
A Mathematical Modeling Approach for Targeted Radionuclide and Chimeric Antigen Receptor T Cell Combination Therapy

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
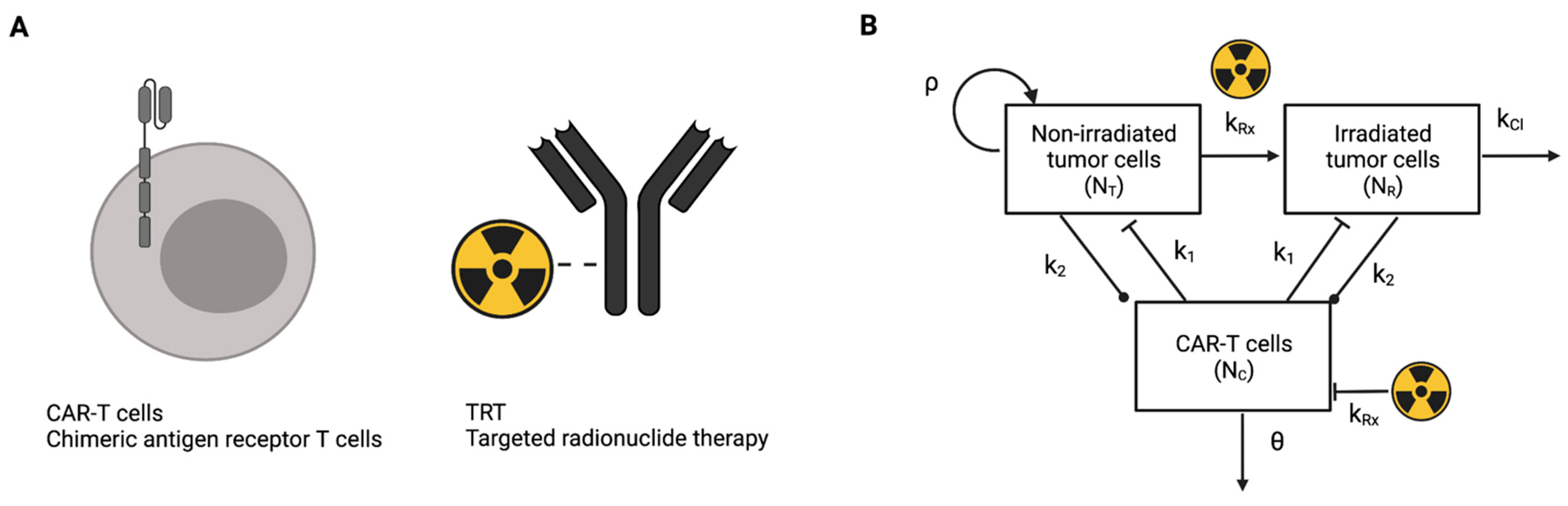
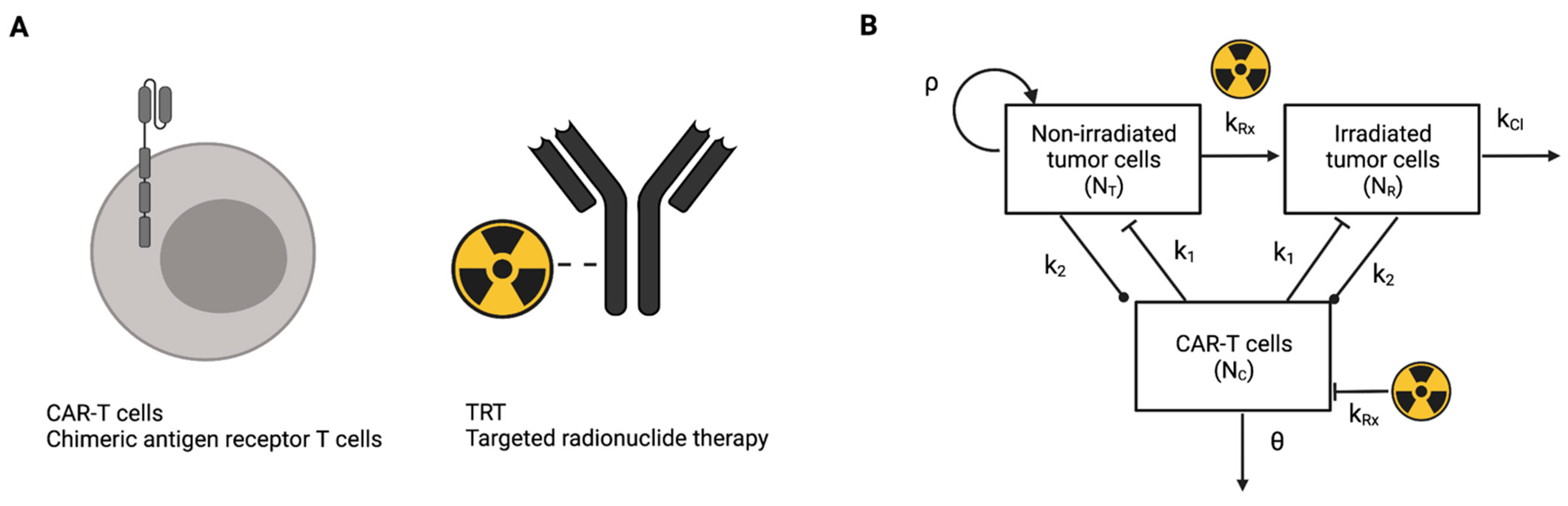
2.1. Mathematical Model
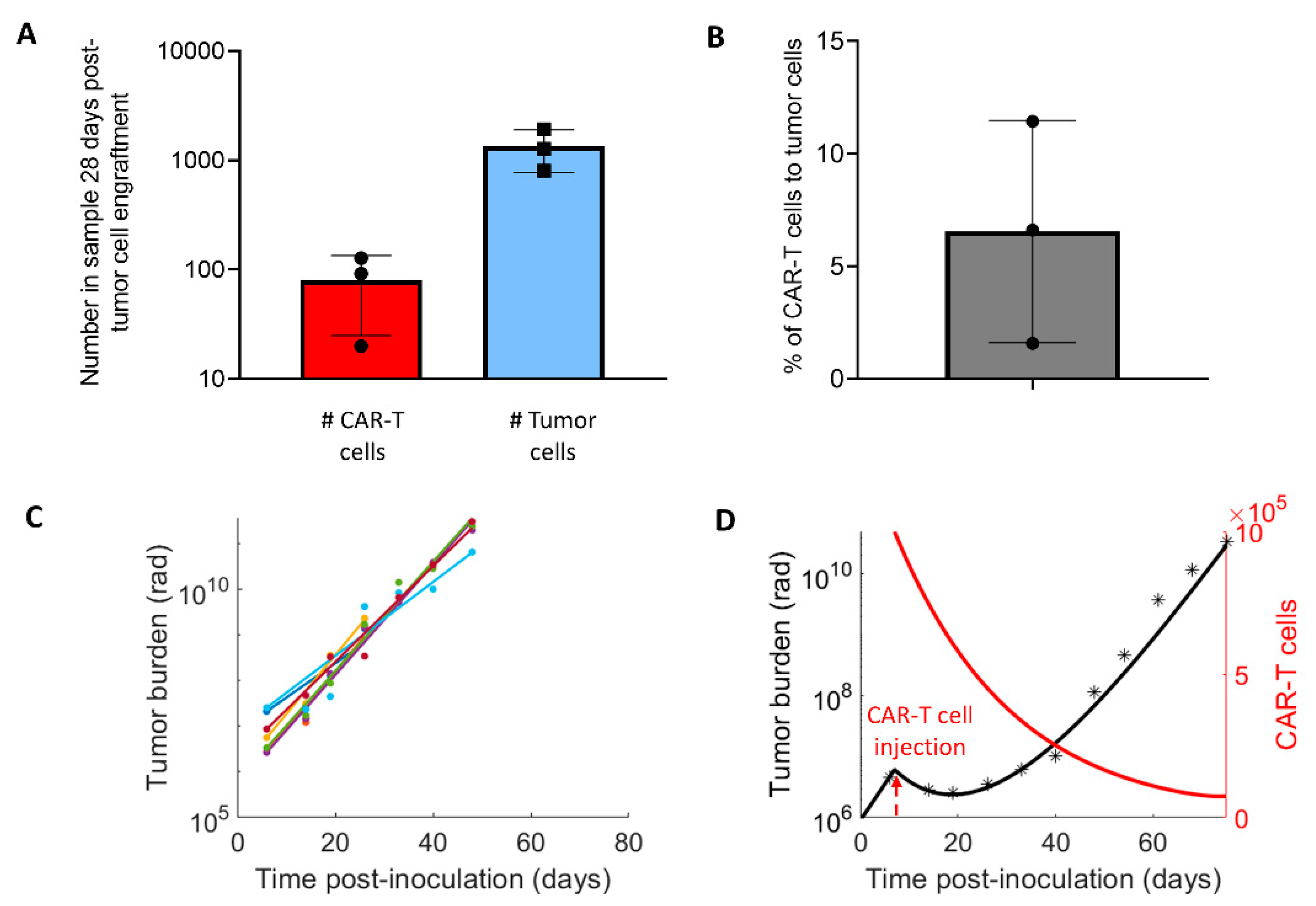
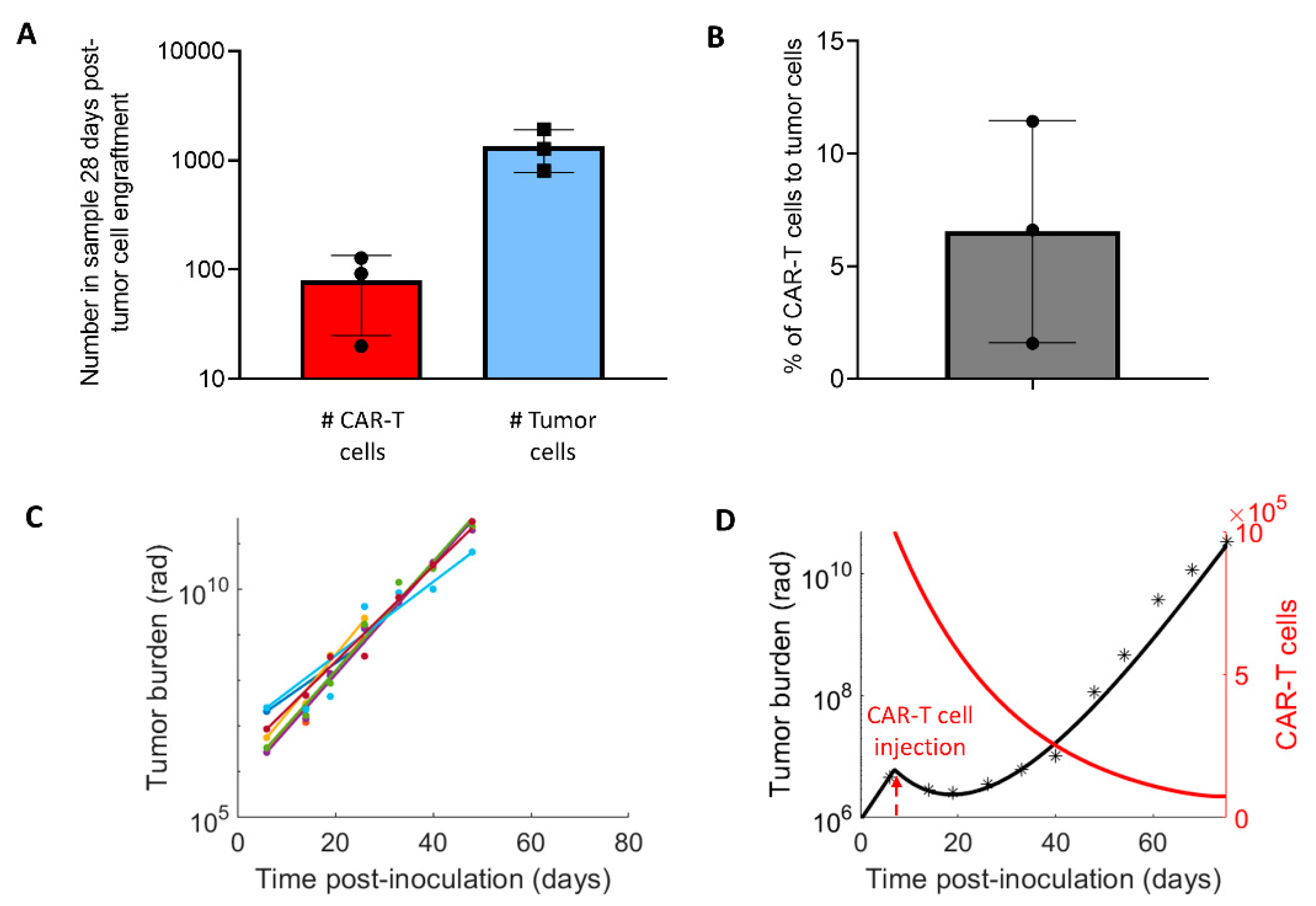
2.2. Experimental Design and Model Parametrization
2.3. Mathematical Model Simulations and Analysis
3. Results
3.1. Parameters for the CAR-T Treatment Model
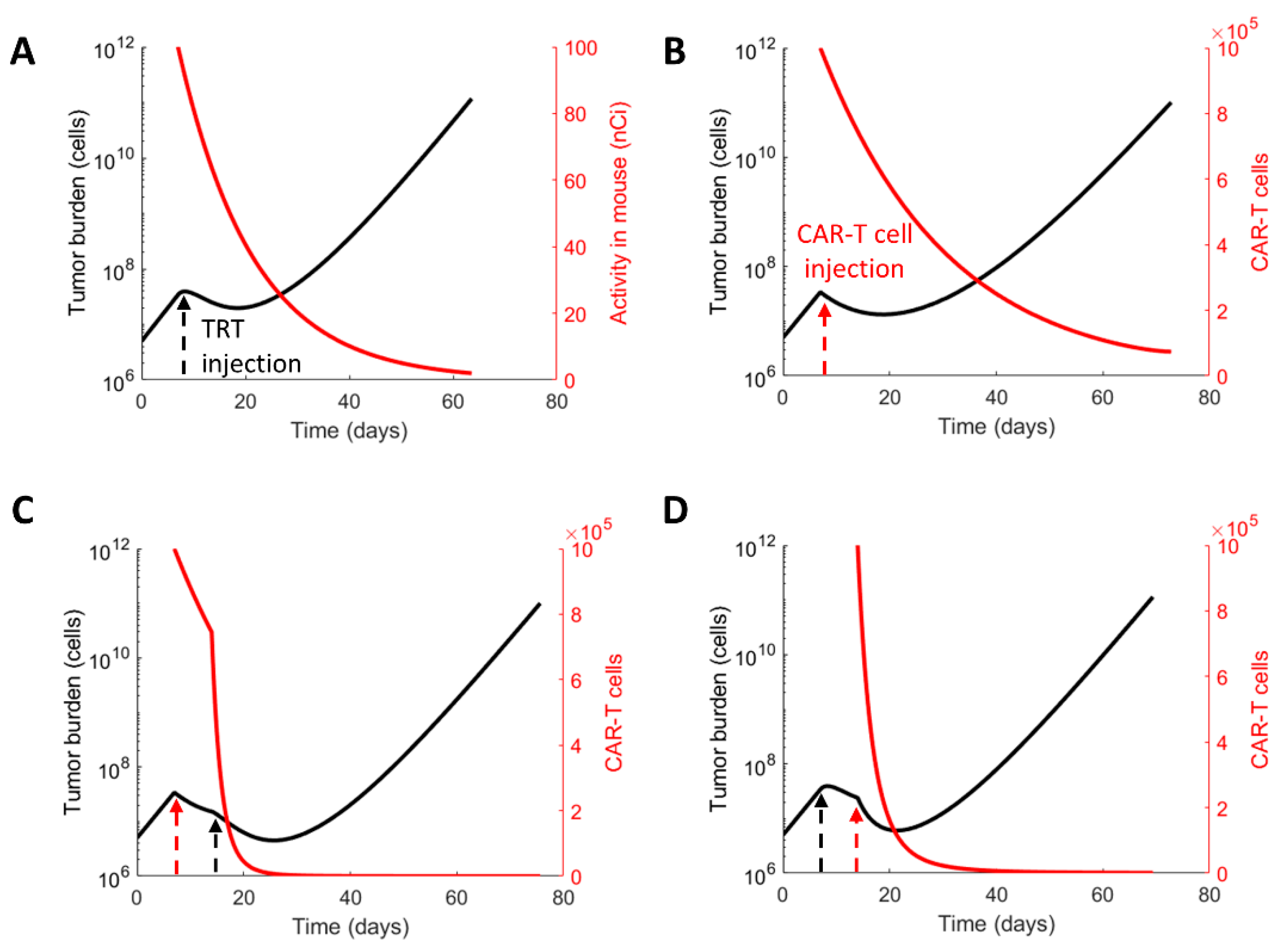
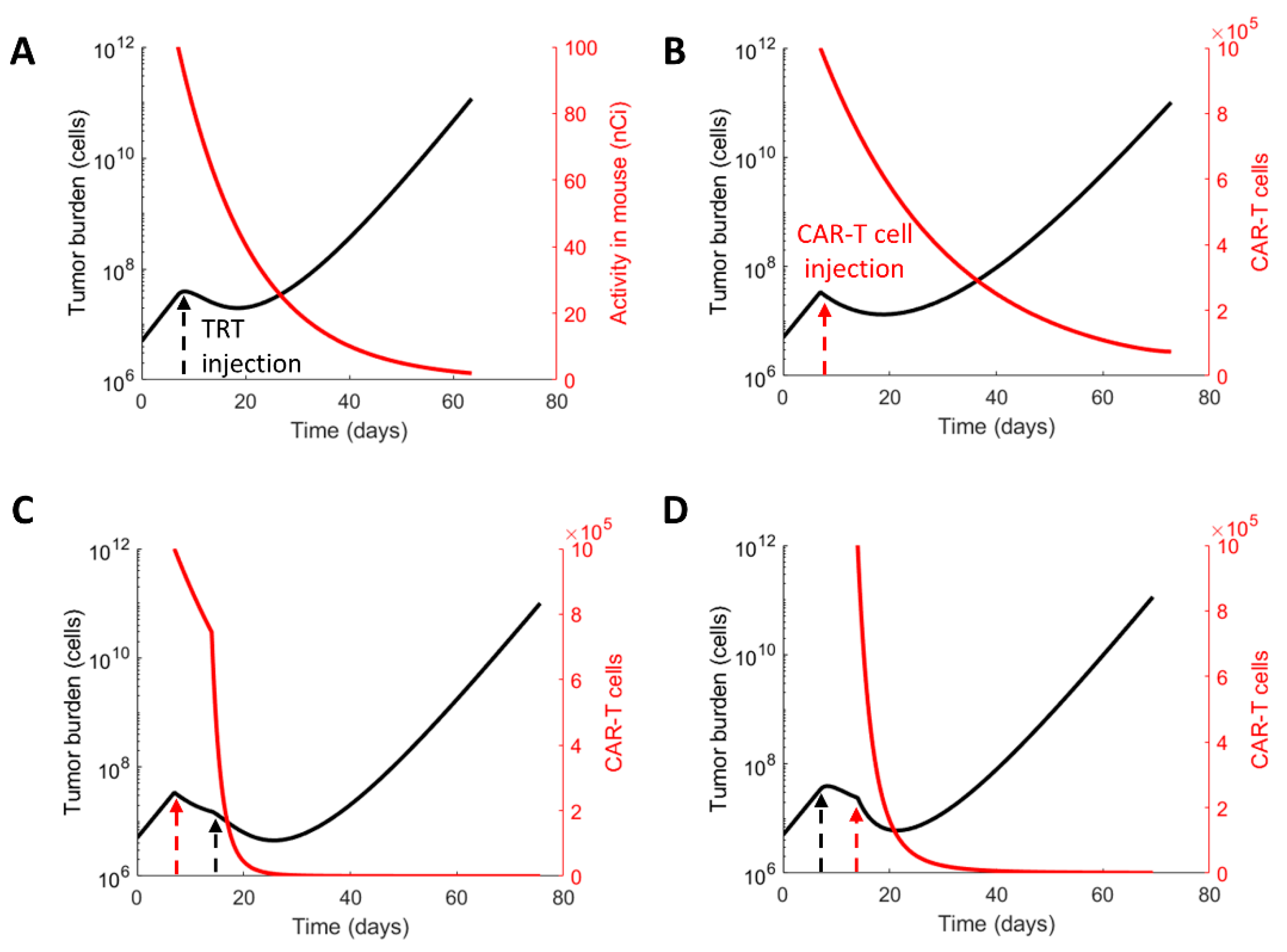
3.2. Evaluating the Therapeutic Regimens
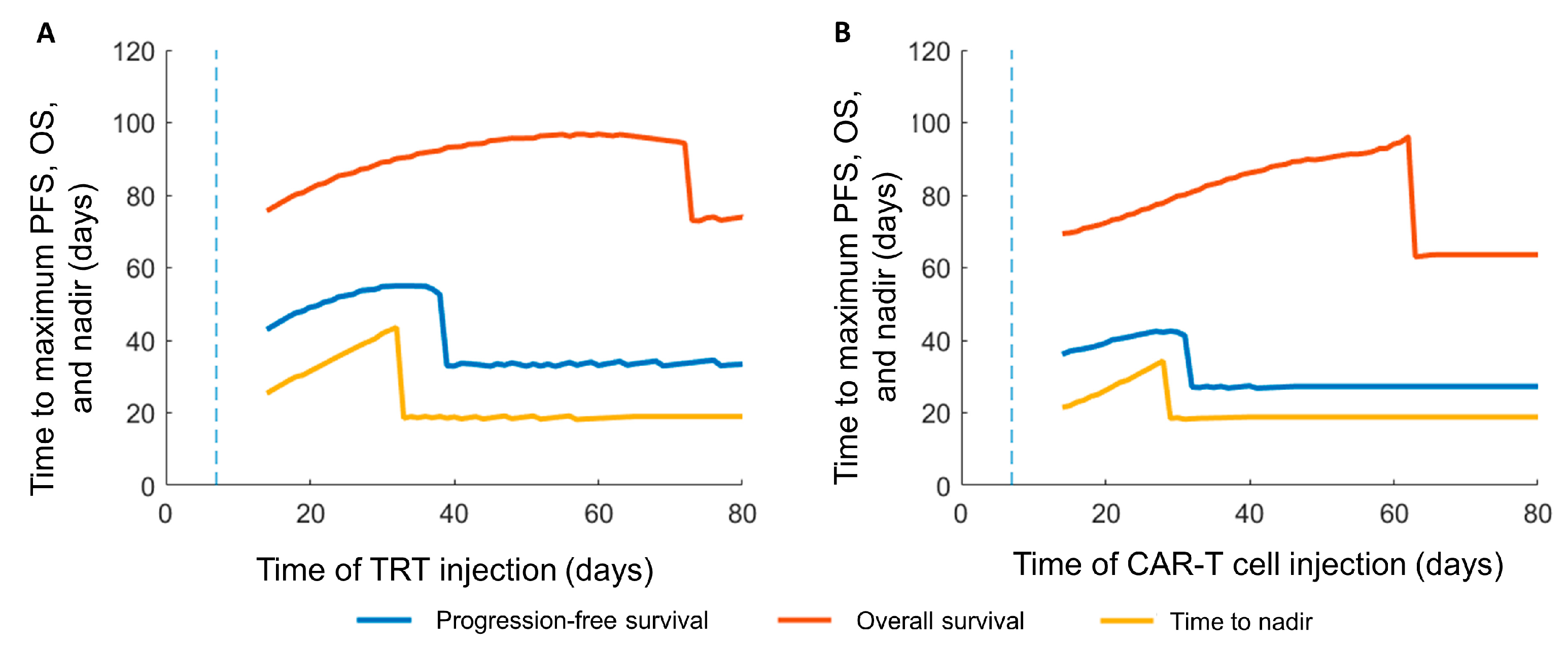
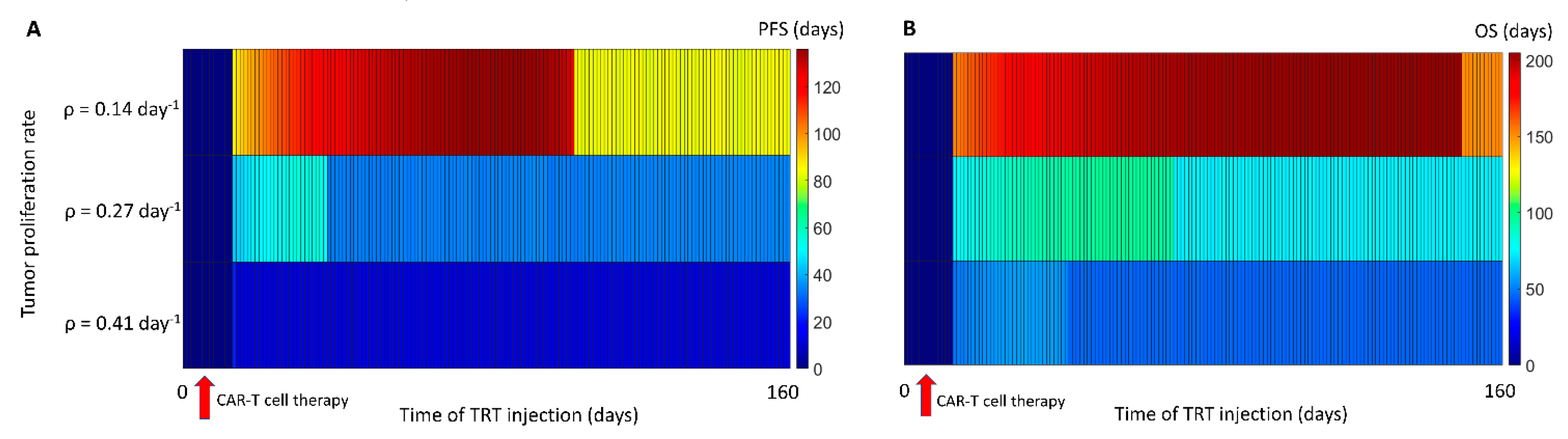
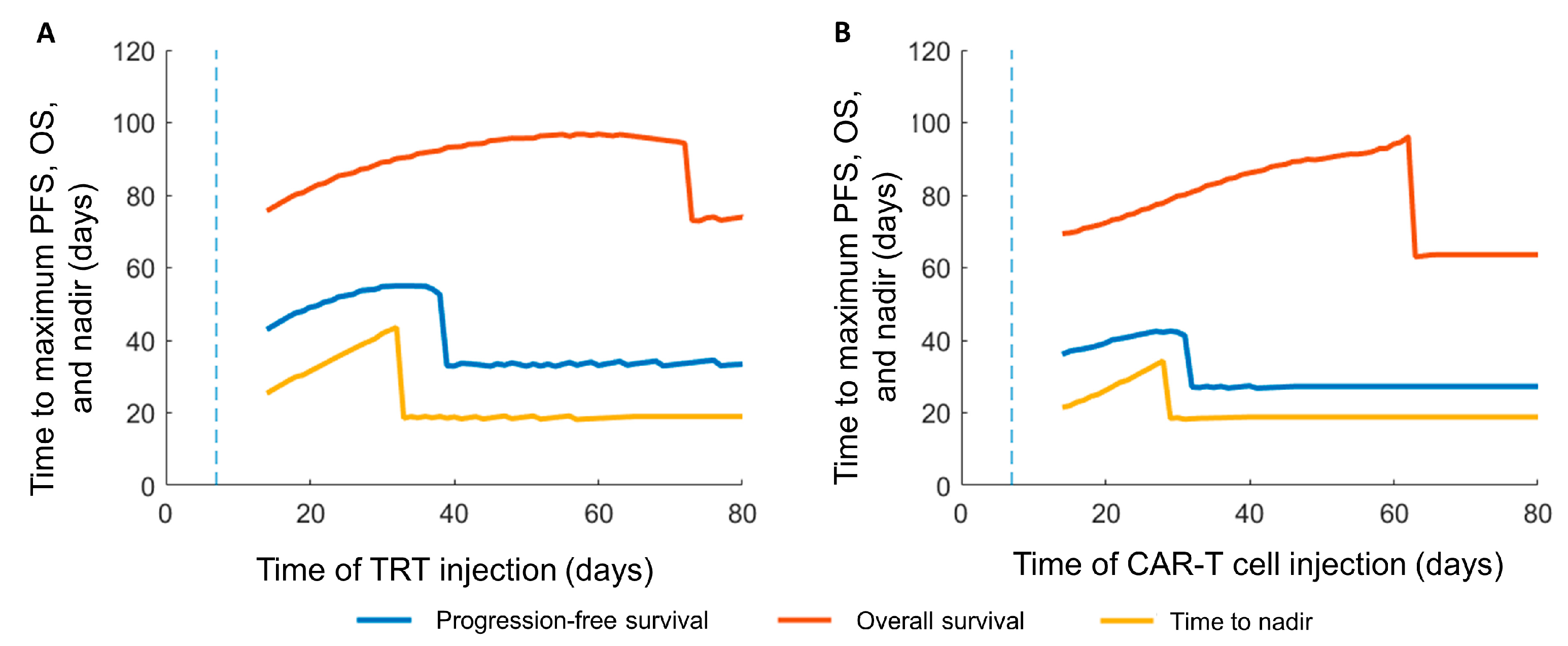
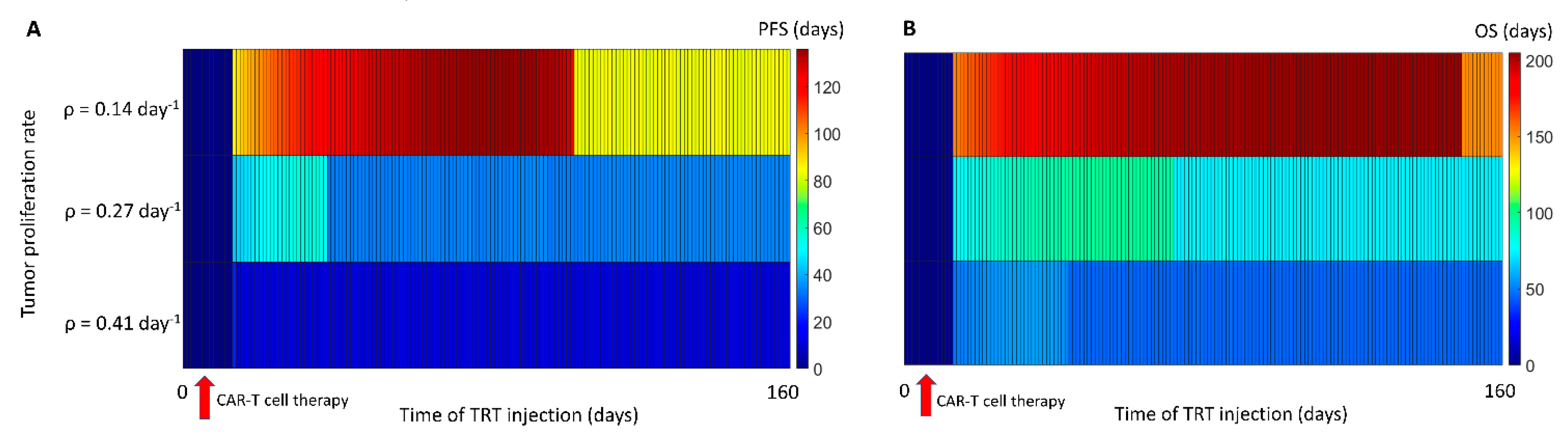
3.3. Sequence of the Therapy and Maximizing Survival
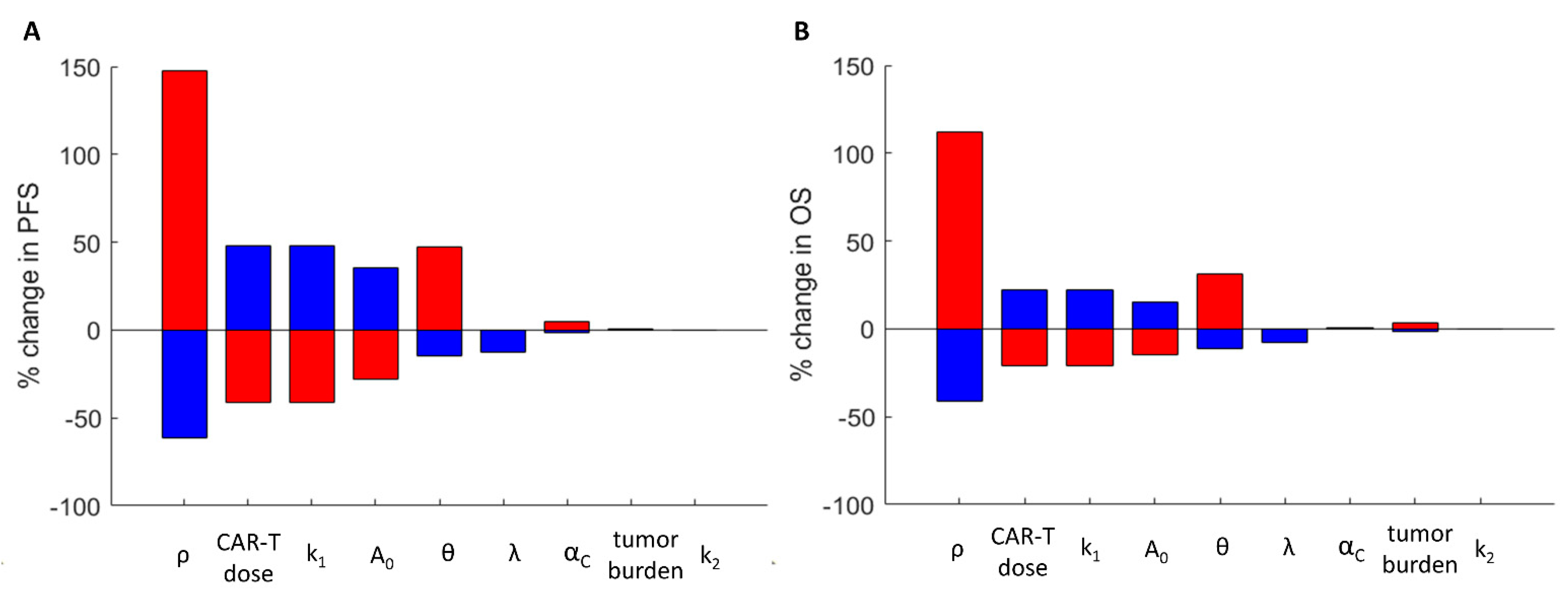
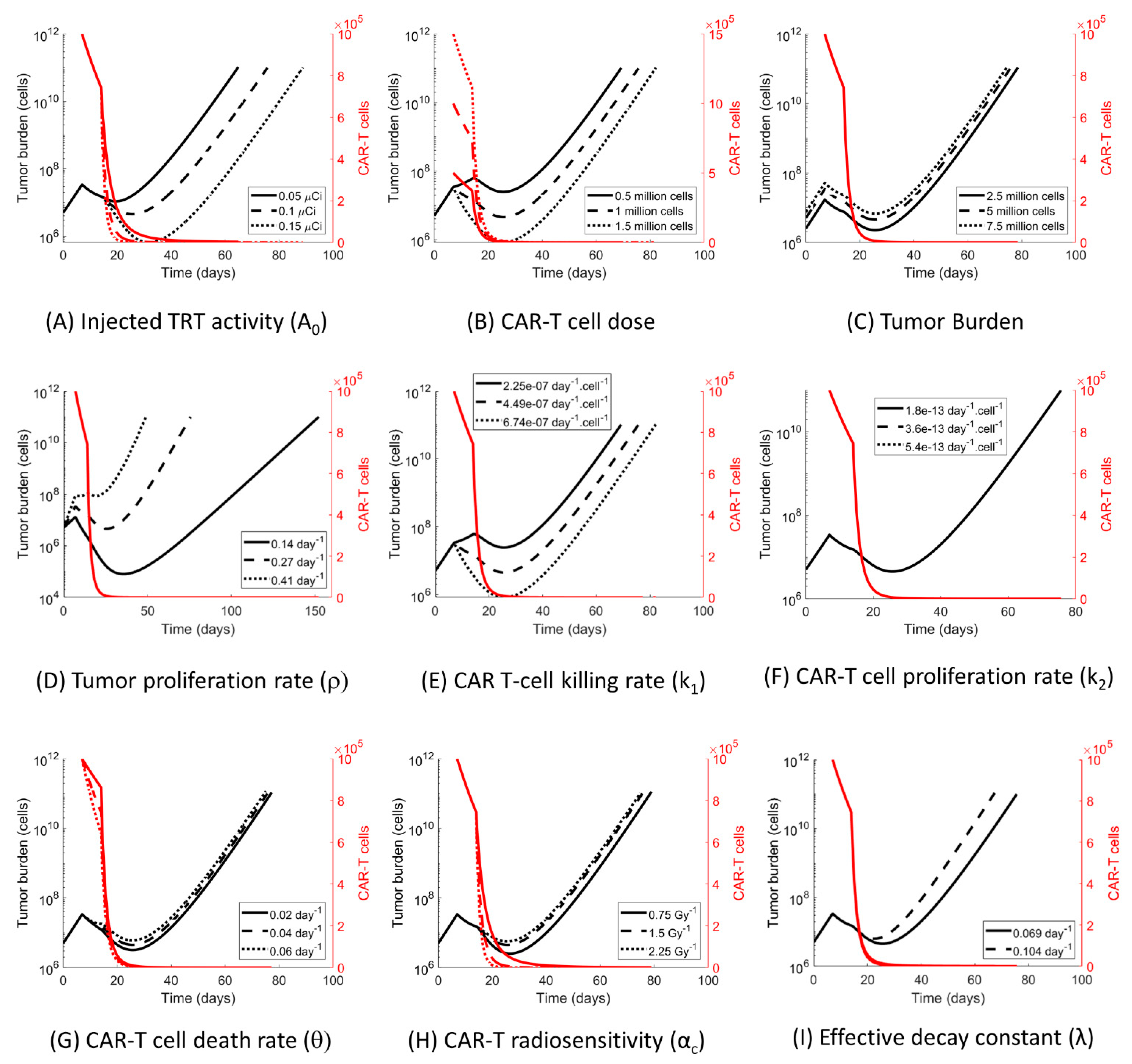
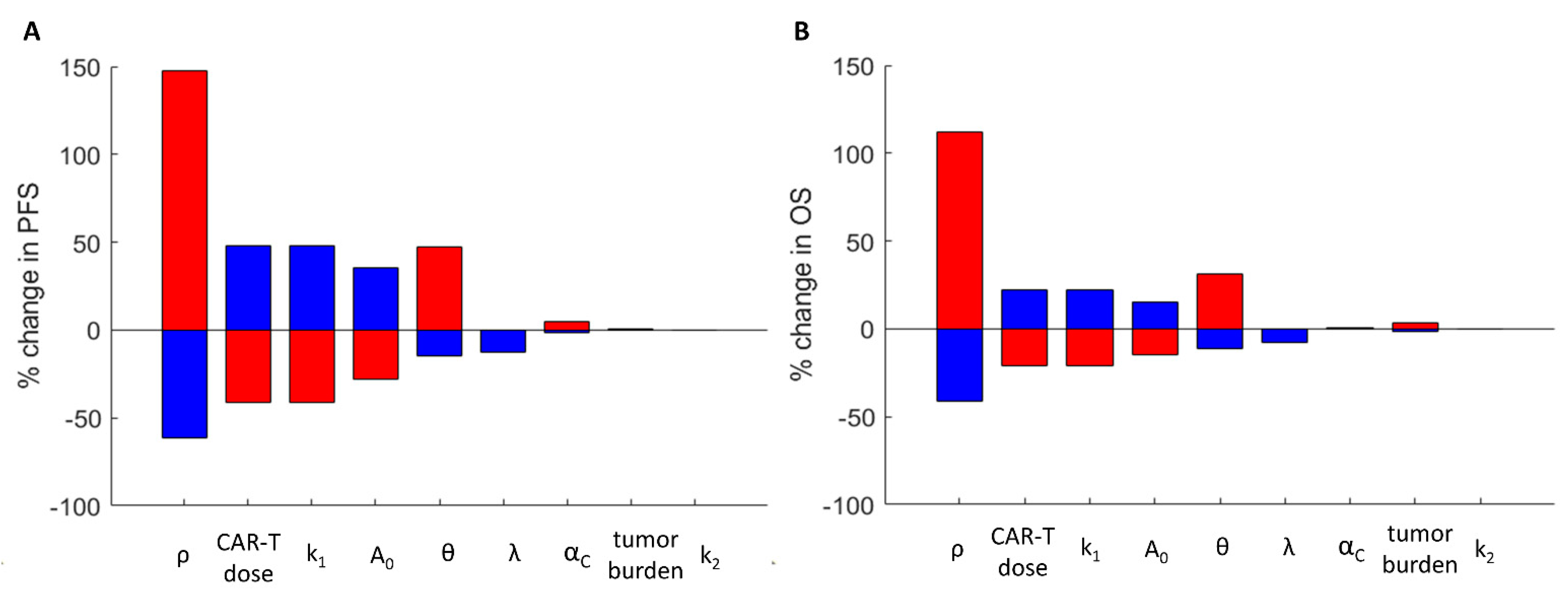
3.4. The Impact of the Model Parameters and TRT-CAR-T Cell Combination Therapy on Tumor Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Formenti, S.C.; Demaria, S. Future of Radiation and Immunotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 3–5. [Google Scholar] [CrossRef]
- Hyrenius-Wittsten, A.; Roybal, K.T. Paving New Roads for CARs. Trends Cancer 2019, 5, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Sur, D.; Havasi, A.D.; Cainap, C.; Samasca, G.; Burz, C.; Balacescu, O.; Lupan, I.; Deleanu, D.; Irimie, A. Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer. J. Clin. Med. 2020, 9, 182. [Google Scholar] [CrossRef] [Green Version]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- James, S.S.; Bednarz, B.; Benedict, S.; Buchsbaum, J.C.; Dewaraja, Y.; Frey, E.; Hobbs, R.; Grudzinski, J.; Roncali, E.; Sgouros, G.; et al. Current Status of Radiopharmaceutical Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 891–901. [Google Scholar] [CrossRef]
- Sahoo, P.; Yang, X.; Abler, D.; Maestrini, D.; Adhikarla, V.; Frankhouser, D.; Cho, H.; Machuca, V.; Wang, D.; Barish, M.; et al. Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data. J. R. Soc. Interface 2020, 17, 20190734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minnix, M.; Adhikarla, V.; Caserta, E.; Poku, E.; Rockne, R.; Shively, J.E.; Pichiorri, F. Comparison of CD38-Targeted alpha- Versus beta-Radionuclide Therapy of Disseminated Multiple Myeloma in an Animal Model. J. Nucl. Med. 2021, 62, 795–801. [Google Scholar] [CrossRef]
- Brenner, D.J.; Hlatky, L.R.; Hahnfeldt, P.J.; Huang, Y.; Sachs, R.K. The linear-quadratic model and most other common radiobiological models result in similar predictions of time-dose relationships. Radiat. Res. 1998, 150, 83–91. [Google Scholar] [CrossRef]
- Neira, S.; Gago-Arias, M.A.; Guiu-Souto, J.; Pardo-Montero, J. A kinetic model of continuous radiation damage to populations of cells: Comparison to the LQ model and application to molecular radiotherapy. Phys. Med. Biol. 2020, 65, 245015. [Google Scholar] [CrossRef]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.-C.; Thomas, S.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018, 24, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caserta, E.; Chea, J.; Minnix, M.; Poku, E.K.; Viola, D.; Vonderfecht, S.; Yazaki, P.; Crow, D.; Khalife, J.; Sanchez, J.F.; et al. Copper 64-labeled daratumumab as a PET/CT imaging tracer for multiple myeloma. Blood 2018, 131, 741–745. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Chaudhury, A.; Zhu, X.; Chu, L.; Goliaei, A.; June, C.H.; Kearns, J.D.; Stein, A.M. Chimeric Antigen Receptor T Cell Therapies: A Review of Cellular Kinetic-Pharmacodynamic Modeling Approaches. J. Clin. Pharmacol. 2020, 60, S147–S159. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Adhikarla, V.; Poku, E.K.; Palmer, J.; Chaudhry, A.; Biglang-Awa, V.E.; Bowles, N.; Nathwani, N.; Rosenzweig, M.; Sahebi, F.; et al. Identifying CD38+ cells in patients with multiple myeloma: First-in-human imaging using copper-64-labeled daratumumab. Blood Adv. 2020, 4, 5194–5202. [Google Scholar] [CrossRef]
- Bernhardt, P.; Svensson, J.; Hemmingsson, J.; van der Meulen, N.; Zeevaart, J.; Konijnenberg, M.; Müller, C.; Kindblom, J. Dosimetric Analysis of the Short-Ranged Particle Emitter (161)Tb for Radionuclide Therapy of Metastatic Prostate Cancer. Cancers 2021, 13, 2011. [Google Scholar] [CrossRef]
- Bernstein, M.B.; Krishnan, S.; Hodge, J.W.; Chang, J.Y. Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 2016, 13, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Jagodinsky, J.C.; Harari, P.M.; Morris, Z.S. The Promise of Combining Radiation Therapy With Immunotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 6–16. [Google Scholar] [CrossRef]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef]
- Pinnix, C.C.; Gunther, J.R.; Dabaja, B.S.; Strati, P.; Fang, P.; Hawkins, M.C.; Adkins, S.; Westin, J.; Ahmed, S.; Fayad, L.; et al. Bridging therapy prior to axicabtagene ciloleucel for relapsed/refractory large B-cell lymphoma. Blood Adv. 2020, 4, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Dandapani, S.; Shouse, D.G.; Yeh, J.; Cao, T.; Wong, J.; Budde, L.E. Bridging Radiation Is an Effective Strategy to Control Lymphoma in Preparation for CAR-T: A City of Hope Experience. Blood 2020, 136, 21–22. [Google Scholar] [CrossRef]
- Samaranayake, C.B.; McCaffrey, E.; Coucher, J.; Lehman, M.; Mai, G.T.; Murphy, M.A. Radiation therapy for augmenting the efficacy of immunotherapy in advanced non-small cell lung cancer: A case-controlled study. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Symbol | Value | Reference/ Comments |
|---|---|---|---|
| Effective decay constant (1/day) | λ | 0.07 | Accounts for biological clearance and physical decay |
| Tumor proliferation rate (1/day) | 0.27 | Mean value obtained from untreated controls | |
| Clearance rate of irradiated tumor cells (1/day) | 0.5 (Held constant) | [10] | |
| CAR-T cell killing rate (1/day/cell) | 4.49 × 10−7 | Optimized from data | |
| CAR-T cell proliferation/ exhaustion rate (1/day/cell) | 3.6 × 10−13 | Optimized from data | |
| CAR-T cell death rate (1/day) | 0.042 | Experimental data and optimization. Range obtained from data is 1–12% | |
| Tumor cell radiosensitivity (1/Gy) * | αT | 1.5 | [10] |
| CAR-T cell radiosensitivity (1/Gy) * | αC | 1.5 | Assumed equal to tumor radiosensitivity |
| Activity to dose conversion factor (Gy/day/μCi) | η | 3.48 | [10] |
| Response Criteria | Control | TRT Only (Day 7) | CAR-T Only (Day 7) | CAR-T before TRT | TRT before CAR-T |
|---|---|---|---|---|---|
| Progression-free survival (PFS) (days) | - | 27 | 33 | 55 | 43 |
| Overall survival (days) | 43 | 64 | 73 | 97 | 96 |
| Time to nadir (days) | - | 19 | 19 | 44 | 34 |
| Interval between therapies for maximizing PFS (days) | - | - | - | 25 | 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikarla, V.; Awuah, D.; Brummer, A.B.; Caserta, E.; Krishnan, A.; Pichiorri, F.; Minnix, M.; Shively, J.E.; Wong, J.Y.C.; Wang, X.; et al. A Mathematical Modeling Approach for Targeted Radionuclide and Chimeric Antigen Receptor T Cell Combination Therapy. Cancers 2021, 13, 5171. https://doi.org/10.3390/cancers13205171
Adhikarla V, Awuah D, Brummer AB, Caserta E, Krishnan A, Pichiorri F, Minnix M, Shively JE, Wong JYC, Wang X, et al. A Mathematical Modeling Approach for Targeted Radionuclide and Chimeric Antigen Receptor T Cell Combination Therapy. Cancers. 2021; 13(20):5171. https://doi.org/10.3390/cancers13205171
Chicago/Turabian StyleAdhikarla, Vikram, Dennis Awuah, Alexander B. Brummer, Enrico Caserta, Amrita Krishnan, Flavia Pichiorri, Megan Minnix, John E. Shively, Jeffrey Y. C. Wong, Xiuli Wang, and et al. 2021. "A Mathematical Modeling Approach for Targeted Radionuclide and Chimeric Antigen Receptor T Cell Combination Therapy" Cancers 13, no. 20: 5171. https://doi.org/10.3390/cancers13205171
APA StyleAdhikarla, V., Awuah, D., Brummer, A. B., Caserta, E., Krishnan, A., Pichiorri, F., Minnix, M., Shively, J. E., Wong, J. Y. C., Wang, X., & Rockne, R. C. (2021). A Mathematical Modeling Approach for Targeted Radionuclide and Chimeric Antigen Receptor T Cell Combination Therapy. Cancers, 13(20), 5171. https://doi.org/10.3390/cancers13205171