
Integrative Metabolomics Reveals Deep Tissue and Systemic Metabolic Remodeling in Glioblastoma

 , , , ,
, , , ,  , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Blood Samples
2.2. Targeted Metabolomics
2.2.1. Reagents
2.2.2. Sample Preparation
2.2.3. Liquid Chromatography and Tandem Mass Spectrometry
2.3. Untargeted Metabolic Phenotyping
2.3.1. Reagents
2.3.2. Sample Preparation
2.3.3. Chromatographic Conditions
2.3.4. Ion Mobility and Mass Spectrometry
2.3.5. Raw Data Processing
2.3.6. Quality Control
2.4. Tissue Metabolic Phenotyping
2.4.1. Reagents
2.4.2. Sample Preparation
2.4.3. Data Acquisition and Data Processing
2.5. Metabolite Annotation and Identification
2.6. Circulating Cell-Free DNA Acquisition
2.7. Data Analysis
3. Results
3.1. Cohort Description
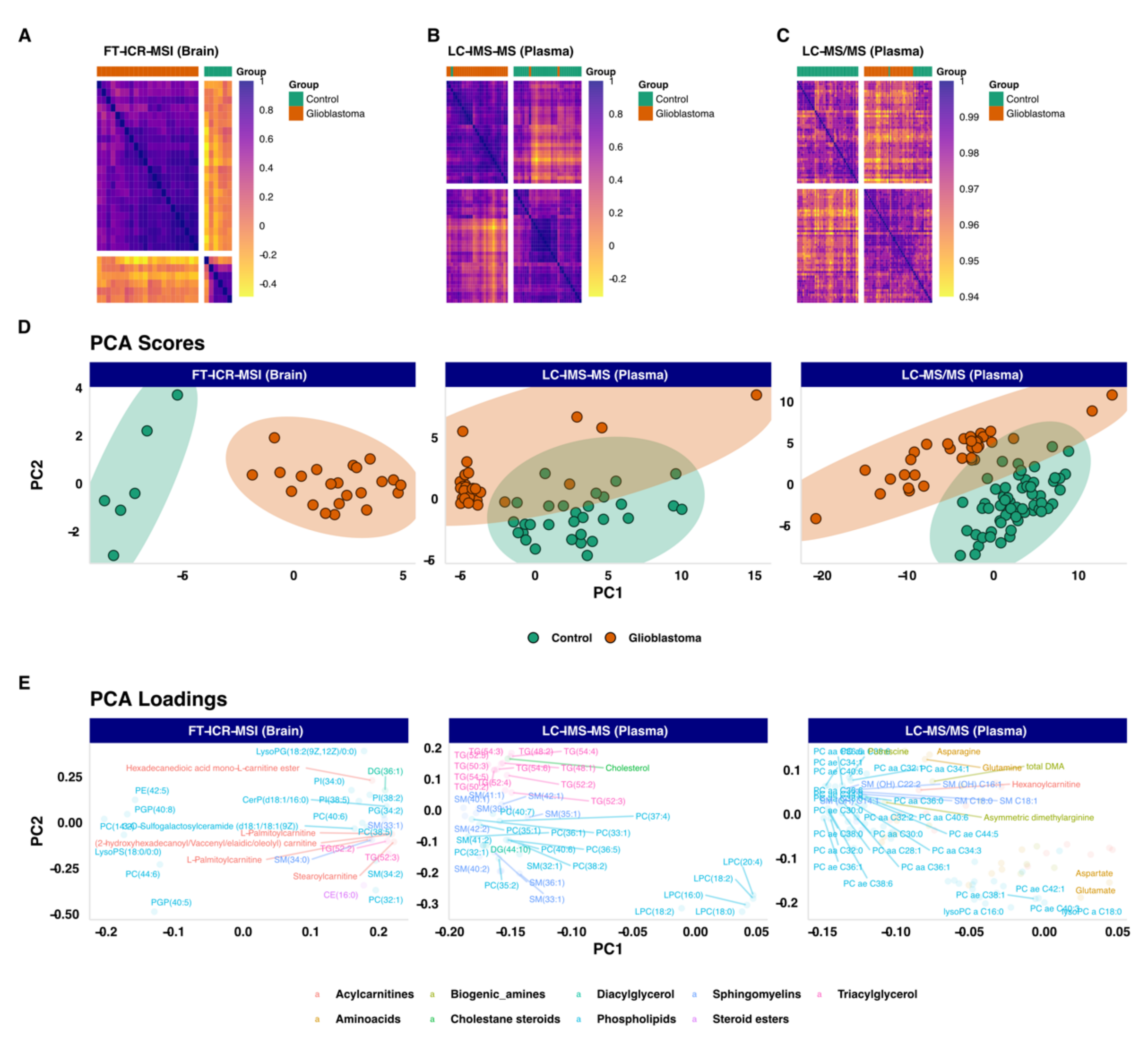
3.2. Unsupervized Exploratory Analysis
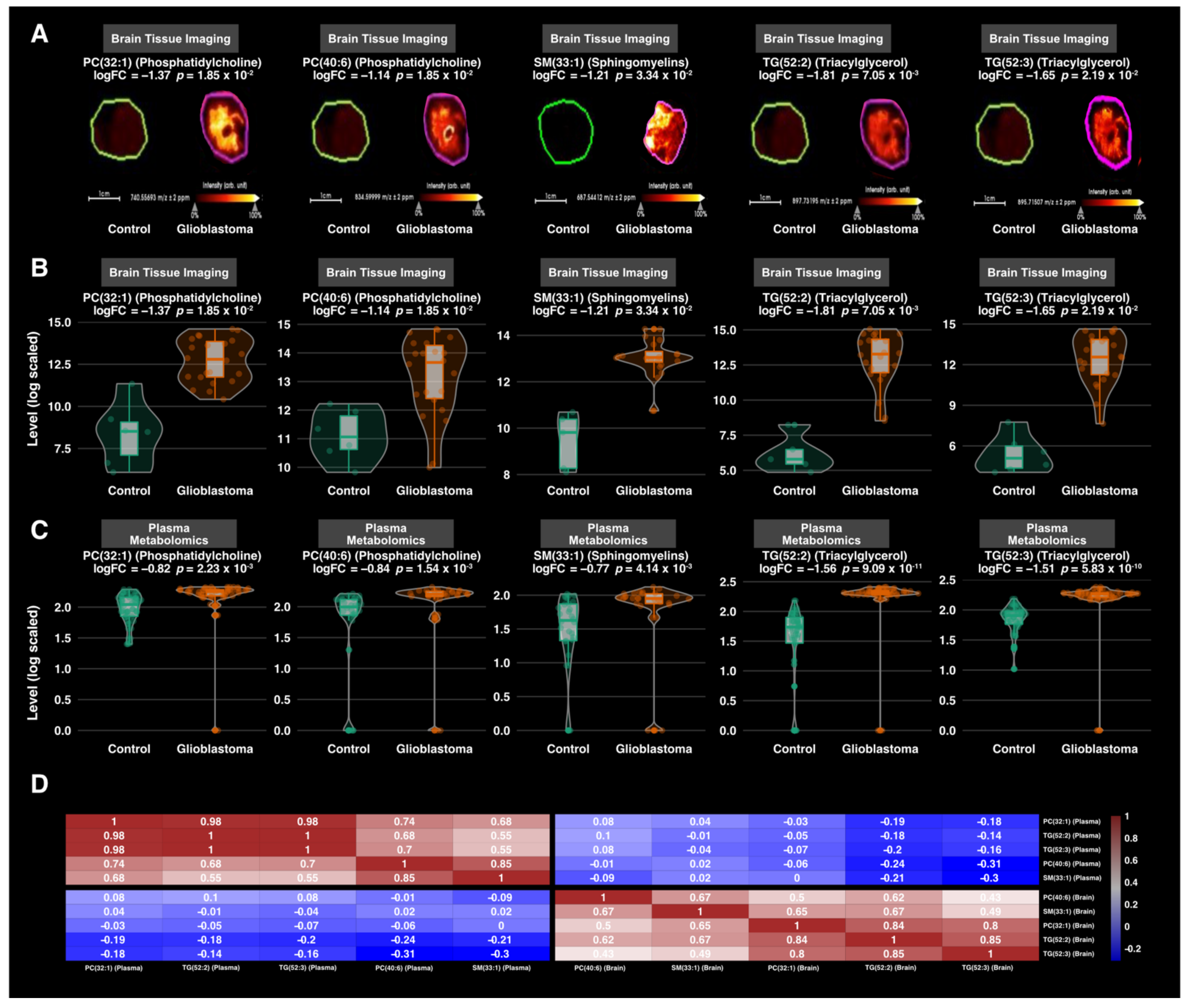
3.3. Differential Expression Analysis
3.4. Correlation Analysis
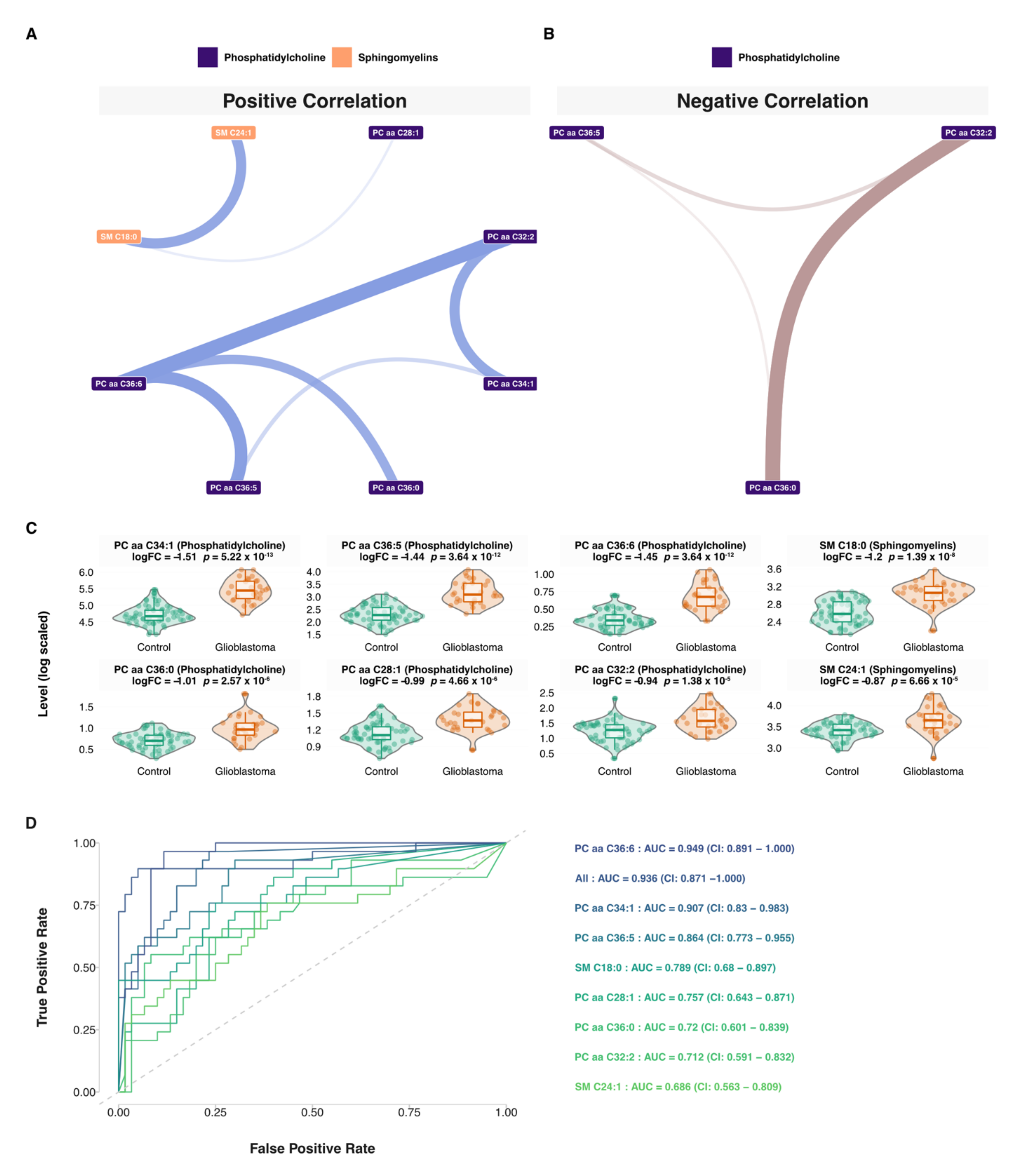
3.5. Network and Predictive Machine Learning Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauchet, L.; Ostrom, Q.T. Epidemiology and molecular epidemiology. Neurosurg. Clin. N. Am. 2019, 30, 1–16. [Google Scholar] [CrossRef]
- Morgan, L.L. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2015, 17, 623–624. [Google Scholar] [CrossRef]
- Khurana, V.; Jain, S.; Smee, R.; Cook, R.; Dobes, M.; Shadbolt, B.; Smith, S.; Dexter, M. Increasing incidence of glioblastoma multiforme and meningioma, and decreasing incidence of Schwannoma (2000–2008): Findings of a multicenter Australian study. Surg. Neurol. Int. 2011, 2, 176. [Google Scholar] [CrossRef]
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain Tumours: Rise in Glioblastoma Multiforme Incidence in England 1995–2015 Suggests an Adverse Environmental or Lifestyle Factor. J. Environ. Public Health 2018, 2018, 7910754. [Google Scholar] [CrossRef]
- Vecht, C.J.; Kerkhof, M.; Duran-Pena, A. Seizure Prognosis in Brain Tumors: New Insights and Evidence-Based Management. Oncol. 2014, 19, 751–759. [Google Scholar] [CrossRef]
- Yan, J.-L.; Li, C.; Boonzaier, N.R.; Fountain, D.M.; Larkin, T.J.; Matys, T.; Van Der Hoorn, A.; Price, S.J. Multimodal MRI characteristics of the glioblastoma infiltration beyond contrast enhancement. Ther. Adv. Neurol. Disord. 2019, 12. [Google Scholar] [CrossRef]
- Law, M.; Yang, S.; Wang, H.; Babb, J.S.; Johnson, G.; Cha, S.; Knopp, E.A.; Zagzag, D. Glioma Grading: Sensitivity, Specificity, and Predictive Values of Perfusion MR Imaging and Proton MR Spectroscopic Imaging Compared with Conventional MR Imaging. Am. J. Neuroradiol. 2003, 24, 1989–1998. [Google Scholar]
- Stupp, R.; Mason, W.P.; Bent, M.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the extent of resection with survival in glioblastoma: A systematic review and meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef]
- Bloch, O.; Han, S.J.; Cha, S.; Sun, M.Z.; Aghi, M.K.; McDermott, M.W.; Berger, M.S.; Parsa, A.T. Impact of extent of re-section for recurrent glioblastoma on overall survival: Clinical article. J. Neurosurg. 2012, 117, 1032–1038. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Malkki, H. Trial watch: Glioblastoma vaccine therapy disappointment in phase iii trial. Nat. Rev. Neurol. 2016, 12, 190. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Ius, T.; Ciani, Y.; Ruaro, M.E.; Isola, M.; Sorrentino, M.; Bulfoni, M.; Candotti, V.; Correcig, C.; Bourkoula, E.; Manini, I.; et al. An nf-kappab signature predicts low-grade glioma prognosis: A precision medicine approach based on patient-derived stem cells. Neuro Oncol. 2018, 20, 776–787. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Borcoman, E.; Kamal, M. Molecular profiling in precision medicine oncology. Nat. Med. 2019, 25, 711–712. [Google Scholar] [CrossRef]
- Ogrinc, N.; Saudemont, P.; Takats, Z.; Salzet, M.; Fournier, I. Cancer surgery 2.0: Guidance by real-time molecular tech-nologies. Trends Mol. Med. 2021. Available online: https://www.cell.com/trends/molecular-medicine/fulltext/S1471-4914(21)00096-4 (accessed on 8 September 2021). [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent glioblastoma: From molec-ular landscape to new treatment perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Aletaha, D. Precision medicine and management of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102405. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Bekri, S. Advances in metabolome information retrieval: Turning chemistry into biology. Part II: Biological information recovery. J. Inherit. Metab. Dis. 2017, 41, 393–406. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2016, 19, 286–302. [Google Scholar] [CrossRef]
- McColl, E.R.; Asthana, R.; Paine, M.F.; Piquette-Miller, M. The age of omics-driven precision medicine. Clin. Pharmacol. Ther. 2019, 106, 477–481. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2018, 20, 110–119. [Google Scholar] [CrossRef]
- Arivaradarajan, P.; Misra, G. Omics Approaches, Technologies and Applications: Integrative Approaches for Understanding Omics Data, 1st ed.; Springer: Singapore, 2018; p. 1. Available online: https://www.springer.com/gp/book/9789811329241 (accessed on 8 September 2021).
- Sarma, A.; Calfee, C.S.; Ware, L.B. Biomarkers and precision medicine: State of the art. Crit Care Clin. 2020, 36, 155–165. [Google Scholar] [CrossRef]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Afonso, C.; Bekri, S. Advances in metabolome information retrieval: Turning chemistry into biology. Part I: Analytical chemistry of the metabolome. J. Inherit. Metab. Dis. 2018, 41, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.; Lindon, J.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.B.; Lin, T.-Y.; Leach, F.E., III; Tolmachev, A.V.; Tolić, N.; Robinson, E.W.; Koppenaal, D.W.; Paša-Tolić, L. 21 tesla fourier transform ion cyclotron resonance mass spectrometer greatly expands mass spectrometry toolbox. J. Am. Soc. Mass Spectrom. 2016, 27, 1929–1936. [Google Scholar] [CrossRef]
- Kooijman, P.C.; Nagornov, K.O.; Kozhinov, A.N.; Kilgour, D.P.A.; Tsybin, Y.; Heeren, R.M.A.; Ellis, S.R. Increased throughput and ultra-high mass resolution in DESI FT-ICR MS imaging through new-generation external data acquisition system and advanced data processing approaches. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Ferey, J.; Marguet, F.; Laquerrière, A.; Marret, S.; Schmitz-Afonso, I.; Bekri, S.; Afonso, C.; Tebani, A. A new optimization strategy for MALDI FTICR MS tissue analysis for untargeted metabolomics using experimental design and data modeling. Anal. Bioanal. Chem. 2019, 411, 3891–3903. [Google Scholar] [CrossRef]
- Ciocan-Cartita, C.A.; Jurj, A.; Buse, M.; Gulei, D.; Braicu, C.; Raduly, L.; Cojocneanu, R.; Pruteanu, L.L.; Iuga, C.A.; Coza, O.; et al. The Relevance of Mass Spectrometry Analysis for Personalized Medicine through Its Successful Application in Cancer “Omics”. Int. J. Mol. Sci. 2019, 20, 2576. [Google Scholar] [CrossRef]
- Duarte, T.T.; Spencer, C.T. Personalized Proteomics: The Future of Precision Medicine. Proteomes 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Rogachev, A.; Alemasov, N.; Ivanisenko, V.; Ivanisenko, N.; Gaisler, E.; Oleshko, O.; Cheresiz, S.; Mishinov, S.; Stupak, V.; Pokrovsky, A. Correlation of Metabolic Profiles of Plasma and Cerebrospinal Fluid of High-Grade Glioma Patients. Metabolites 2021, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Caflisch, L.; Lodi, A.; Brenner, A.J.; Tiziani, S. Metabolomic signature of brain cancer. Mol. Carcinog. 2017, 56, 2355–2371. [Google Scholar] [CrossRef]
- Heiland, D.H.; Wörner, J.; Haaker, J.G.; Delev, D.; Pompe, N.; Mercas, B.; Franco, P.; Gäbelein, A.; Heynckes, S.; Pfeifer, D.; et al. The integrative metabolomic-transcriptomic landscape of glioblastome multiforme. Oncotarget 2017, 8, 49178–49190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, J.; Weinstein, S.J.; Kitahara, C.M.; Karoly, E.D.; Sampson, J.N.; Albanes, D. A prospective study of serum metab-olites and glioma risk. Oncotarget 2017, 8, 70366–70377. [Google Scholar] [CrossRef]
- Hvinden, I.C.; Berg, H.E.; Sachse, D.; Skaga, E.; Skottvoll, F.S.; Lundanes, E.; Sandberg, C.J.; Vik-Mo, E.O.; Rise, F.; Wilson, S.R. Nuclear Magnetic Resonance Spectroscopy to Identify Metabolite Biomarkers of Nonresponsiveness to Targeted Therapy in Glioblastoma Tumor Stem Cells. J. Proteome Res. 2019, 18, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Kurake, N.; Ishikawa, K.; Tanaka, H.; Hashizume, H.; Nakamura, K.; Kajiyama, H.; Toyokuni, S.; Kikkawa, F.; Mizuno, M.; Hori, M. Non-thermal plasma-activated medium modified metabolomic profiles in the glycolysis of U251SP glioblastoma. Arch. Biochem. Biophys. 2018, 662, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Wibom, C.; Surowiec, I.; Mörén, L.; Bergström, P.; Johansson, M.; Antti, H.; Bergenheim, A.T. Metabolomic Patterns in Glioblastoma and Changes during Radiotherapy: A Clinical Microdialysis Study. J. Proteome Res. 2010, 9, 2909–2919. [Google Scholar] [CrossRef]
- Yu, D.; Xuan, Q.; Zhang, C.; Hu, C.; Li, Y.; Zhao, X.; Liu, S.; Ren, F.; Zhang, Y.; Zhou, L.; et al. Metabolic Alterations Related to Glioma Grading Based on Metabolomics and Lipidomics Analyses. Metabolites 2020, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Jaroch, K.; Modrakowska, P.; Bojko, B. Glioblastoma Metabolomics—In Vitro Studies. Metabolites 2021, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, S.L.; Guggenbichler, W.; Weinberger, K.M.; Graber, A.; Stoeggl, W.M. Device for quantitative analysis of a drug or metabolite profile. Google Patents. 2012. Available online: https://patents.google.com/patent/RU2008102835A/en (accessed on 8 September 2021).
- Ramsay, S.L.; Stoeggl, W.M.; Weinberger, K.M.; Graber, A.; Guggenbichler, W. Apparatus and method for analyzing a metabolite profile. Google Patents. 2012. Available online: https://patents.google.com/patent/US8265877B2/en (accessed on 8 September 2021).
- Bush, M.F.; Campuzano, I.D.G.; Robinson, C.V. Ion Mobility Mass Spectrometry of Peptide Ions: Effects of Drift Gas and Calibration Strategies. Anal. Chem. 2012, 84, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tu, J.; Xiong, X.; Shen, X.; Zhu, Z.-J. LipidCCS: Prediction of Collision Cross-Section Values for Lipids with High Precision To Support Ion Mobility–Mass Spectrometry-Based Lipidomics. Anal. Chem. 2017, 89, 9559–9566. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. Metlin: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. Hmdb 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801. [Google Scholar] [CrossRef]
- Zhou, Z.; Luo, M.; Chen, X.; Yin, Y.; Xiong, X.; Wang, R.; Zhu, Z.-J. Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Fontanilles, M.; Marguet, F.; Beaussire, L.; Magne, N.; Pépin, L.-F.; Alexandru, C.; Tennevet, I.; Hanzen, C.; Langlois, O.; Jardin, F.; et al. Cell-free DNA and circulating tert promoter mutation for disease monitoring in newly-diagnosed glio-blastoma. Acta Neuropathol. Commun. 2020, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; Van Der Werf, M.J. Centering, scaling, and trans-formations: Improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar] [CrossRef]
- Ritchie, M.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Liang, F.; Song, Q.; Qiu, P. An Equivalent Measure of Partial Correlation Coefficients for High-Dimensional Gaussian Graphical Models. J. Am. Stat. Assoc. 2015, 110, 1248–1265. [Google Scholar] [CrossRef]
- Thistlethwaite, L.R.; Petrosyan, V.; Li, X.; Miller, M.J.; Elsea, S.H.; Milosavljevic, A. CTD: An information-theoretic algorithm to interpret sets of metabolomic and transcriptomic perturbations in the context of graphical models. PLoS Comput. Biol. 2021, 17, e1008550. [Google Scholar] [CrossRef]
- Wright, M.N.; Ziegler, A. ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77, 1–17. [Google Scholar] [CrossRef]
- Kuhn, M. Caret: Classification and Regression Training. 2020. Available online: https://cran.r-project.org/web/packages/caret/caret.pdf (accessed on 8 September 2021).
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing Website. 2020. Available online: https://www.gbif.org/zh/tool/81287/r-a-language-and-environment-for-statistical-computing (accessed on 8 September 2021).
- Kampa, J.M.; Kellner, U.; Marsching, C.; Ramallo Guevara, C.; Knappe, U.J.; Sahin, M.; Giampà, M.; Niehaus, K.; Bednarz, H. Glioblastoma multiforme: Metabolic differences to peritumoral tissue and idh-mutated gliomas revealed by mass spec-trometry imaging. Neuropathology 2020, 40, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Barritault, M.; Picart, T.; Poncet, D.; Fenouil, T.; D’Hombres, A.; Gabut, M.; Guyotat, J.; Jouanneau, E.; Ameli, R.; Joubert, B.; et al. Avoiding New Biopsies by Identification of IDH1 and TERT Promoter Mutation in Nondiagnostic Biopsies From Glioma Patients. Neurosurgery 2020, 87, E513–E519. [Google Scholar] [CrossRef]
- Bobeff, E.J.; Szczesna, D.; Bieńkowski, M.; Janczar, K.; Chmielewska-Kassassir, M.; Wiśniewski, K.; Papierz, W.; Wozniak, L.A.; Jaskólski, D.J. Plasma amino acids indicate glioblastoma with ATRX loss. Amino Acids 2021, 53, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.B. Acetylcholine. WormBook 2007, 1–21. [Google Scholar] [CrossRef]
- Sonkar, K.; Ayyappan, V.; Tressler, C.; Adelaja, O.; Cai, R.; Cheng, M.; Glunde, K. Focus on the glycerophosphocholine pathway in choline phospholipid metabolism of cancer. NMR Biomed. 2018, 32, e4112. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Kumabe, T.; Shirane, R.; Yoshimoto, T. Correlation between Choline Level Measured by Proton MR Spectroscopy and Ki-67 Labeling Index in Gliomas. Am. J. Neuroradiol. 2000, 21, 659–665. [Google Scholar]
- Lima, E.; Otaduy, M.; Tsunemi, M.H.; Pincerato, R.; Cardoso, E.; Rosemberg, S.; de Aguiar, P.H.P.; Cerri, G.; Leite, C. The Effect of Paramagnetic Contrast in Choline Peak in Patients with Glioblastoma Multiforme Might Not Be Significant. Am. J. Neuroradiol. 2012, 34, 80–84. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini Rev. Med. Chem. 2007, 7, 984–990. [Google Scholar] [CrossRef]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid processing in the brain: A key regulator of systemic metabolism. Front. Endocrinol. 2017, 8, 60. [Google Scholar] [CrossRef]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2018, 18, 960–969. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 2016, 19, 43–54. [Google Scholar] [CrossRef]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing fatty acid-induced lipotoxicity and its therapeutic potential in glioblastoma using stimulated Raman microscopy. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3, 10–18103. [Google Scholar]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth via Autophagic Release of Stored Fatty Acids. iScience 2020, 23, 101569. [Google Scholar] [CrossRef]
- Antal, O.; Péter, M.; Hackler, L.; Mán, I.; Szebeni, G.J.; Ayaydin, F.; Hideghéty, K.; Vigh, L.; Kitajka, K.; Balogh, G.; et al. Lipidomic analysis reveals a radiosensitizing role of gamma-linolenic acid in glioma cells. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2015, 1851, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Panzarini, E.; Tacconi, S.; Carata, E.; Mariano, S.; Tata, A.; Dini, L. Molecular Characterization of Temozolomide-Treated and Non Temozolomide-Treated Glioblastoma Cells Released Extracellular Vesicles and Their Role in the Macrophage Response. Int. J. Mol. Sci. 2020, 21, 8353. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef] [PubMed]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Rev. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Control (Brain), n = 6 1 | Control (Plasma), n = 60 1 | Glioblastoma, n = 22 1 | p-Value 2 |
|---|---|---|---|---|
| Sex | 0.023 | |||
| Female | 2 (33%) | 30 (50%) | 4 (18%) | |
| Male | 4 (67%) | 30 (50%) | 18 (82%) | |
| Age (Years) | 39 (25, 46) | 34 (27, 42) | 62 (54, 66) | <0.001 |
| ATRX_mutation | ||||
| Absent | 1 (6.2%) | |||
| Present | 15 (94%) | |||
| Unknown | 6 | |||
| IDH_mutation | ||||
| Absent | 15 (88%) | |||
| Present | 2 (12%) | |||
| Unknown | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilard, V.; Ferey, J.; Marguet, F.; Fontanilles, M.; Ducatez, F.; Pilon, C.; Lesueur, C.; Pereira, T.; Basset, C.; Schmitz-Afonso, I.; et al. Integrative Metabolomics Reveals Deep Tissue and Systemic Metabolic Remodeling in Glioblastoma. Cancers 2021, 13, 5157. https://doi.org/10.3390/cancers13205157
Gilard V, Ferey J, Marguet F, Fontanilles M, Ducatez F, Pilon C, Lesueur C, Pereira T, Basset C, Schmitz-Afonso I, et al. Integrative Metabolomics Reveals Deep Tissue and Systemic Metabolic Remodeling in Glioblastoma. Cancers. 2021; 13(20):5157. https://doi.org/10.3390/cancers13205157
Chicago/Turabian StyleGilard, Vianney, Justine Ferey, Florent Marguet, Maxime Fontanilles, Franklin Ducatez, Carine Pilon, Céline Lesueur, Tony Pereira, Carole Basset, Isabelle Schmitz-Afonso, and et al. 2021. "Integrative Metabolomics Reveals Deep Tissue and Systemic Metabolic Remodeling in Glioblastoma" Cancers 13, no. 20: 5157. https://doi.org/10.3390/cancers13205157
APA StyleGilard, V., Ferey, J., Marguet, F., Fontanilles, M., Ducatez, F., Pilon, C., Lesueur, C., Pereira, T., Basset, C., Schmitz-Afonso, I., Di Fioré, F., Laquerrière, A., Afonso, C., Derrey, S., Marret, S., Bekri, S., & Tebani, A. (2021). Integrative Metabolomics Reveals Deep Tissue and Systemic Metabolic Remodeling in Glioblastoma. Cancers, 13(20), 5157. https://doi.org/10.3390/cancers13205157