
Cell Biology of Giant Cell Tumour of Bone: Crosstalk between m/wt Nucleosome H3.3, Telomeres and Osteoclastogenesis


 ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
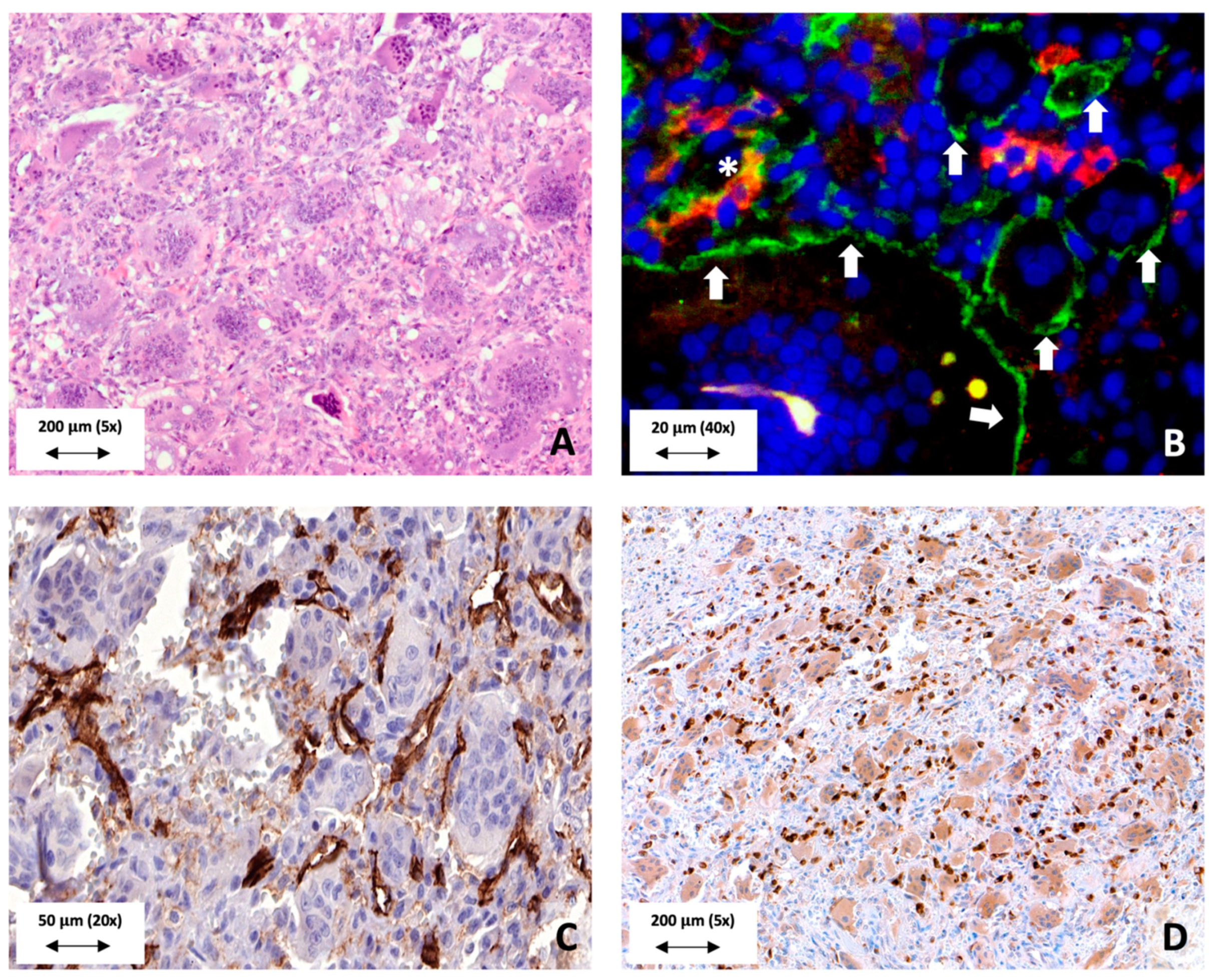
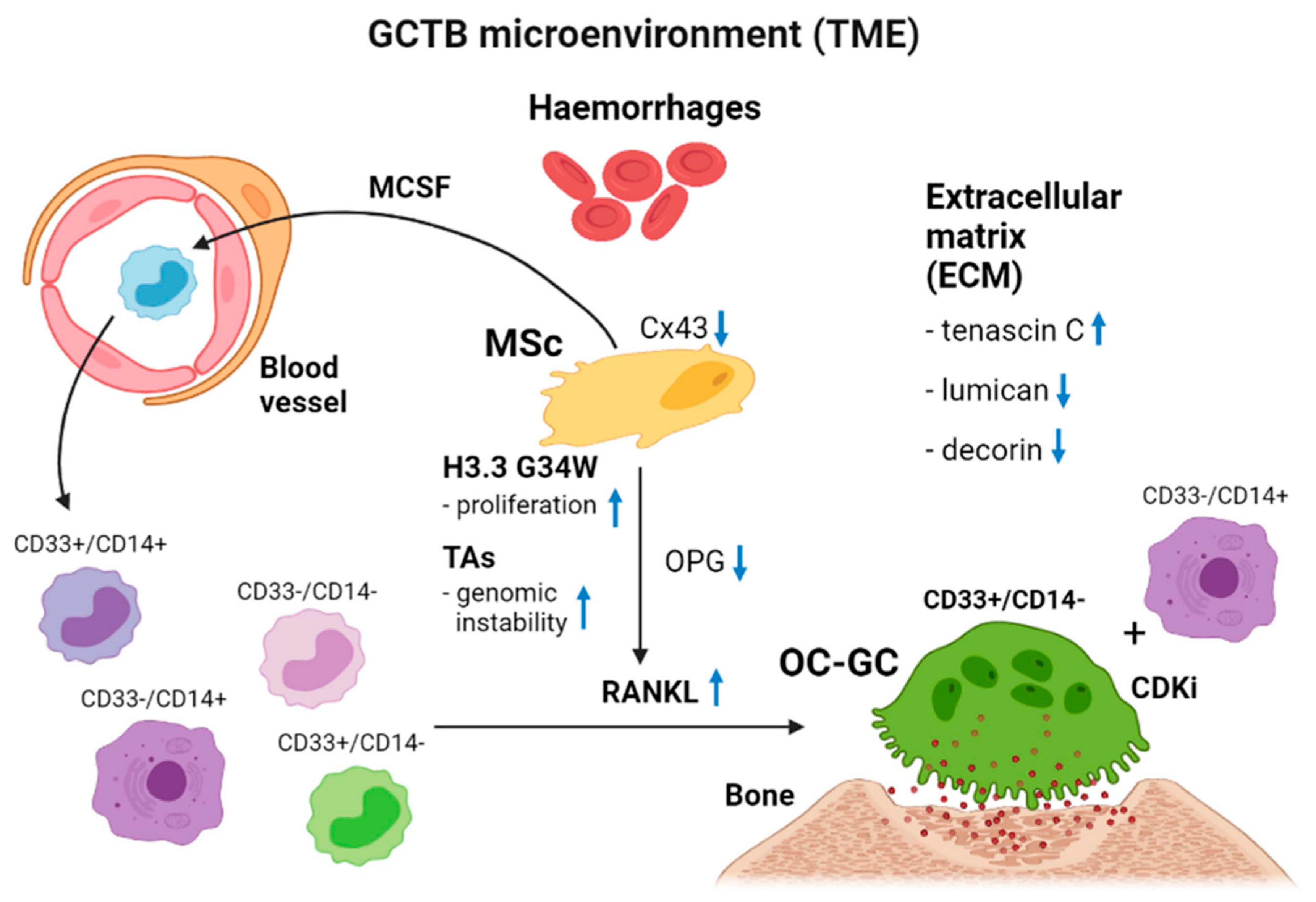
1. Cellular Makeup of GCTB
2. Cytogenetics of GCTB
3. Histones
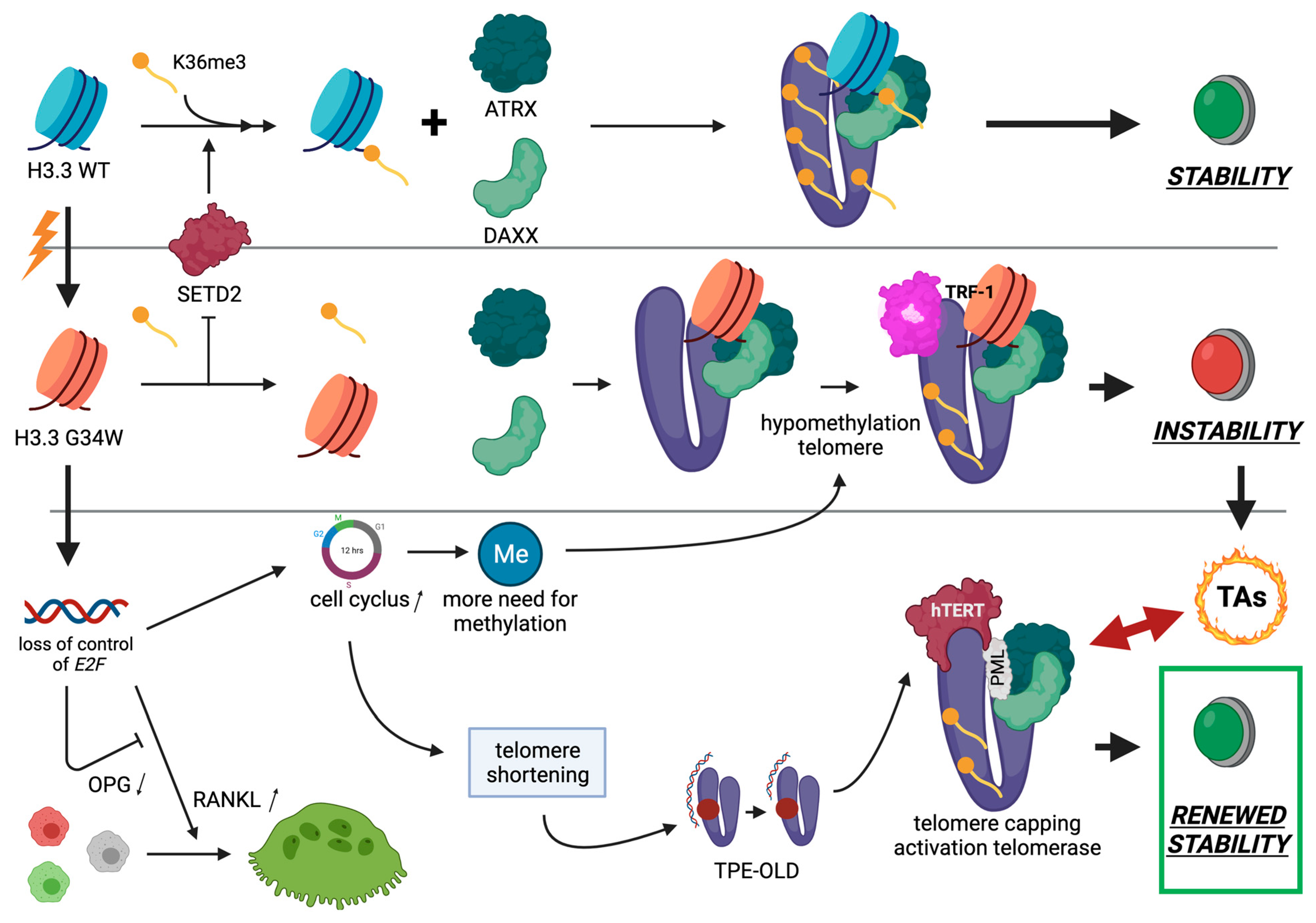
3.1. Histone H3.3
3.2. H3.3 Histone and Telomeres
3.3. H3.3-G34W, Telomere Biology and GCTB
3.4. H3.3-G34W and Malignant GCTB
3.5. H3.3-G34W, Histomorphology and Denosumab
4. Summary
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van der Heijden, L.; Dijkstra, P.D.S.; Blay, J.-Y.; Gelderblom, H. Giant cell tumour of bone in the denosumab era. Eur. J. Cancer 2017, 77, 75–83. [Google Scholar] [CrossRef]
- Liplaa, A.; Kroep, J.R.; van der Heijden, L.; Jutte, P.C.; Hogendoorn, P.C.W.; Dijsktra, S.; Gelderblom, H. Adjuvant Zoledronic Acid in High-Risk Giant Cell Tumor of Bone: A Multicenter Randomized Phase II Trial. Oncologist 2019, 24, 889–e421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakkers, R.J.B.; VAN DER Heul, R.O.; Kroon, H.M.; Taminiau, A.H.M.; Hogendoorn, P.C.W. Late Malignant Transformation of a Benign Giant-Cell Tumor of Bone. A Case Report. J. Bone Jt. Surg-Am. Vol. 1997, 79, 259–262. [Google Scholar] [CrossRef]
- Forsyth, R.G.; De Boeck, G.; Baelde, J.J.; Taminiau, A.H.; Uyttendaele, D.; Roels, H.; Praet, M.M.; Hogendoorn, P.C. CD33+ CD14− Phenotype Is Characteristic of Multinuclear Osteoclast-Like Cells in Giant Cell Tumor of Bone. J. Bone Miner. Res. 2009, 24, 70–77. [Google Scholar] [CrossRef]
- Maggiani, F.; Forsyth, R.; Hogendoorn, P.C.W.; Krenacs, T.; Athanasou, N.A. The immunophenotype of osteoclasts and macrophage polykaryons. J. Clin. Pathol. 2011, 64, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Branstetter, D.G.; Nelson, S.D.; Manivel, J.C.; Blay, J.-Y.; Chawla, S.; Thomas, D.; Jun, S.; Jacobs, I. Denosumab Induces Tumor Reduction and Bone Formation in Patients with Giant-Cell Tumor of Bone. Clin. Cancer Res. 2012, 18, 4415–4424. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.; Henshaw, R.; Seeger, L.; Choy, E.; Blay, J.-Y.; Ferrari, S.; Kroep, J.; Grimer, R.; Reichardt, P.; Rutkowski, P.; et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: Interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013, 14, 901–908. [Google Scholar] [CrossRef]
- Lindeman, J.H.; Hanemaaijer, R.; Mulder, A.; Dijkstra, P.S.; Szuhai, K.; Bromme, D.; Verheijen, J.H.; Hogendoorn, P.C. Cathepsin K Is the Principal Protease in Giant Cell Tumor of Bone. Am. J. Pathol. 2004, 165, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Maros, E.M.; Balla, P.; Micsik, T.; Sapi, Z.; Szendroi, M.; Wenz, H.; Groden, C.; Forsyth, R.G.; Picci, P.; Krenacs, T. Cell cycle regulatory protein expression in multinucleated giant cells of giant cell tumor of bone: Do they proliferate? Pathol. Oncol. Res. 2021, 27. [Google Scholar] [CrossRef]
- Alberghini, M.; Kliskey, K.; Krenacs, T.; Picci, P.; Kindblom, L.; Forsyth, R.; Athanasou, N.A. Morphological and immunophenotypic features of primary and metastatic giant cell tumour of bone. Virchows Arch. 2009, 456, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, L.; Conti, A.; Chiechi, A.; Novello, C.; Magagnoli, G.; Astolfi, A.; Pession, A.; Krenacs, T.; Alberghini, M.; Picci, P.; et al. Differential gene expression in classic giant cell tumours of bone: Tenascin C as biological risk factor for local relapses and metastases. Histopathology 2010, 57, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adhes. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasou, N.A.; Bansal, M.; Forsyth, R.; Reid, R.P.; Sapi, Z. Giant cell tumour of bone. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D.M., Bridge, J.A., Hogendoorn, P.C.W., Mertens, F., Eds.; Lyon International Agency for Research on Cancer (IARC): Lyon, France, 2013; p. 321e4. [Google Scholar]
- Balla, P.; Maros, M.E.; Barna, G.; Antal, I.; Papp, G.; Sapi, Z.; Athanasou, N.A.; Benassi, M.S.; Picci, P.; Krenacs, T. Prognostic Impact of Reduced Connexin43 Expression and Gap Junction Coupling of Neoplastic Stromal Cells in Giant Cell Tumor of Bone. PLoS ONE 2015, 10, e0125316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paznekas, W.A.; Karczeski, B.; Vermeer, S.; Lowry, R.B.; Delatycki, M.; Laurence, F.; Koivisto, P.A.; Van Maldergem, L.; Boyadjiev, S.A.; Bodurtha, J.N.; et al. GJA1mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 2009, 30, 724–733. [Google Scholar] [CrossRef]
- Bellido, T.; Plotkin, L.I. Novel actions of bisphosphonates in bone: Preservation of osteoblast and osteocyte viability. Bone 2011, 49, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, U.N.; Goodman, M.; Chung, W.-W.; Swalski, P.; Pal, R.; Finkelstein, S. Molecular analysis of primary and recurrent giant cell tumors of bone. Cancer Genet. Cytogenet. 2005, 158, 126–136. [Google Scholar] [CrossRef]
- Gorunova, L.; von Steyern, F.V.; Storlazzi, C.T.; Bjerkehagen, B.; Follerås, G.; Heim, S.; Mandahl, N.; Mertens, F. Cytogenetic analysis of 101 giant cell tumors of bone: Nonrandom patterns of telomeric associations and other structural aberrations. Genes Chromosom. Cancer 2009, 48, 583–602. [Google Scholar] [CrossRef]
- Forsyth, R.; De Boeck, G.; Bekaert, S.; De Meyer, T.; Taminiau, A.; Uyttendaele, D.; Roels, H.; Praet, M.; Hogendoorn, P.; Hogendoorn, P. Telomere biology in giant cell tumour of bone. J. Pathol. 2007, 214, 555–563. [Google Scholar] [CrossRef]
- Aschacher, T.; Wolf, B.; Ascacher, O.; Enzmann, F.; Laszlo, V.; Messner, B.; Türkcan, A.; Weis, S.; Spiegl-Kreinecker Holzman, K.; Laufer, G.; et al. Long interspersed element-1 ribonucleoproetin particles protect telomeric ends in alternative lengthening of telomeres dependent cells. Neoplasia 2020, 22, 61–75. [Google Scholar] [CrossRef]
- Lisaingo, K.; Uringa, E.-J.; Lansdorp, P.M. Resolution of telomere associations by TRF1 cleavage in mouse embryonic stem cells. Mol. Biol. Cell 2014, 25, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Cleven, A.H.; Höcker, S.; Bruijn, I.B.-D.; Szuhai, K.; Cleton-Jansen, A.-M.; Bovée, J.V. Mutation Analysis of H3F3A and H3F3B as a Diagnostic Tool for Giant Cell Tumor of Bone and Chondroblastoma. Am. J. Surg. Pathol. 2015, 39, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Venneker, S.; Szuhai, K.; Hogendoorn, P.; Bovée, J.V.M.G. Mutation-driven epigenetic alterations as a defining hallmark of central cartilaginous tumours, giant cell tumour of bone and chondroblastoma. Virchows Arch. 2019, 476, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskovszky, L.; Szuhai, K.; Krenács, T.; Hogendoorn, P.C.W.; Szendrői, M.; Benassi, M.S.; Kopper, L.; Füle, T.; Sápi, Z. Genomic instability in giant cell tumor of bone. A study of 52 cases using DNA ploidy, relocalization FISH, and array-CGH analysis. Genes Chromosom. Cancer 2009, 48, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Antal, I.; Sapi, Z.; Szendrői, M. The prognostic significance of DNA cytophotometry and proliferation index (Ki-67) in giant cell tumors of bone. Int. Orthop. 1999, 23, 315–319. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chormatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Koning, L.; Corpet, A.; Haber, J.E.; Almouzni, G. Histone chaperons: An escort network regulating histone traffic. Nat. Struct. Mol. Biol. 2007, 14, 997–1007. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 Complexes Mediate Nucleosome Assembly Pathways Dependent or Independent of DNA Synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Drané, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.; Elsaesser, S.J.; Noh, K.-M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.; George, A.; Morgan, K.A.; Mann, J.R.; Choo, K.A. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C. Feisengeld G. Nucelosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Smith, M.; Kanno, T.; Dasenbrock, H.; Nishiyama, A.; Ozato, K. Inducible Deposition of the Histone Variant H3.3 in Interferon-stimulated Genes. J. Biol. Chem. 2009, 284, 12217–12225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.T.; et al. Dynamics of histone H3 deposition in vivo reval a nucleosome gap–filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneiderman, J.I.; Orsi, G.; Hughes, K.T.; Loppin, B.; Ahmad, K. Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc. Natl. Acad. Sci. USA 2012, 109, 19721–19726. [Google Scholar] [CrossRef] [Green Version]
- Deal, R.B.; Henikoff, J.G.; Henikoff, S. Genome-Wide Kinetics of Nucleosome Turnover Determined by Metabolic Labeling of Histones. Science 2010, 328, 1161–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.; Listovsky, T.; Guilbaud, G.; Sarkies, P.; Sale, J.E. Histone H3.3 Is Required to Maintain Replication Fork Progression after UV Damage. Curr. Biol. 2014, 24, 2195–2201. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, D.; Turniansky, R.; Green, E.M. Choose Your Own Adventure: The Role of Histone Modifications in Yeast Cell Fate. J. Mol. Biol. 2016, 429, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Jezek, M.; Green, E.M. Histone modifications and the maintenance of telomere integrity. Cells 2019, 8, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maros, M.E.; Schnaidt, S.; Balla, P.; Kelemen, Z.; Sapi, Z.; Szendroi, M.; Laszlo, T.; Forsyth, R.; Picci, P.; Krenacs, T. In situ cell cycle analysis in giant cell tumor of bone reveals patients with elevated risk of reduced progression-free survival. Bone 2019, 127, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Shi, J.; Shi, X.; Li, W.; Wen, H. Histone H3.3 G34 mutations alter H3K36 and H3K27 methylation in cis. J. Mol. Biol. 2018, 430, 1562–1565. [Google Scholar] [CrossRef]
- Goldberg, A.; Banaszynski, L.A.; Noh, K.-M.; Lewis, P.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, L.H.; Ren, H.; Williams, E.; McGhie, J.; Ahn, S.; Sim, M.; Tam, A.; Earle, E.; Anderson, M.A.; Mann, J.; et al. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 2008, 19, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, F.T.M.; McGhie, J.D.; Chan, F.L.; Tang, M.C.; Anderson, M.A.; Mann, J.R.; Choo, K.H.A.; Wong, L.H. PML bodies provide an important platform for the maintenance of telomeric chromatin integrity in embryonic stem cells. Nucleic Acids Res. 2013, 41, 4447–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, M.J.; Lower, K.M.; Voon, H.P.; Hughes, J.R.; Garrick, D.; Virpakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H. Epigenetic regulation of telomere chromatin integrity in pluripotent embryonic stem cells. Epigenomics 2010, 2, 639–655. [Google Scholar] [CrossRef]
- Elsaesser, S.J.; Goldberg, A.; Allis, C.D. New functions for an old variant: No substitute for histone H3.3. Curr. Opin. Genet. Dev. 2010, 20, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Gibbons, R.; Yan, Z.; Yang, D.; McDowell, T.L.; Sechi, S.; Qin, J.; Zhou, S.; Higgs, D.; Wang, W. The ATRX syndrome protein forms a chromatin-remodelling complex with Daxx and localizes in promyelocytemic leukemia nuclear bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 10635–10640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDowell, T.L.; Gibbons, R.J.; Sutherland, H.; O’Rourke, D.M.; Bickmore, W.A.; Pombo, A.; Turley, H.; Gatter, K.; Picketts, D.J.; Buckle, V.J.; et al. Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc. Natl. Acad. Sci. USA 1999, 96, 13983–13988. [Google Scholar] [CrossRef] [Green Version]
- Udugama, M.; Chang, F.T.M.; Chan, F.L.; Tang, M.C.; Pickett, H.A.; McGhie, J.D.R.; Mayne, L.; Collas, P.; Mann, J.R.; Wong, L.H. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 2015, 43, 10227–10237. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.; Polo, S.; Almouzni, G. Transcription Recovery after DNA Damage Requires Chromatin Priming by the H3.3 Histone Chaperone HIRA. Cell 2013, 155, 94–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsik, P.; Baude, A.; Mancarella, D.; Öz, S.; Kühn, A.; Toth, R.; Hey, J.; Toprak, U.H.; Lim, J.; Nguyen, V.H.; et al. Globally altered epigenetic landscape and delayed osteogenic differentiation in H3.3-G34W-mutant giant cell tumor of bone. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Berger, S.L. Epigenetics of aging and aging-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S17–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardat, M.; Déjardin, J. Telomere chromatin establishment and its maintenance during mammalian development. Chromosoma 2017, 127, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Bitterman, K.J.; Medvedik, O.; Sinclair, D.A. Longevety regulation in Saccharomyces cerevisiae: Linking metabolism, genome stability, and heterochromatin. Microbiol. Mol. Biol. Rev. 2003, 67, 376–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. From The Cover: Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Kim, H.; Huang, R.; Lu, W.; Tang, M.; Shi, F.; Yang, D.; Zhang, X.; Huang, J.; Liu, D.; et al. The Daxx/Atrx Complex Protects Tandem Repetitive Elements during DNA Hypomethylation by Promoting H3K9 Trimethylation. Cell Stem Cell 2015, 17, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Khelifi, A.F.; D’Alcontres, M.S.; Salomoni, P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005, 12, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Salamoni, P.; Khelifi, A.F. DAXX: Death or survival protein? Trends Cell. Biol. 2006, 16, 97–104. [Google Scholar] [CrossRef]
- Bérubé, N.G.; Healy, J.; Medina, C.F.; Wu, S.; Hodgson, T.; Jagla, M.; Picketts, D.J. Patient mutations alter ATRX targeting to PML nuclear bodies. Eur. J. Hum. Genet. 2007, 16, 192–201. [Google Scholar] [CrossRef]
- Iwase, S.; Xiang, B.; Ghosh, S.; Ren, T.; Lewis, P.W.; Cochrane, J.C.; Allis, C.D.; Picketts, D.J.; Patel, D.J.; Li, H.; et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 2011, 18, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Salomoni, P.; Ferguson, B.; Wyllie, A.; Rich, T.; Ferguson, B. New insights into the role of PML in tumour suppression. Cell Res. 2008, 18, 622–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maizels, N.; Gray, L.T. The G4 genome. PLoS Genet. 2013, 9, e1003468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, K.; Halder, R.; Chowdhury, S. Genome-wide analysis predicts DNA structural motifs as nucleosome exclusion signals. Mol. BioSyst. 2009, 5, 1703–1712. [Google Scholar] [CrossRef]
- Wong, H.M.; Huppert, J.L. Stable G-quadruplexes are found outside nucleosome-bound regions. Mol. Biosyst. 2009, 5, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Morohashi, N.; Nishimura, Y.; Kurumizaka, H.; Shimizu, M. Telomeric repeats act as nucleosome-disfavouring sequences in vivo. Nucleic Acids Res. 2013, 42, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric Repeat Containing RNA and RNA Surveillance Factors at Mammalian Chromosome Ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Higgs, D.; Gibbons, R. The chromatin remodeller ATRX: A repeat offender in human disease. Trends Biochem. Sci. 2013, 38, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.A.; Solomon, L.A.; Li, J.R.; Jiang, Y.; Edwards, M.; Shin-ya, K.; Beier, F.; Bérube, N. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J. Clin. Investig. 2013, 123, 2049–2063. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; De Lange, T.; De, S.; Petrini, J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- García-Cao, M.; O’Sullivan, R.; Peters, A.H.F.M.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2003, 36, 94–99. [Google Scholar] [CrossRef]
- Galati, A.; Micheli, E.; Alicata, C.; Ingegnere, T.; Cicconi, A.; Pusch, M.C.; Giraud-Panis, M.-J.; Gilson, E.; Cacchione, S. TRF1 and TRF2 binding to telomeres is modulated by nucleosomal organization. Nucleic Acids Res. 2015, 43, 5824–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, H.S.; Bataille, A.R.; Zhang, L.; Pugh, B.F. Subnucleosomal Structures and Nucleosome Asymmetry across a Genome. Cell 2014, 159, 1377–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Gan, H.; Lee, J.-H.; Han, J.; Wang, Z.; Riester, S.M.; Jinglong, W.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRCS activity by gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Lopez de Silanes, I.; Grana, O.; De Bonis, M.L.; Dominguez, O.; Pisano, D.G.; Blasco, M.A. Identification of TERRA locus unveils a telomere protetction role through associationto nearly all chromosomes. Nat. Commun. 2014, 5, 4723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA Binding to TRF2 Facilitates Heterochromatin Formation and ORC Recruitment at Telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, J.J.; Lopez-Silanes, I.; Megias, D.; Fraga, M.F.; Castells-Garcia Blasco, M.A. TERRA recruitment of polycomb to telomeres is essential for histonetrimethylation marks at telomere heterochromatin. Nat. Commun. 2018, 9, 1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.T.; Mayerson, J.; Nowak, N.J.; Suster, D.; Mohammed, N.; Long, S.; Auer, H.; Jones, S.; McKeegan, C.; Young, G.; et al. 20q11.1 amplification in giant-cell tumor of bone: Array CGH, FISH, and association with outcome. Genes Chromosom. Cancer 2006, 45, 957–966. [Google Scholar] [CrossRef]
- Chow, T.T.; Shi, X.; Wei, J.H.; Guan, J.; Stadler, G.; Huang, B.; Blackburn, E.H. Local enrichtment of HP1alpha at telomeres alters their structure and regulation of telomere protection. Nat. Commun. 2018, 9, 3583. [Google Scholar] [CrossRef] [Green Version]
- Gottschling, D.E.; Aparicio, O.; Billington, B.L.; Zakian, V.A. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 1990, 63, 751–762. [Google Scholar] [CrossRef]
- Misteli, T. The long reach telomeres. Genes Dev. 2014, 28, 2445–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef] [Green Version]
- Mukerherjee, A.K.; Sharma, S.; Sengupta, S.; Saha, D.; Kumar PHussain, T.; Srivastava, V.; Deb Roy, S.; Shay, J.W.; Chowdhurry, S. Telomere length-dependent transcription and epigenetic modifications in promotors remote from telomere ends. PLoS Genet. 2018, 14, e1007782. [Google Scholar]
- Robin, J.; Ludlow, A.T.; Batten, K.; Gaillard, M.-C.; Stadler, G.; Magdinier, F.; Wright, W.; Shay, J.W. SORBS2transcription is activated by telomere position effect–over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy. Genome Res. 2015, 25, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.; Stadler, G.; Mender, I.; Lai, T.-P.; Zhang, N.; Wright, W.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmeri, E.; Picci, P.; Reichardt, P.; Downey, G. Malignancy in Giant Cell Tumor of Bone: A Review of the Literature. Technol. Cancer Res. Treat. 2019, 18, 1–9. [Google Scholar]
- Aponte-Tinao, L.A.; Piuzzi, N.S.; Roitman, P.; Farfalli, G.L. A High-grade Sarcoma Arising in a Patient with Recurrent Benign Giant Cell Tumor of the Proximal Tibia While Receiving Treatment with Denosumab. Clin. Orthop. Relat. Res. 2015, 473, 3050–3055. [Google Scholar] [CrossRef]
- Broehm, C.J.; Garbrecht, E.L.; Wood, J.; Bocklage, T. Two Cases of Sarcoma Arising in Giant Cell Tumor of Bone Treated with Denosumab. Case Rep. Med. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, S.; Righi, A.; Vanel, D.; Honoki, K.; Donati, D.M.; Errani, C. Development of high-grade osteosarcoma in a patient with recurrent giant cell tumor of the ischium while receiving treatment with denosumab. Jpn. J. Clin. Oncol. 2017, 47, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- WHO classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, WHO classification of tumours series, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 3, pp. 440–446. [Google Scholar]
- Fittall, M.W.; Lyskjaer, I.; Ellery, P.; Lombard, P.; Ijaz, J.; Strobi, A.C.; Oukrif, D.; Tarabichi, M.; Sill, M.; Koelsche, C.; et al. Drivers un-derpinning the malignant transformation of giant cell tumour of bone. J. Pathol. 2020, 252, 433–440. [Google Scholar] [CrossRef]
- War, A.R.; Dang, K.; Jiang, S.; Xiao, Z.; Miao, Z.; Yang, T.; Li, Y.; Qian, A. Role of cancer stem cells in the development of giant cell tumor of bone. Cancer Cell Int. 2020, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hardouin, S.N.; Guo, N.; Romeo, P.H.; Nagy, A.; Aubin, J.E. Impaireed mesenchymal stem cell differentiation and osteoclastogenesis in mice deficient for Igf2-P2 transcripts. Development 2011, 138, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.-C.; Wu, G.-H.; Zhou, L.-L.; Yang, X.-J.; Liu, J.-T. Leptin improves osteoblast differentiation of human bone marrow stroma stem cells. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 3507–3513. [Google Scholar]
- Mancini, I.; Righi, A.; Gambarotti, M.; Picci, P.; Dei Tos, A.P.; Billing, S.D.; Simi, L.; Franchi, A. Phenotypic and molecular differences between giant-cell tumour of soft tissue and its bone counterpart. Histopathology 2017, 71, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Garmilla, P.; Riancho, J.A. Do epigenetic marks govern bone mass and homeostasis? Curr. Genom. 2012, 13, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Ghayor, C.; Weber, F.E. Epigentic regulation of bone remodelling and its impacts in osteoporosis. Int. J. Mol. Sci. 2016, 17, 1446. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, A.; Pelka, S.; Gray, D.S.; Moghe, P.V. Engeneering lineage potency and plasticity of stem cells using epigenetic molecules. Sci. Rep. 2018, 8, 16289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, E.; Wysocka, J. Modification of Enhancer Chromatin: What, How, and Why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2015, 32, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, R.; Gonzales-Cope, M.; Chronis, C.; Bonora, G.; McKee, R.; Huang, C.; Patel, S.; Lopez, D.; Mishra, N.; Pellegrini, M.; et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct Epigenomic Landscapes of Pluripotent and Lineage-Committed Human Cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Gambacurta, A.; Merlini, G.; Ruggiero, C.; Diedenhofen, G.; Battista, N.; Bari, M.; Balsamo, M.; Piccirillo, S.; Valentini, G.; Mascetti, G.; et al. Human osteogenic differentiation in Space: Proteomic and epigenetic clues to better understand osteoporosis. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Niu, N.; Li, L.; Shao, R.; Ouyang, H.; Zou, W. H3K36 trimethylation mediated by SETD2 regulates the fate of bone marrow mesenchymal stem cells. PLoS Biol. 2018, 16, e2006522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazaei, S.; De Jay, N.; Deshmukh Hendrikse, L.D.; Jawhar, W.; Chen, C.C.L.; Mikael, L.G.; Faury, D.; Marchione, D.M.; Lanoix, J.; Bonneil, É.; et al. H3.3 G34W promotes growth and impedes differentiation of osteoblast-like mesenchymal progenitors in giant cell tumor of bone. Cancer Discov. 2020, 10, 1968–1987. [Google Scholar] [CrossRef]
- Hemingway, F.; Kashima, T.G.; Mahendra, G.; Dhongre, A.; Hogendoorn, P.; Mertens, F.; Athanasou, N.A. Smooth muscle actin expression in primary bone tumours. Virchows Arch. 2012, 460, 525–534. [Google Scholar] [CrossRef]
- Roitman, P.D.; Jauk, F.; Farfalli, G.L.; Albergo, J.I.; Aponte-Tinao, L.A. Denosumab-treated giant cell tumor of bone. Its histological spectrum and potential diagnotisc pitfalls. Hum. Pathol. 2017, 63, 89–97. [Google Scholar]
- Van der Heijden, L.; van de Sande, M.A.; Hogendoorn, P.C.; Gelderblom, H.; Dijkstra, P.D.S. Neoadjuvant denosumab for extensive giant cell tumor in os ischium: A case report. Acta Othop. 2015, 86, 393–395. [Google Scholar] [CrossRef]
- Chawla, S.; Blay, J.-Y.; Rutkowski, P.; Le Cesne, A.; Reichardt, P.; Gelderblom, H.; Grimer, R.J.; Choy, E.; Skubitz, K.; Seeger, L.; et al. Denosumab in patients with giant-cell tumour of bone: A multicentre, open-label, phase 2 study. Lancet Oncol. 2019, 20, 1719–1729. [Google Scholar] [CrossRef]
- Strauss, S.J.; Frezza, A.M.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; Bovalot, S.; et al. ESMO guidelines Committee, EUROCAN, GENTURIS and ARNPaedCan. Bone sarcomas: ESMO-EUROCAN-GENTURIS-ERNPaedCan clinical practice guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 29 (Suppl. S4), iv79–iv95. [Google Scholar]
- Song, I.; Kim, K.; Kim, J.H.; Lee, Y.-K.; Jung, H.-J.; Byun, H.-O.; Yoon, G.; Kim, N. GATA4 negatively regulates osteoblast differentiation by downregulation of Runx2. BMB Rep. 2014, 47, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, I.; Furuya, M.; Matsuo, K.; Kawabata, Y.; Tanaka, R.; Ohashi, K. Giant cell tumours of bone treated with denosumab: Histological, immunohistochemical and H3F3A mutation analyses. Histopathology 2017, 72, 914–922. [Google Scholar] [CrossRef]
- Lim, J.; Park, J.H.; Baude, A.; Yoo, Y.; Lee, Y.K.; Schmidt, C.R.; Park, J.B.; Fellenberg, J.; Zustin, J.; Haller, F.; et al. The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Nakashima, T.; Kobayashi, Y.; Yamasaki, S.; Kawakami, A.; Eguchi, K.; Sasaki, H.; Sakai, H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: Modulation of the expression by osteetropic factors and cytokines. Biochem. Biophys. Res. Commun. 2000, 275, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Conceição, A.L.G.; Babeto; Candido, N.M.; Franco, F.C.; Zuccari, D.A.P.D.C.; Bonilha, J.L.; Cordeiro, J.A.; Calmon, M.; Rahal, P. Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. J. Cancer 2015, 6, 593–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [Green Version]
- Sugamori, Y.; Mise-Omata, S.; Maeda, C.; Aoki, S.; Tabata, Y.; Murali, R.; Yasuda, H.; Udagawa, N.; Suzuki, H.; Honma, M.; et al. Peptide drugs accelerate BMP-2-induced calvarial bone regeneration and stimulate osteoblast differentiation through mTORC1 signaling. BioEssays 2016, 38, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef]
- Martin, T.J.; Sims, N.A. RANKL/OPG: Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Guo, C.; Xu, X.; Gao, J.; Zhang, J.; Chen, T.; Cui, D. Osteogenic growth peptide enhances the proliferation of bone marrow mesenchymal stem cells from osteoprotegerin-deficient mice by CDK2/cyclin A. Acta Biochim. Biophys. Sin. 2010, 42, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T. Regulation of osteoblast differentiation by transcription factors. J. Cell. Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forsyth, R.G.; Krenács, T.; Athanasou, N.; Hogendoorn, P.C.W. Cell Biology of Giant Cell Tumour of Bone: Crosstalk between m/wt Nucleosome H3.3, Telomeres and Osteoclastogenesis. Cancers 2021, 13, 5119. https://doi.org/10.3390/cancers13205119
Forsyth RG, Krenács T, Athanasou N, Hogendoorn PCW. Cell Biology of Giant Cell Tumour of Bone: Crosstalk between m/wt Nucleosome H3.3, Telomeres and Osteoclastogenesis. Cancers. 2021; 13(20):5119. https://doi.org/10.3390/cancers13205119
Chicago/Turabian StyleForsyth, Ramses G., Tibor Krenács, Nicholas Athanasou, and Pancras C. W. Hogendoorn. 2021. "Cell Biology of Giant Cell Tumour of Bone: Crosstalk between m/wt Nucleosome H3.3, Telomeres and Osteoclastogenesis" Cancers 13, no. 20: 5119. https://doi.org/10.3390/cancers13205119
APA StyleForsyth, R. G., Krenács, T., Athanasou, N., & Hogendoorn, P. C. W. (2021). Cell Biology of Giant Cell Tumour of Bone: Crosstalk between m/wt Nucleosome H3.3, Telomeres and Osteoclastogenesis. Cancers, 13(20), 5119. https://doi.org/10.3390/cancers13205119