyleMansoori, Behzad, Mikkel Green Terp, Ali Mohammadi, Christina Bøg Pedersen, Henrik Jørn Ditzel, Behzad Baradaran, and Morten Frier Gjerstorff. 2021. "HMGA2 Supports Cancer Hallmarks in Triple-Negative Breast Cancer" Cancers 13, no. 20: 5197. https://doi.org/10.3390/cancers13205197
APA StyleMansoori, B., Terp, M. G., Mohammadi, A., Pedersen, C. B., Ditzel, H. J., Baradaran, B., & Gjerstorff, M. F. (2021). HMGA2 Supports Cancer Hallmarks in Triple-Negative Breast Cancer. Cancers, 13(20), 5197. https://doi.org/10.3390/cancers13205197