
Global Methylome Scores Correlate with Histological Subtypes of Colorectal Carcinoma and Show Different Associations with Common Clinical and Molecular Features

 , , , , ,
, , , , , 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tumor Samples
2.2. DNA Extraction
2.3. Treatment with Bisulfite and DNA Methylation Assay
2.4. Analysis of Methylation Data
2.5. Validation of Global Methylation Patterns in an Independent Series
2.6. Oncogene Mutation Status and Microsatellite Instability
2.7. Assessment of CpG Island Methylation Phenotype (CIMP)
2.8. Statistical Analysis
3. Results
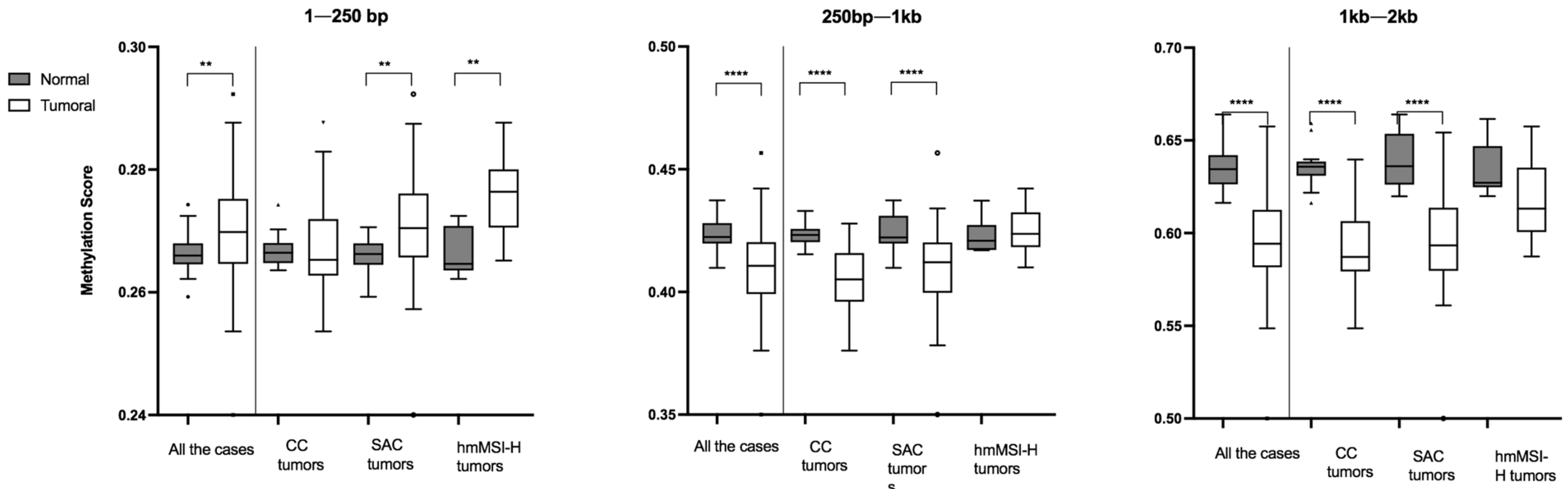
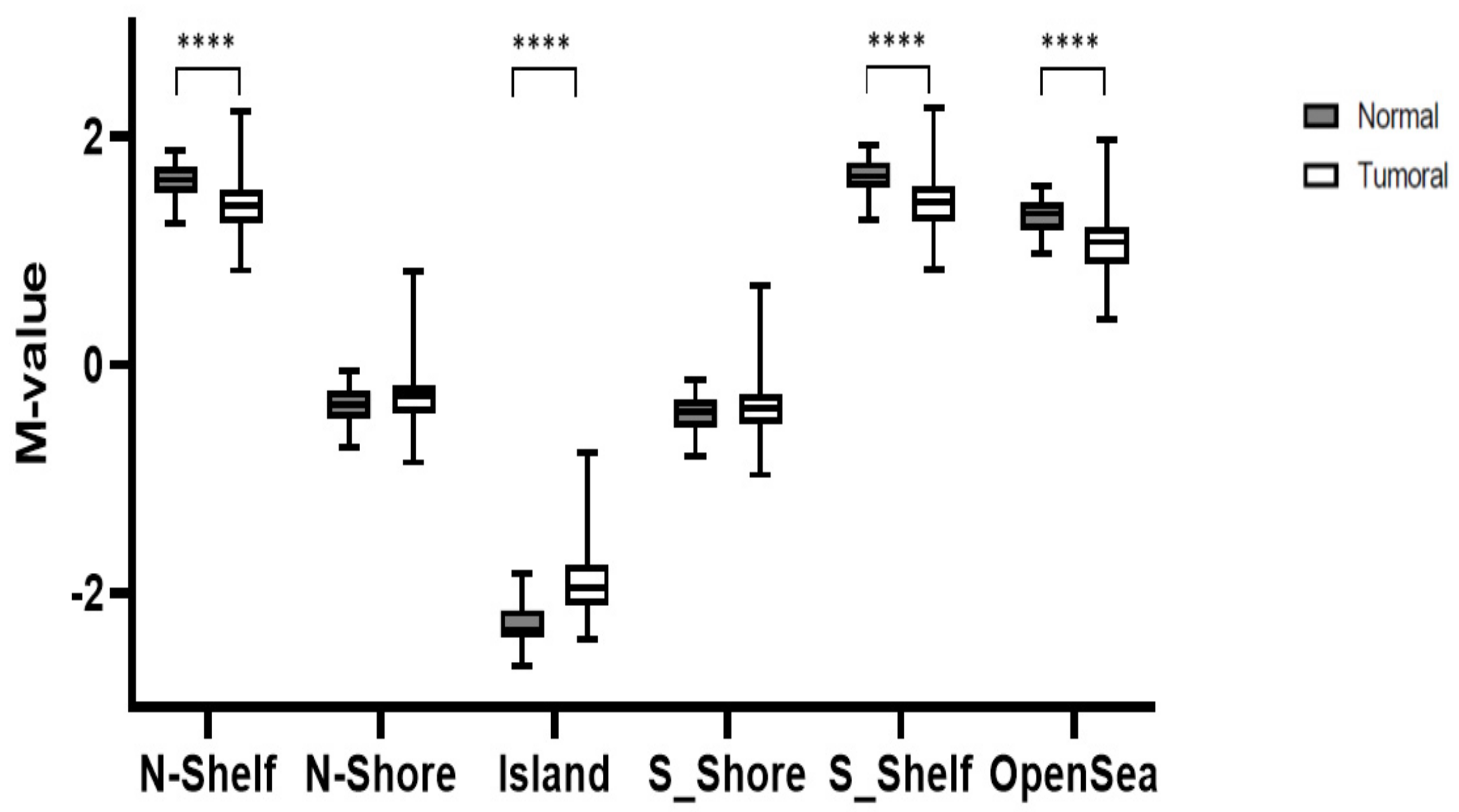
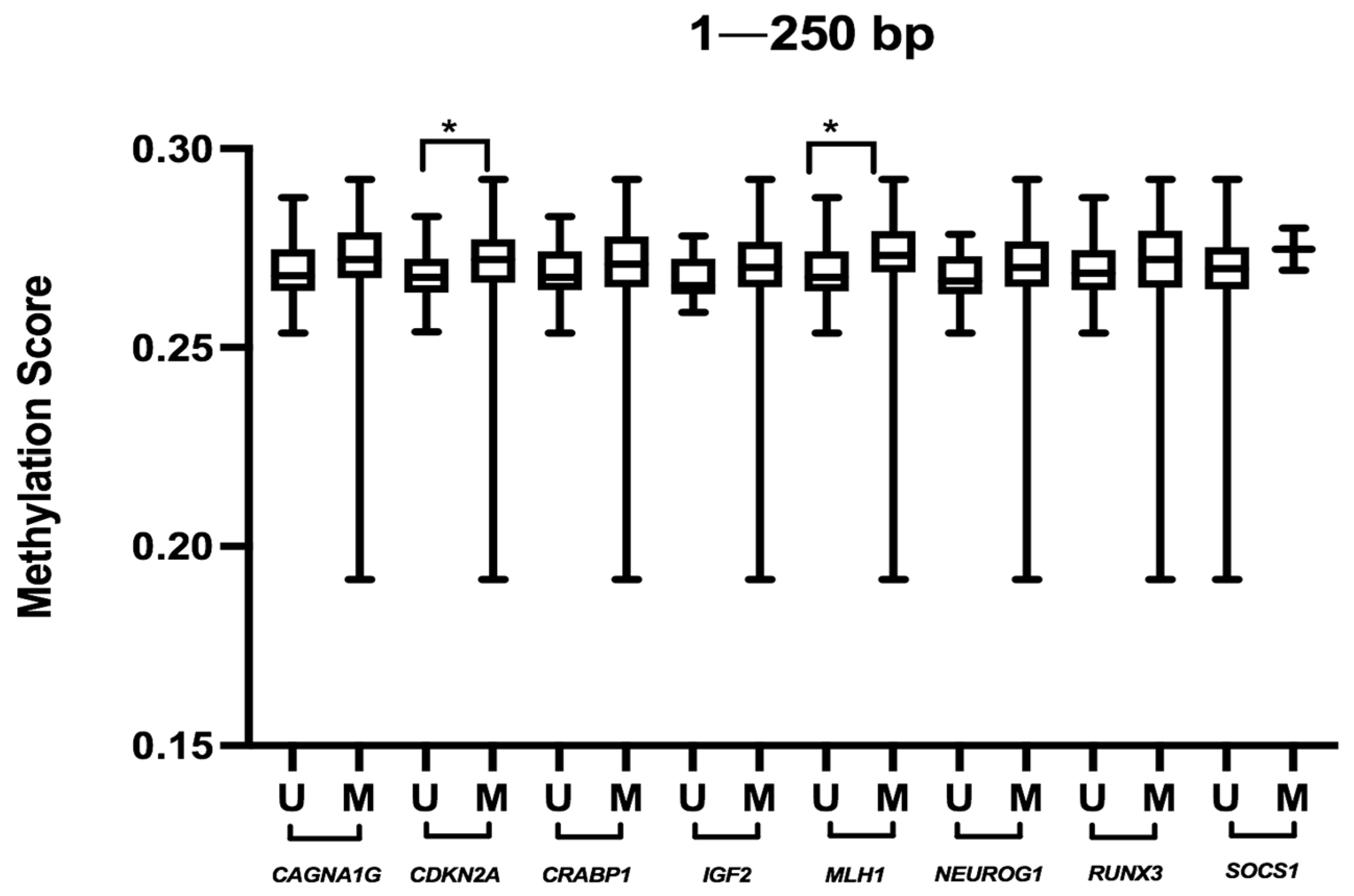
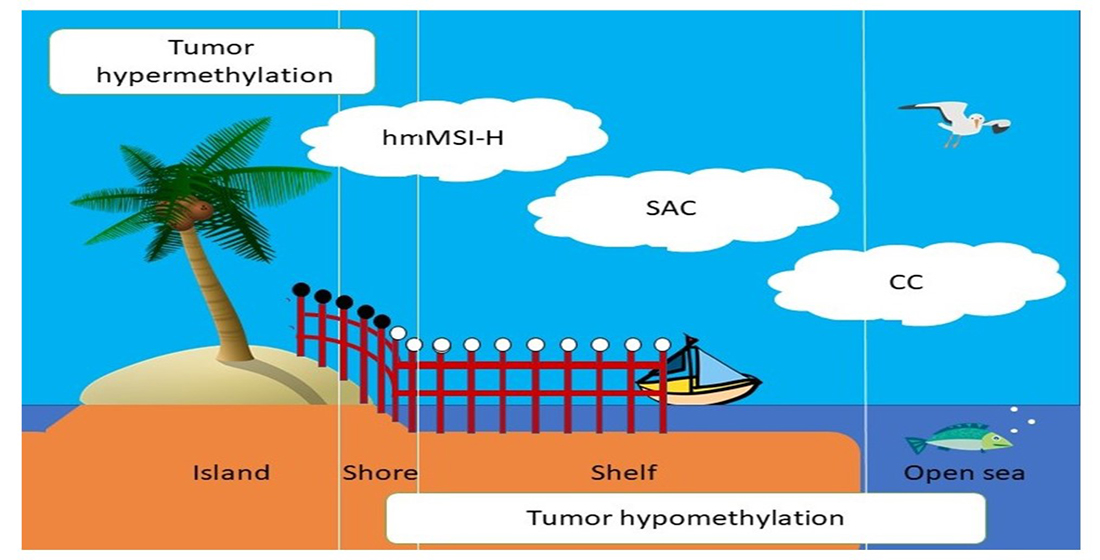
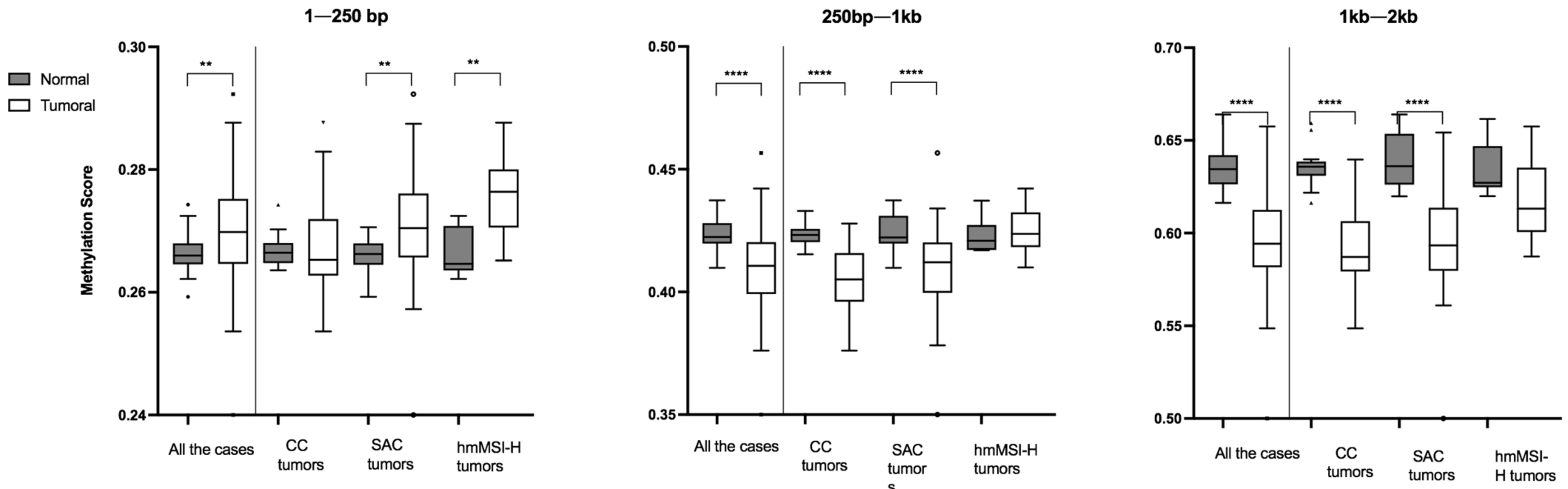
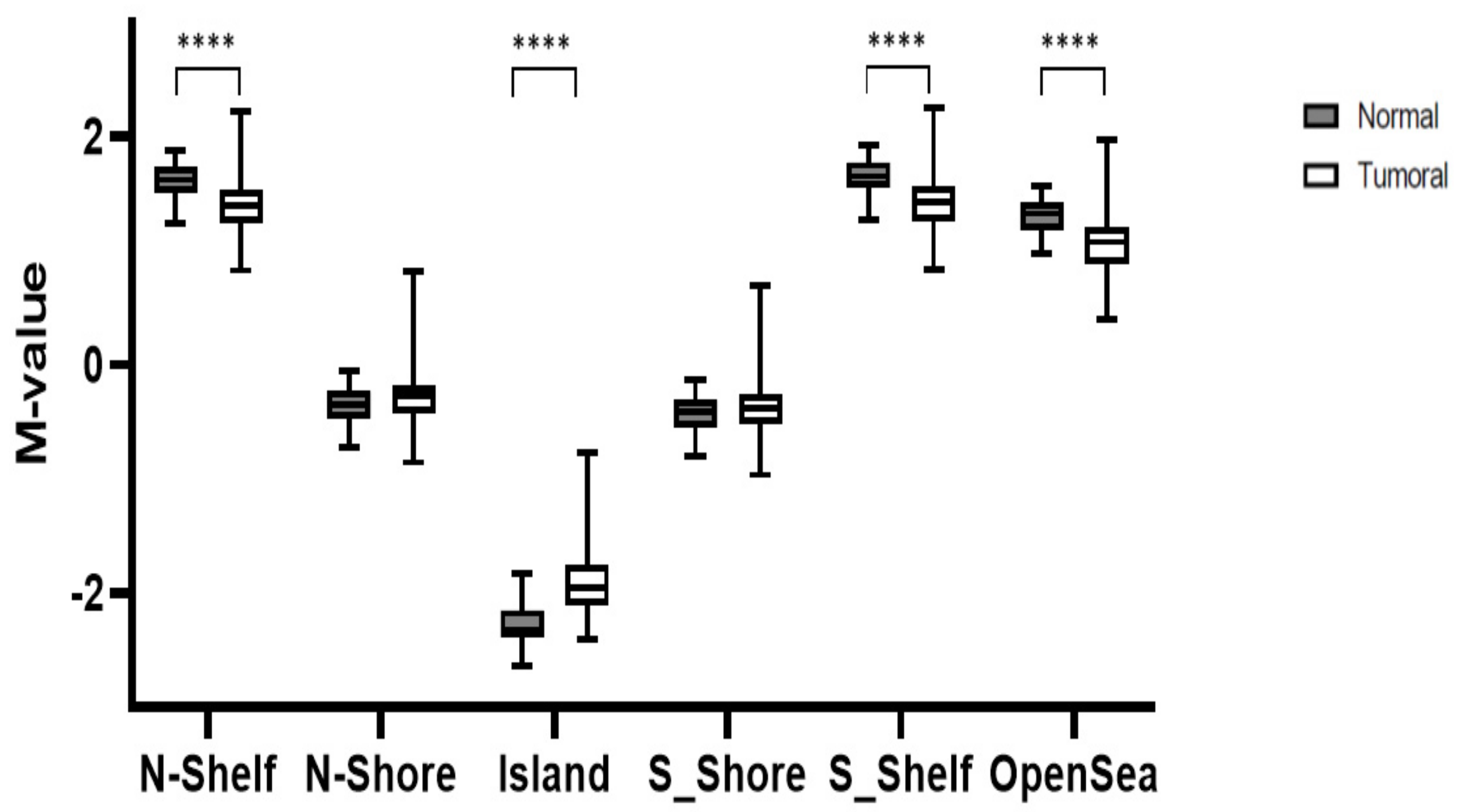
3.1. Global Methylation Levels in Normal Tissue and Their Comparison with Tumoral Tissue
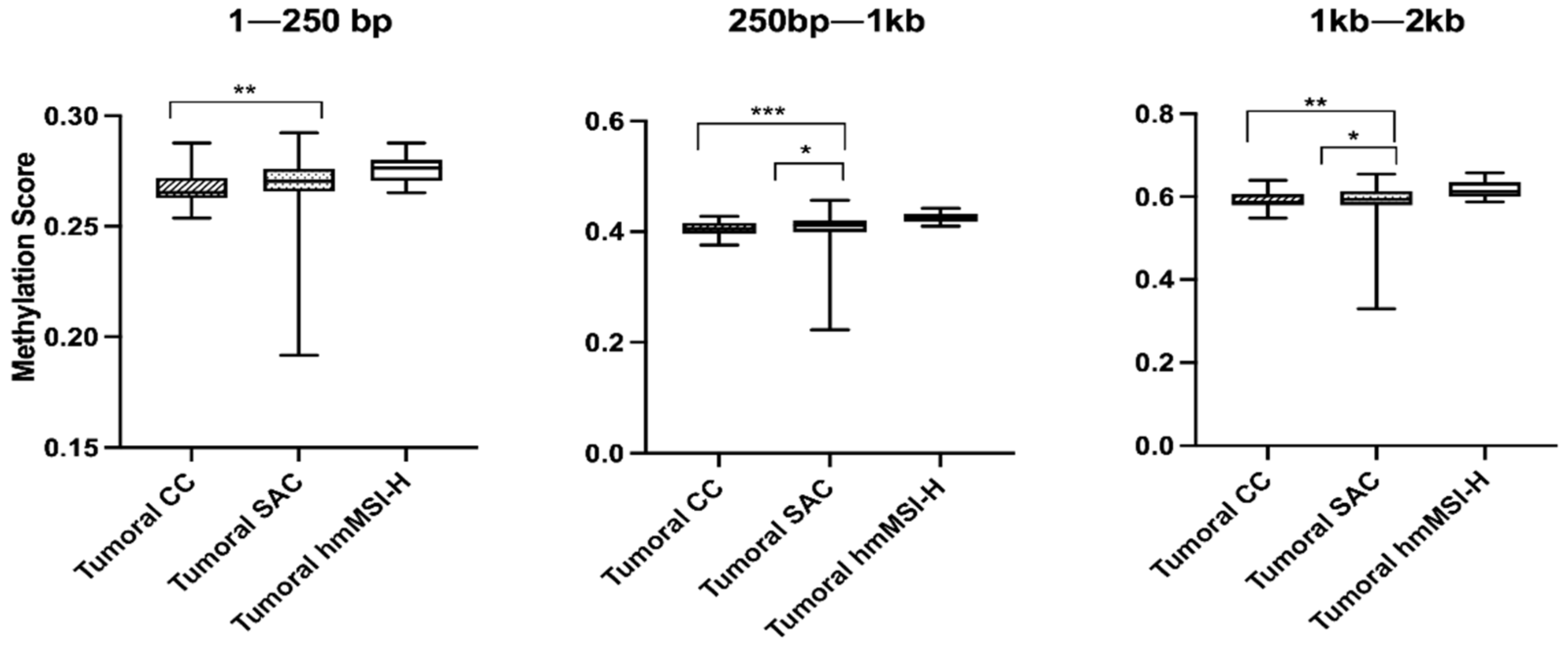
3.2. Relationship between Methylation Scores and Tumoral Tissue Features
3.3. Relationship between Methylation Scores and Molecular Markers (MSI, KRAS, BRAF, CIMP)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| bp | Base pair |
| BRAF | V-Raf Murine Sarcoma Viral Oncogene Homolog B1 |
| CC | Conventional carcinoma |
| CACNA1G | Calcium voltage-gated channel subunit alpha 1G |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CIMP | CpG island methylation phenotype |
| CRC | Colorectal cancer |
| dHPLC | Denaturing high-performance liquid chromatography |
| DNA | Deoxyribonucleic acid |
| HG | High grade |
| hm-MSI-H | Colorectal cancer showing histological and molecular features of high level of microsatellite instability |
| IGF2 | Insulin-like growth factor 2 |
| kb | Kilobase |
| KRAS | V-Ki-Ras2 Kirsten Rat Sarcoma 2 Viral Oncogene |
| LG | Low grade |
| LINE-1 | Long interspersed nuclear element 1 |
| lncRNA | Long non-coding RNA |
| MLH1 | MutL Homolog 1 |
| MS-MLPA | Methylation-specific multiplex ligation-dependent probe amplification |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| NEUROG1 | Neurogenin 1 |
| RNA | Ribonucleic acid |
| RUNX3 | Runt-related transcription factor 3 |
| SAC | Serrated adenocarcinoma |
| SNP | Single nucleotide polymorphism |
| SOCS1 | Supressor of Cytokine Signaling 1 |
| TNM | Tumour, Node, Metastasis stage |
| TSS | transcription starting site |
| UTR | Untranslated region |
| WHO | World Health Organization |
| WT | Wild type |
References
- Vaiopoulos, A.G.; Athanasoula, K.C.; Papavassiliou, A.G. Epigenetic Modifications in Colorectal Cancer: Molecular Insights and Therapeutic Challenges. Biochim. Biophys. Acta. 2014, 1842, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of Colorectal Cancer: Biomarker and Therapeutic Potential. Nat. Rev. Gastroenterol Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Carmona, F.J.; Esteller, M. Epigenomics of Human Colon Cancer. Mutat. Res. 2010, 693, 53–60. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA Hypomethylation in Cancer Cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- ENCODE Project Consortium An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- O’Brien, M.J.; Yang, S.; Mack, C.; Xu, H.; Huang, C.S.; Mulcahy, E.; Amorosino, M.; Farraye, F.A. Comparison of Microsatellite Instability, CpG Island Methylation Phenotype, BRAF and KRAS Status in Serrated Polyps and Traditional Adenomas Indicates Separate Pathways to Distinct Colorectal Carcinoma End Points. Am. J. Surg. Pathol. 2006, 30, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.-K.; Yu, J. Promoter Hypermethylation of Tumour Suppressor Genes as Potential Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2015, 16, 2472–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellizzi, A.M.; Frankel, W.L. Colorectal Cancer Due to Deficiency in DNA Mismatch Repair Function: A Review. Adv. Anat. Pathol. 2009, 16, 405–417. [Google Scholar] [CrossRef]
- Stefanius, K.; Ylitalo, L.; Tuomisto, A.; Kuivila, R.; Kantola, T.; Sirniö, P.; Karttunen, T.J.; Mäkinen, M.J. Frequent Mutations of KRAS in Addition to BRAF in Colorectal Serrated Adenocarcinoma. Histopathology 2011, 58, 679–692. [Google Scholar] [CrossRef] [Green Version]
- García-Solano, J.; Conesa-Zamora, P.; Carbonell, P.; Trujillo-Santos, J.; Torres-Moreno, D.D.; Pagán-Gómez, I.; Rodríguez-Braun, E.; Pérez-Guillermo, M. Colorectal Serrated Adenocarcinoma Shows a Different Profile of Oncogene Mutations, MSI Status and DNA Repair Protein Expression Compared to Conventional and Sporadic MSI-H Carcinomas. Int. J. Cancer 2012, 131, 1790–1799. [Google Scholar] [CrossRef]
- Conesa-Zamora, P.; García-Solano, J.; Turpin, M.D.C.; Sebastián-León, P.; Torres-Moreno, D.; Estrada, E.; Tuomisto, A.; Wilce, J.; Mäkinen, M.J.; Pérez-Guillermo, M.; et al. Methylome Profiling Reveals Functions and Genes Which Are Differentially Methylated in Serrated Compared to Conventional Colorectal Carcinoma. Clin. Epigenet. 2015, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Solano, J.; Turpin, M.C.; Torres-Moreno, D.; Huertas-López, F.; Tuomisto, A.; Mäkinen, M.J.; Conesa, A.; Conesa-Zamora, P. Two Histologically Colorectal Carcinomas Subsets from the Serrated Pathway Show Different Methylome Signatures and Diagnostic Biomarkers. Clin. Epigenet. 2018, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- García-Solano, J.; Pérez-Guillermo, M.; Conesa-Zamora, P.; Acosta-Ortega, J.; Trujillo-Santos, J.; Cerezuela-Fuentes, P.; Mäkinen, M.J. Clinicopathologic Study of 85 Colorectal Serrated Adenocarcinomas: Further Insights into the Full Recognition of a New Subset of Colorectal Carcinoma. Hum. Pathol. 2010, 41, 1359–1368. [Google Scholar] [CrossRef]
- García-Solano, J.; Conesa-Zamora, P.; Trujillo-Santos, J.; Mäkinen, M.J.; Pérez-Guillermo, M. Tumour Budding and Other Prognostic Pathological Features at Invasive Margins in Serrated Colorectal Adenocarcinoma: A Comparative Study with Conventional Carcinoma. Histopathology 2011, 59, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- García-Solano, J.; Turpin-Sevilla, M.D.C.; García-García, F.; Carbonell-Muñoz, R.; Torres-Moreno, D.; Conesa, A.; Conesa-Zamora, P. Differences in Gene Expression Profiling and Biomarkers between Histological Colorectal Carcinoma Subsets from the Serrated Pathway. Histopathology 2019, 75, 496–507. [Google Scholar] [CrossRef]
- Esteban-Gil, A.; Pérez-Sanz, F.; García-Solano, J.; Alburquerque-González, B.; Parreño-González, M.A.; Legaz-García, M.D.C.; Fernández-Breis, J.T.; Rodriguez-Braun, E.; Pimentel, P.; Tuomisto, A.; et al. ColPortal, an Integrative Multiomic Platform for Analysing Epigenetic Interactions in Colorectal Cancer. Sci. Data 2019, 6, 255. [Google Scholar] [CrossRef] [Green Version]
- Kondelova, A.; Alburquerque-González, B.; Vychytilova-Faltejskova, P.; García-Solano, J.; Prochazka, V.; Kala, Z.; Pérez, F.; Slaby, O.; Conesa-Zamora, P. MiR-181a-2* Expression Is Different amongst Carcinomas from the Colorectal Serrated Route. Mutagenesis 2020, 35, 233–241. [Google Scholar] [CrossRef]
- Mäkinen, M.J. Colorectal Serrated Adenocarcinoma. Histopathology 2007, 50, 131–150. [Google Scholar] [CrossRef]
- Redston, M. CHAPTER 23-Epithelial Neoplasms of the Large Intestine. In Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas, 2nd ed.; Odze, R.D., Goldblum, J.R., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2009; pp. 597–637. ISBN 978-1-4160-4059-0. [Google Scholar]
- Arends, M.; Ozde, R.D. Tumours of the Colon and Rectum. In WHO Classification of Tumours: Digestive System Tumours; Bosman, F., Carneiro, F., Hruban, R., Theise, N., Eds.; WHO Classification of Tumours: Geneva, Switzerland, 2019; Volume 1, pp. 157–162. [Google Scholar]
- Conesa-Zamora, P.; García-Solano, J.; García-García, F.; Turpin, M.D.C.; Trujillo-Santos, J.; Torres-Moreno, D.; Oviedo-Ramírez, I.; Carbonell-Muñoz, R.; Muñoz-Delgado, E.; Rodriguez-Braun, E.; et al. Expression Profiling Shows Differential Molecular Pathways and Provides Potential New Diagnostic Biomarkers for Colorectal Serrated Adenocarcinoma. Int. J. Cancer 2013, 132, 297–307. [Google Scholar] [CrossRef]
- Fennell, L.; Dumenil, T.; Wockner, L.; Hartel, G.; Nones, K.; Bond, C.; Borowsky, J.; Liu, C.; McKeone, D.; Bowdler, L.; et al. Integrative Genome-Scale DNA Methylation Analysis of a Large and Unselected Cohort Reveals 5 Distinct Subtypes of Colorectal Adenocarcinomas. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 269–290. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. A Language and Environment for Statistical Computing. R Found Stat Comput. Available online: https://www.R-project.org (accessed on 17 July 2020).
- Davis, S.; Du, P.; Bilke, S.; Triche, T.; Bootwalla, M. Type Package Title Handle Illumina Methylation Data. Version 2.6.1. Date 2013-04-05. 2013. Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.393.8030&rep=rep1&type=pdf (accessed on 17 July 2020).
- Schalkwyk, L.C.; Pidsley, R.; Wong, C.C.; Toulemeimat, N.; Defrance, M.; Maksimovic, J. WateRmelon: Illumina 450 Methylation Array Normalization and Metrics. R Package Version 1.4.0.
- Beta-Mixture Quantile Normalization Method for Correcting Probe Design Bias in Illumina Infinium 450 k DNA Methylation Data|Bioinformatics|Oxford Academic. Available online: https://academic.oup.com/bioinformatics/article/29/2/189/204142?login=true (accessed on 21 July 2021).
- Hansen, K.D.; Aryee, M.J. The Minfi User’s Guide Analyzing Illumina 450 k Methylation Arrays, R Package. 2012, pp. 1–21. Available online: https://bioc.ism.ac.jp/packages/3.0/bioc/vignettes/minfi/inst/doc/minfi.pdf (accessed on 15 July 2020).
- Hansen, K.D. IlluminaHumanMethylation450kanno.Ilmn12.Hg19 Annotation for Illumina’s 450 k Methylation Arrays. R Package. 2016. [Google Scholar]
- Carbonell, P.; Turpin, M.C.; Torres-Moreno, D.; Molina-Martínez, I.; García-Solano, J.; Perez-Guillermo, M.; Conesa-Zamora, P. Comparison of Allelic Discrimination by DHPLC, HRM, and TaqMan in the Detection of BRAF Mutation V600E. J. Mol. Diagn. 2011, 13, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Kraft, P.; Loda, M.; Fuchs, C.S. Evaluation of Markers for CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer by a Large Population-Based Sample. J. Mol. Diagn. 2007, 9, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG Island Methylator Phenotype Underlies Sporadic Microsatellite Instability and Is Tightly Associated with BRAF Mutation in Colorectal Cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-Value and M-Value Methods for Quantifying Methylation Levels by Microarray Analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmer, F.; Brinkman, A.B.; Assenov, Y.; Matarese, F.; Kaan, A.; Sabatino, L.; Villanueva, A.; Huertas, D.; Esteller, M.; Lengauer, T.; et al. Comparative Genome-Wide DNA Methylation Analysis of Colorectal Tumor and Matched Normal Tissues. Epigenetics 2012, 7, 1355–1367. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Hu, B.; Choi, A.-J.; Gopalan, B.; Lee, B.H.; Kalady, M.F.; Church, J.M.; Ting, A.H. Unique DNA Methylome Profiles in CpG Island Methylator Phenotype Colon Cancers. Genome Res. 2012, 22, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Lee, J.; Sidransky, D. DNA Methylation Markers in Colorectal Cancer. Cancer Metastasis Rev. 2010, 29, 181–206. [Google Scholar] [CrossRef]
- Kerachian, M.A.; Kerachian, M. Long Interspersed Nucleotide Element-1 (LINE-1) Methylation in Colorectal Cancer. Clin. Chim. Acta. 2019, 488, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fennell, L.J.; Bettington, M.L.; Walker, N.I.; Dwine, J.; Leggett, B.A.; Whitehall, V.L.J. DNA Methylation Changes That Precede Onset of Dysplasia in Advanced Sessile Serrated Adenomas. Clin. Epigenet. 2019, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Cen, S.; Ding, G.; Wu, W. Mucinous Colorectal Adenocarcinoma: Clinical Pathology and Treatment Options. Cancer Commun. 2019, 39, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visone, R.; Bacalini, M.G.; Di Franco, S.; Ferracin, M.; Colorito, M.L.; Pagotto, S.; Laprovitera, N.; Licastro, D.; Di Marco, M.; Scavo, E.; et al. DNA Methylation of Shelf, Shore and Open Sea CpG Positions Distinguish High Microsatellite Instability from Low or Stable Microsatellite Status Colon Cancer Stem Cells. Epigenomics 2019, 11, 587–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CC (n = 32) | SAC (n = 40) | hmMSI-H (n = 10) | p-Value | |
|---|---|---|---|---|---|
| Sex | Female | 15 (46.9%) | 21 (52.5%) | 8 (80%) | 0.191 |
| Male | 17 (53.1%) | 19 (47.5%) | 2 (20%) | ||
| Age | ≤71 | 15 (46.9%) | 17 (42.5%) | 4 (40%) | 0.952 |
| ≥72 | 17 (53.1%) | 23 (57.5%) | 6 (60%) | ||
| Location | Proximal | 19 (59.4%) | 28 (70%) | 10 (100%) | 0.038 |
| Distal | 13 (40.6%) | 12 (30%) | 0 (0%) | ||
| T | Tis | 1 (3.1%) | 0 (0%) | 0 (0%) | 0.453 |
| T2 | 3 (9.4%) | 8 (20%) | 0 (0%) | ||
| T3 | 21 (65.6%) | 23 (57.5%) | 6 (60%) | ||
| T4 | 7 (21.9%) | 9 (22.5%) | 4 (40%) | ||
| N | N0 | 19 (59.4%) | 18 (45%) | 4 (40%) | 0.720 |
| N1 | 5 (15.6%) | 9 (22.5%) | 2 (20%) | ||
| N2 | 8 (25.0%) | 8 (25%) | 4 (40%) | ||
| M | M0 | 28 (90.3%) | 31 (81.6%) | 6 (60%) | 0.101 |
| M1 | 3 (9.7%) | 7 (18.4%) | 4 (40%) | ||
| Grade | LG | 30 (93.8%) | 30 (75%) | 7 (70%) | 0.050 |
| HG | 2 (6.3%) | 10 (25%) | 3 (30%) | ||
| Tumor budding | LG | 20 (80.3%) | 12 (54.5%) | 7 (70%) | 0.116 |
| HG | 4 (16.7%) | 10 (54.5%) | 3 (30%) | ||
| Tumor budding No. | Absent | 10 (41.7%) | 8 (36.4%) | 7 (70%) | 0.029 |
| <10 | 11 (45.8%) | 4 (18.2%) | 0 (0%) | ||
| 10–19 | 2 (8.3%) | 5 (22.7%) | 1 (10%) | ||
| >20 | 1 (4.2%) | 5 (22.7%) | 2 (20%) | ||
| Mucin | Absent | 18 (75%) | 3 (13.6%) | 4 (40%) | <0.001 * |
| <50% | 5 (20.8%) | 7 (31.8%) | 3 (3%) | ||
| 50–75% | 0 (0%) | 2 (9.1%) | 2 (20%) | ||
| >75% | 1 (4.2%) | 10 (45.5%) | 1 (10%) | ||
| BRAF | WT | 31 (96.9%) | 28 (70%) | 6 (60%) | 0.003 * |
| Mutated | 1 (3.1%) | 12 (30%) | 4 (40%) | ||
| KRAS | WT | 17 (53.1%) | 25 (62.5%) | 8 (80%) | 0.335 |
| Mutated | 15 (46.9%) | 15 (37.5%) | 2 (20%) | ||
| MSI | MSS/MSI-L | 32 (100%) | 33 (82.5%) | 3 (30%) | <0.001 * |
| MSI-H | 0 (0%) | 7 (17.5%) | 7 (70%) | ||
| CIMP Weisemberger et al. | Low CIMP | 31 (96.9%) | 25 (65.8%) | 3 (30%) | <0.001 * |
| High CIMP | 1 (3.1%) | 13 (34.2%) | 7 (70%) | ||
| CIMP Ogino et al. | Low CIMP | 32 (100%) | 29 (76.3%) | 3 (30%) | <0.001 * |
| High CIMP | 0 (0%) | 9 (23.7%) | 7 (70%) | ||
| Variable | 250 bp | 1 kb | 2 kb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Median | IQR | p | Median | IQR | p | Median | IQR | p | ||
| All | 35 | 0.266 | 0.003 | 0.422 | 0.008 | 0.634 | 0.016 | ||||
| Age | ≤71 | 14 | 0.267 | 0.004 | 1.000 | 0.424 | 0.009 | 0.678 | 0.638 | 0.016 | 0.263 |
| >72 | 21 | 0.266 | 0.004 | 0.422 | 0.009 | 0.634 | 0.022 | ||||
| Sex | Female | 17 | 0.267 | 0.005 | 0.184 | 0.422 | 0.010 | 0.335 | 0.631 | 0.017 | 0.245 |
| Male | 18 | 0.265 | 0.004 | 0.423 | 0.008 | 0.637 | 0.016 | ||||
| Location | Proximal | 22 | 0.265 | 0.003 | 0.335 | 0.422 | 0.011 | 0.335 | 0.634 | 0.022 | 1.000 |
| Distal | 13 | 0.268 | 0.005 | 0.424 | 0.007 | 0.636 | 0.012 | ||||
| Type | CC | 14 | 0.266 | 0.003 | 0.775 | 0.423 | 0.005 | 0.805 | 0.636 | 0.008 | 0.752 |
| SAC | 15 | 0.266 | 0.004 | 0.422 | 0.011 | 0.636 | 0.027 | ||||
| hmMSI-H | 6 | 0.265 | 0.007 | 0.421 | 0.010 | 0.627 | 0.022 | ||||
| Variable | 250 bp | 1 kb | 2 kb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Median | IQR | p | Median | IQR | p | Median | IQR | p | ||
| All | 82 | 0.2698 | 0.0106 | 0.4106 | 0.0211 | 0.5943 | 0.0309 | ||||
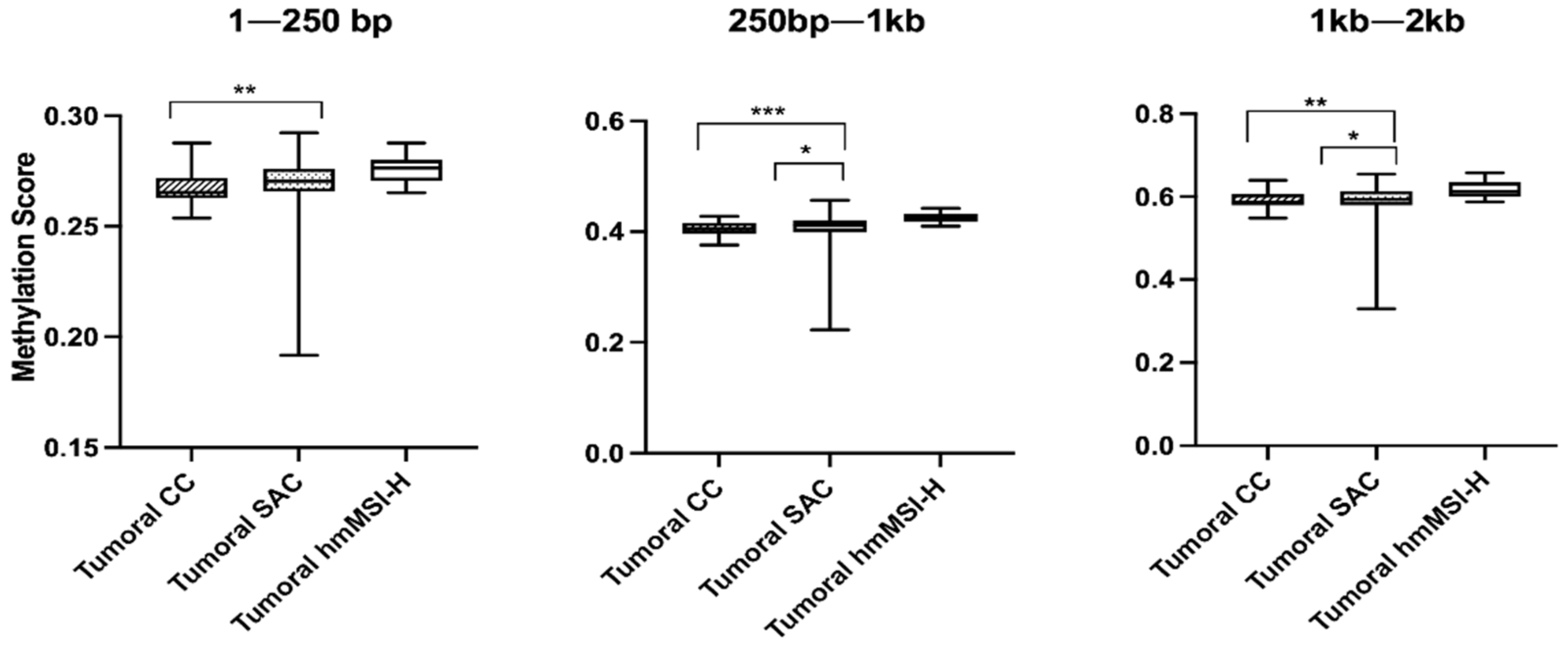
| Type | CC | 32 | 0.2653 | 0.0090 | 0.0060 | 0.4051 | 0.1980 | 0.0010 * | 0.5871 | 0.0271 | 0.0120 |
| SAC | 40 | 0.2704 | 0.0104 | 0.4121 | 0.0205 | 0.5933 | 0.0347 | ||||
| hmMSI-H | 10 | 0.2763 | 0.0094 | 0.4236 | 0.1410 | 0.6131 | 0.0347 | ||||
| Nationality | Spanish | 56 | 0.2698 | 0.0106 | 0.6320 | 0.4098 | 0.0207 | 0.9440 | 0.5956 | 0.0308 | 0.6320 |
| Finnish | 26 | 0.2698 | 0.0116 | 0.4121 | 0.0239 | 0.5915 | 0.0294 | ||||
| Age | ≤71 | 36 | 0.2703 | 0.0137 | 0.8740 | 0.4090 | 0.2530 | 0.9630 | 0.5933 | 0.3140 | 0.8010 |
| ≥72 | 46 | 0.2694 | 0.0094 | 0.4106 | 0.1870 | 0.5955 | 0.3140 | ||||
| Sex | female | 44 | 0.2695 | 0.0103 | 0.6220 | 0.4065 | 0.2277 | 0.4080 | 0.5921 | 0.0293 | 0.5330 |
| Male | 38 | 0.2707 | 0.0111 | 0.4136 | 0.0207 | 0.5959 | 0.0302 | ||||
| Location | Proximal | 57 | 0.2701 | 0.0107 | 0.6980 | 0.4095 | 0.2200 | 0.3020 | 0.5939 | 0.0333 | 0.2880 |
| Distal | 25 | 0.2695 | 0.0109 | 0.4163 | 0.2190 | 0.5961 | 0.2090 | ||||
| T | Tis | 1 | |||||||||
| T2 | 11 | 0.2701 | 0.1510 | 0.3770 | 0.4040 | 0.0222 | 0.1620 | 0.5969 | 0.0271 | 0.0510 | |
| T3 | 50 | 0.2700 | 0.0960 | 0.4106 | 0.0222 | 0.5893 | 0.0303 | ||||
| T4 | 20 | 0.2700 | 0.0137 | 0.4151 | 0.2340 | 0.6048 | 0.2790 | ||||
| N | N0 | 41 | 0.2695 | 0.1040 | 0.9870 | 0.4040 | 0.0256 | 0.8010 | 0.5939 | 0.3580 | 0.9660 |
| N1 | 16 | 0.2708 | 0.0091 | 0.4113 | 0.0124 | 0.5955 | 0.0210 | ||||
| N2 | 25 | 0.2692 | 0.0140 | 0.4139 | 0.0230 | 0.5927 | 0.0295 | ||||
| M | M0 | 65 | 0.2694 | 0.0109 | 0.4960 | 0.4078 | 0.0226 | 0.1780 | 0.5939 | 0.0308 | 0.2130 |
| M1 | 14 | 0.2704 | 0.0119 | 0.4164 | 0.0177 | 0.6013 | 0.0351 | ||||
| Grade | LG | 67 | 0.2695 | 0.0102 | 0.8530 | 0.4092 | 0.0205 | 0.5450 | 0.5939 | 0.0304 | 0.5770 |
| HG | 15 | 0.2701 | 0.0143 | 0.4163 | 0.0255 | 0.6023 | 0.0356 | ||||
| Tumor budding | LG | 39 | 0.2687 | 0.0124 | 0.8530 | 0.4092 | 0.0207 | 0.5450 | 0.5939 | 0.0252 | 0.5770 |
| HG | 17 | 0.2709 | 0.0098 | 0.4166 | 0.0414 | 0.6021 | 0.0435 | ||||
| Mucin | Absent | 26 | 0.2654 | 0.0120 | 0.2820 | 0.4034 | 0.0199 | 0.0320 | 0.5864 | 0.0320 | 0.1070 |
| Present | 15 | 0.2709 | 0.0093 | 0.4159 | 0.0197 | 0.6019 | 0.0299 | ||||
| Variable | 250 bp | 1 kb | 2 kb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Median | IQR | p | Median | IQR | p | Median | IQR | p | ||
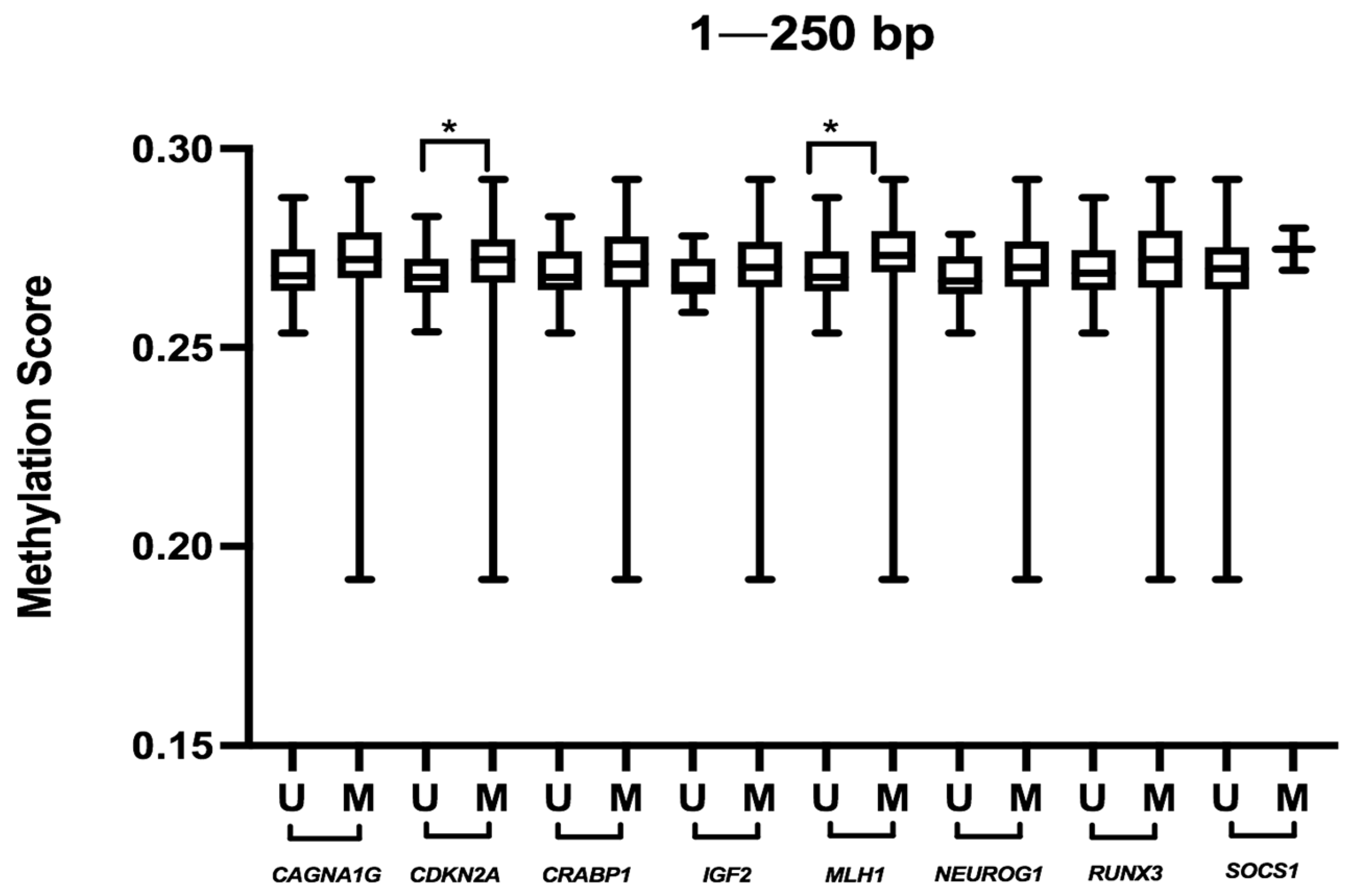
| BRAF | WT | 65 | 0.2690 | 0.0104 | 0.1720 | 0.4092 | 0.0203 | 0.2060 | 0.0254 | 0.0270 | 0.2800 |
| Mutated | 17 | 0.2731 | 0.0143 | 0.4166 | 0.0254 | 0.6023 | 0.0474 | ||||
| KRAS | WT | 50 | 0.2702 | 0.0097 | 0.9550 | 0.4093 | 0.0226 | 0.9920 | 0.5950 | 0.0376 | 0.5880 |
| Mutated | 32 | 0.2691 | 0.0126 | 0.4114 | 0.0184 | 0.5913 | 0.0254 | ||||
| MSI | MSS/MSI-L | 68 | 0.2686 | 0.0103 | 0.0250 | 0.4093 | 0.0222 | 0.1150 | 0.5915 | 0.0317 | 0.1430 |
| MSI-H | 14 | 0.2739 | 0.0111 | 0.4155 | 0.0256 | 0.6027 | 0.0313 | ||||
| CIMP Weisemberger et al. | Low CIMP | 59 | 0.2681 | 0.0105 | 0.0380 | 0.4079 | 0.0222 | 0.3170 | 0.5902 | 0.0320 | 0.4410 |
| High CIMP | 21 | 0.2731 | 0.0108 | 0.4152 | 0.0201 | 0.5974 | 0.0282 | ||||
| CIMP Ogino et al. | Low CIMP | 64 | 0.2681 | 0.0100 | 0.0300 | 0.4093 | 0.0661 | 0.4070 | 0.5915 | 0.0313 | 0.8290 |
| High CIMP | 16 | 0.2739 | 0.0108 | 0.4155 | 0.0203 | 0.5972 | 0.0305 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turpín-Sevilla, M.d.C.; Pérez-Sanz, F.; García-Solano, J.; Sebastián-León, P.; Trujillo-Santos, J.; Carbonell, P.; Estrada, E.; Tuomisto, A.; Herruzo, I.; Fennell, L.J.; et al. Global Methylome Scores Correlate with Histological Subtypes of Colorectal Carcinoma and Show Different Associations with Common Clinical and Molecular Features. Cancers 2021, 13, 5165. https://doi.org/10.3390/cancers13205165
Turpín-Sevilla MdC, Pérez-Sanz F, García-Solano J, Sebastián-León P, Trujillo-Santos J, Carbonell P, Estrada E, Tuomisto A, Herruzo I, Fennell LJ, et al. Global Methylome Scores Correlate with Histological Subtypes of Colorectal Carcinoma and Show Different Associations with Common Clinical and Molecular Features. Cancers. 2021; 13(20):5165. https://doi.org/10.3390/cancers13205165
Chicago/Turabian StyleTurpín-Sevilla, María del Carmen, Fernando Pérez-Sanz, José García-Solano, Patricia Sebastián-León, Javier Trujillo-Santos, Pablo Carbonell, Eduardo Estrada, Anne Tuomisto, Irene Herruzo, Lochlan J. Fennell, and et al. 2021. "Global Methylome Scores Correlate with Histological Subtypes of Colorectal Carcinoma and Show Different Associations with Common Clinical and Molecular Features" Cancers 13, no. 20: 5165. https://doi.org/10.3390/cancers13205165
APA StyleTurpín-Sevilla, M. d. C., Pérez-Sanz, F., García-Solano, J., Sebastián-León, P., Trujillo-Santos, J., Carbonell, P., Estrada, E., Tuomisto, A., Herruzo, I., Fennell, L. J., Mäkinen, M. J., Rodríguez-Braun, E., Whitehall, V. L. J., Conesa, A., & Conesa-Zamora, P. (2021). Global Methylome Scores Correlate with Histological Subtypes of Colorectal Carcinoma and Show Different Associations with Common Clinical and Molecular Features. Cancers, 13(20), 5165. https://doi.org/10.3390/cancers13205165