
HCV Activates Somatic L1 Retrotransposition—A Potential Hepatocarcinogenesis Pathway

 , , ,
, , ,  , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
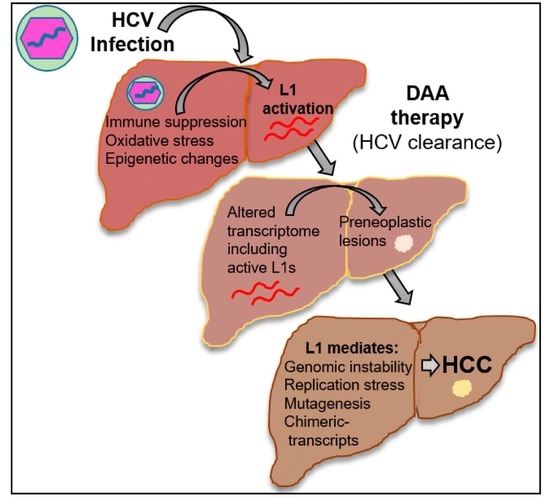
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Chemical Inhibitors
2.2. Patient Samples
2.3. Plasmid Transfection
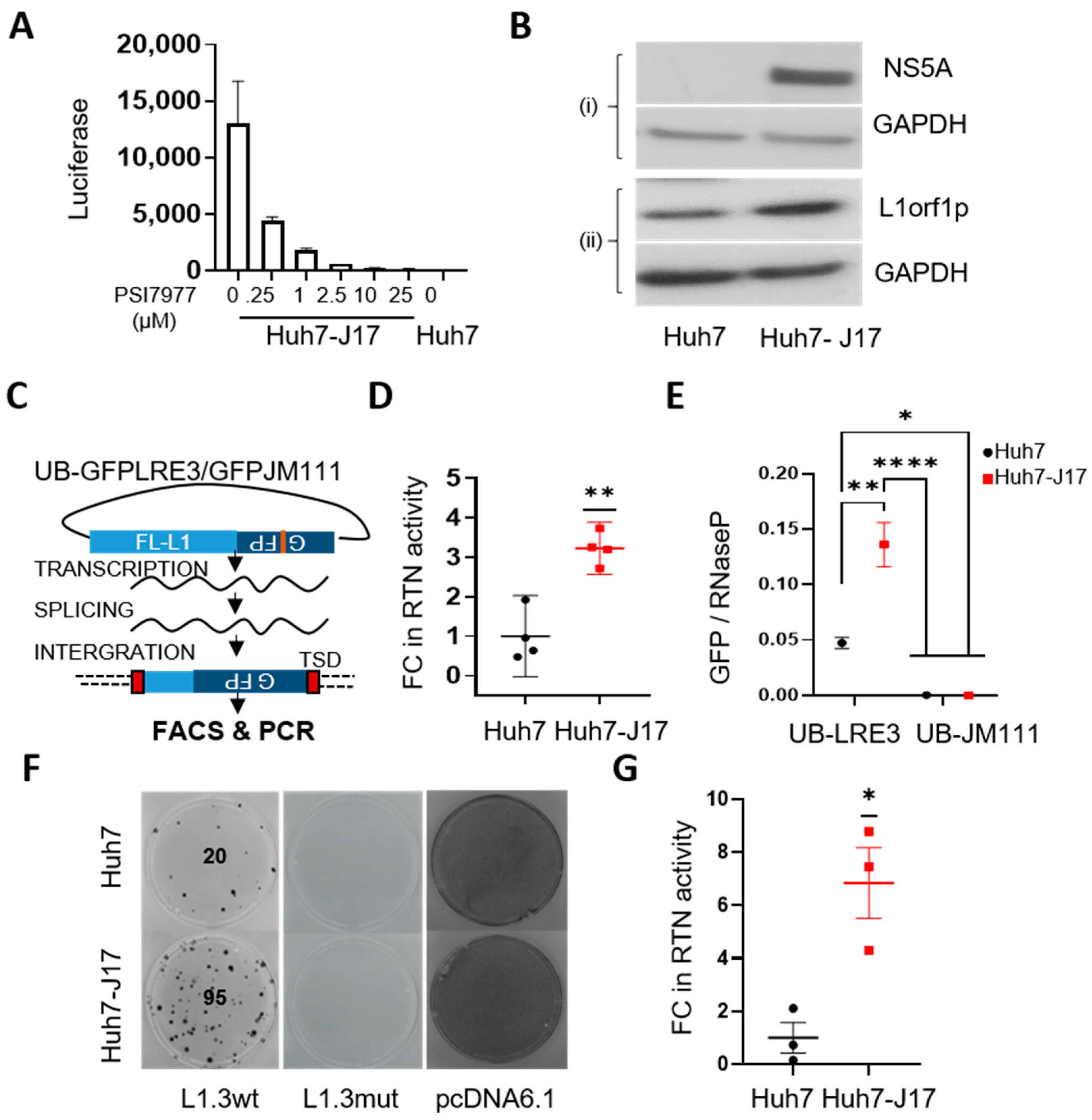
2.4. Retrotransposition Assay
2.5. DNA Damage Repair Plasmid Re-Joining Assays
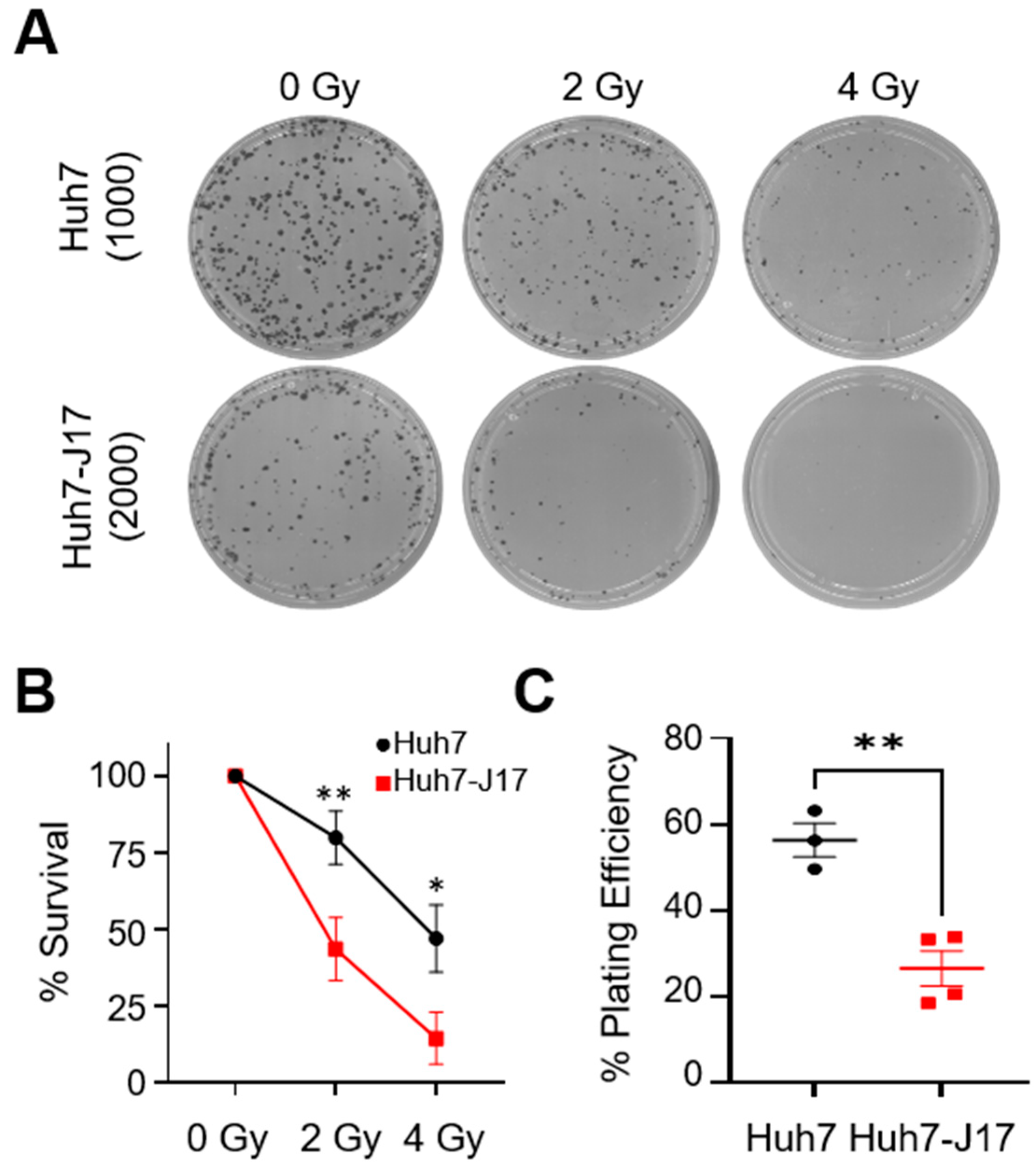
2.6. X-ray Irradiation Sensitivity Assay
conditions) × 100.
2.7. Immunohistochemistry (IHC)
2.8. Western Blot Analysis
2.9. Luciferase Assay
2.10. Bioinformatics
2.11. Statistical Analysis
3. Results
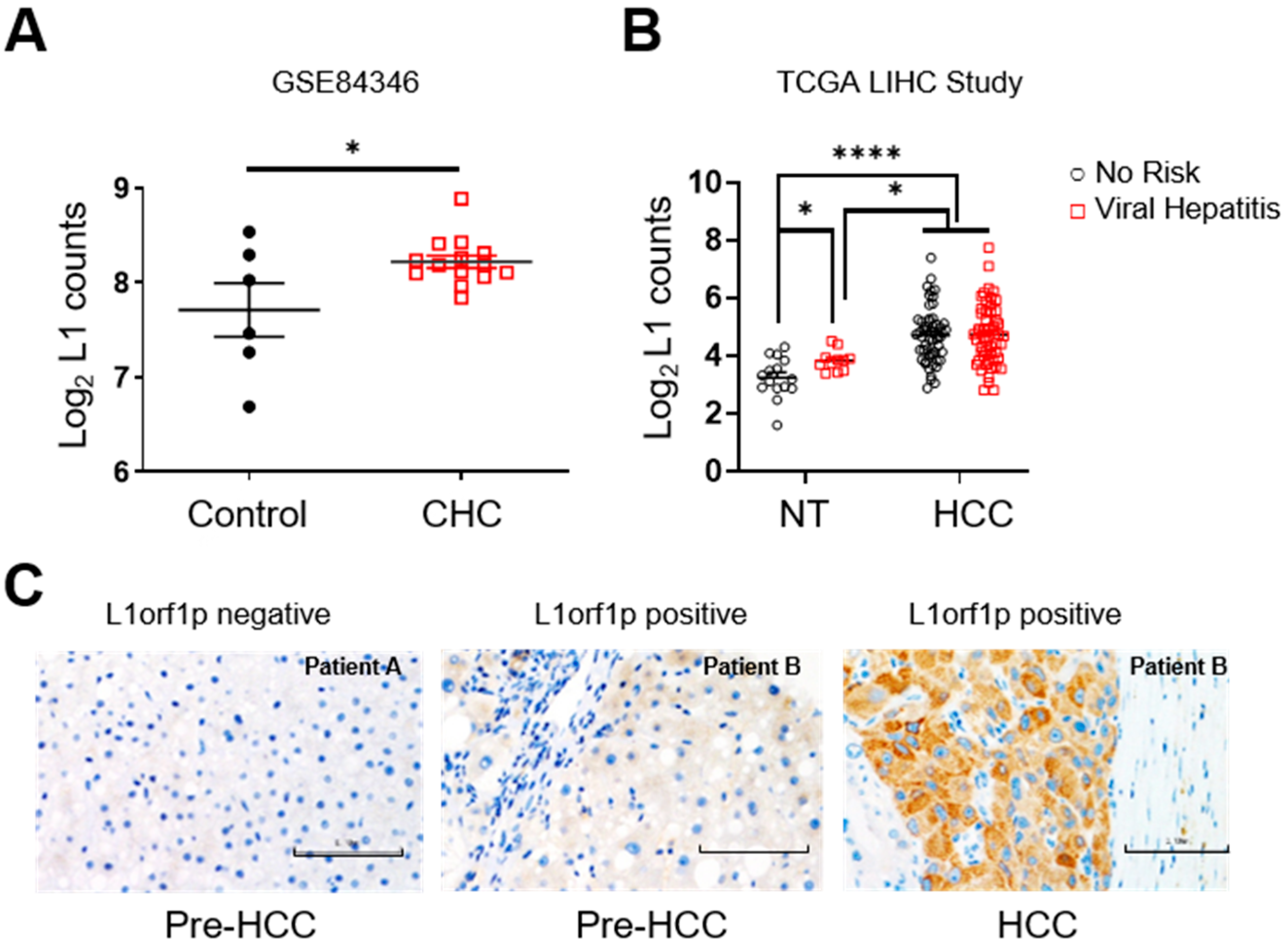
3.1. L1 Expression Is Upregulated in Non-Tumour Tissue of Patients with Chronic Hepatitis
3.2. HCV Activates L1 Retrotransposition
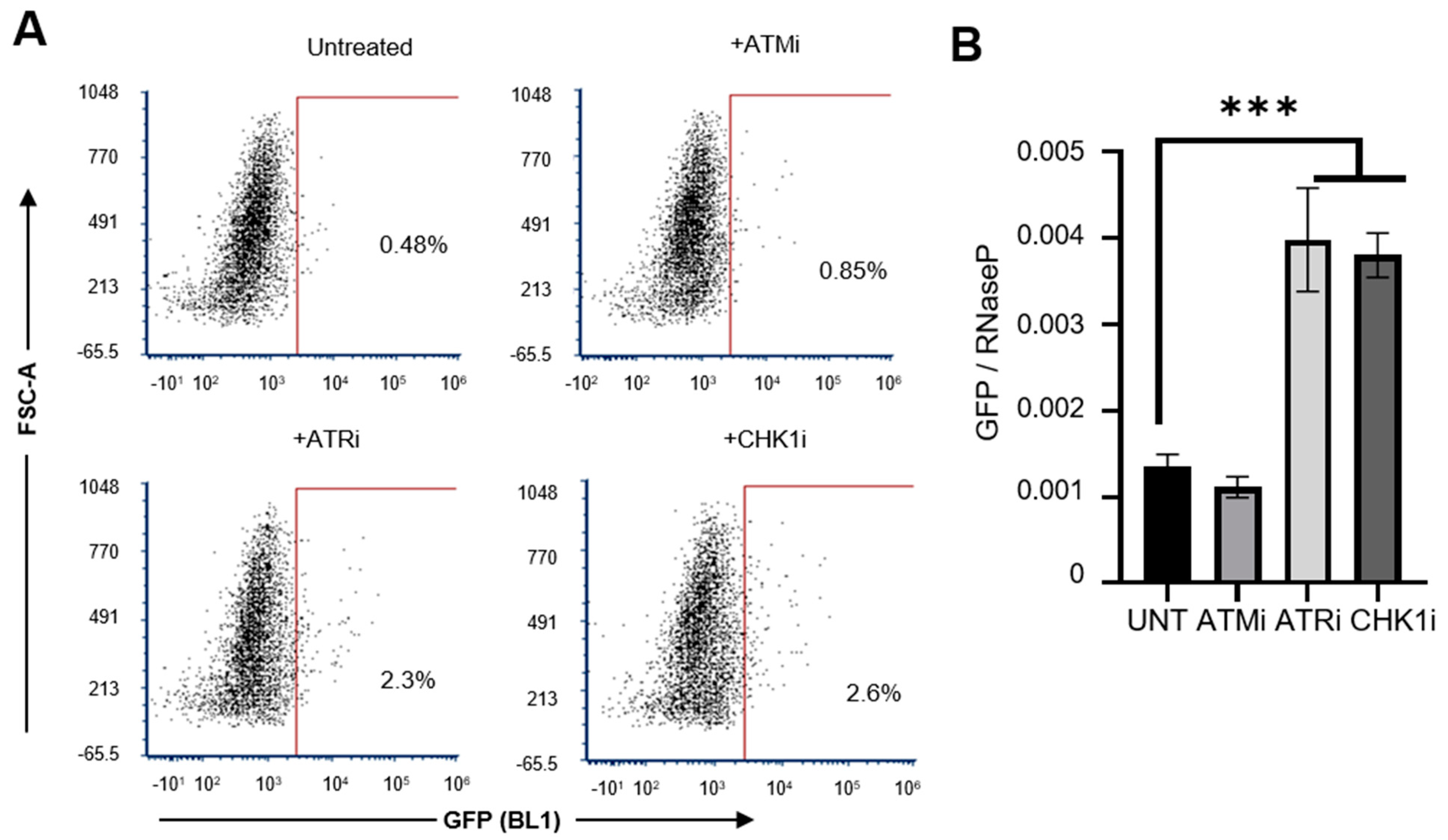
3.3. HCV Potentially Upregulates L1 Retrotransposition via the Inhibition of DNA Damage Repair Pathways
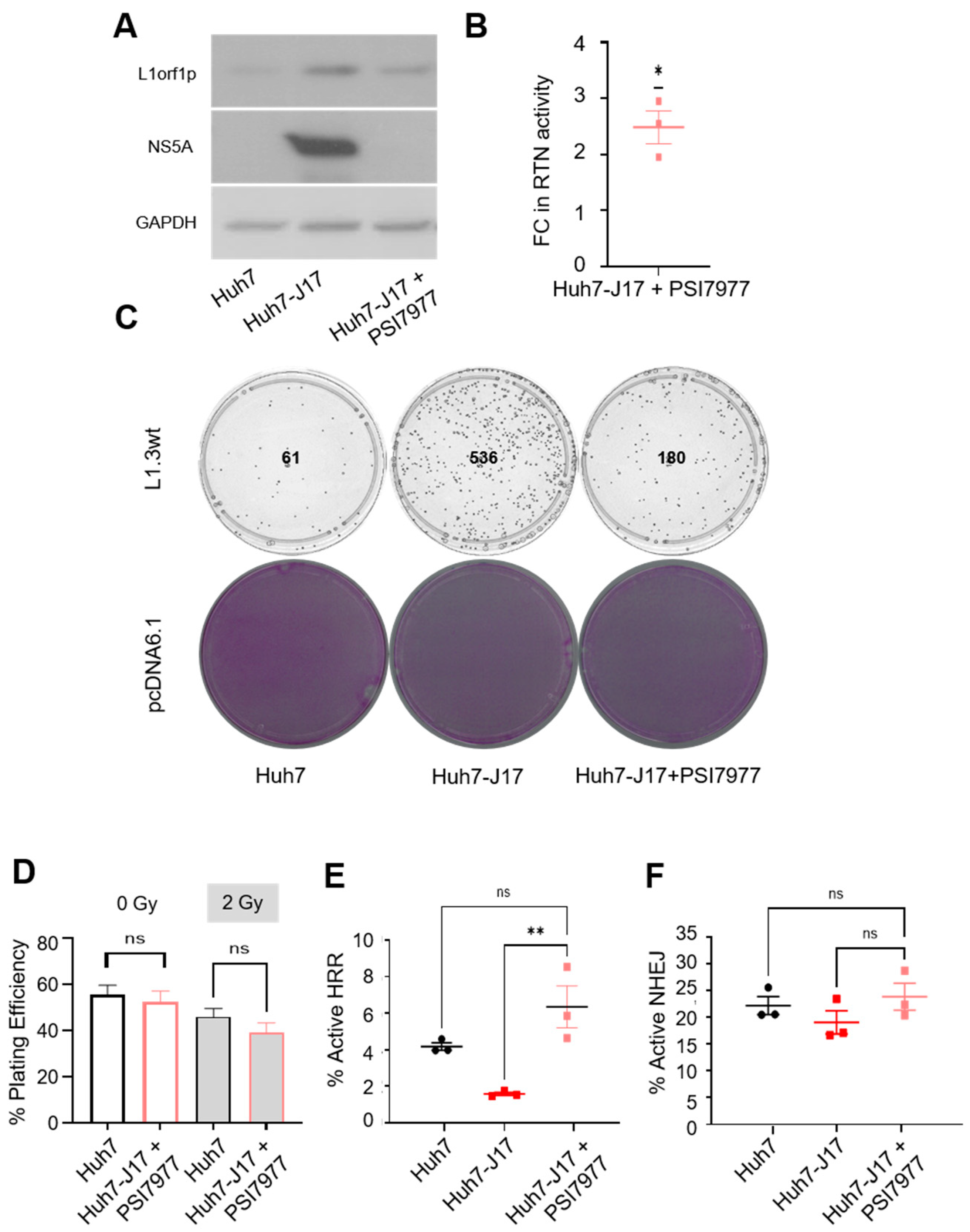
3.4. Influence on L1 Retrotransposition and Its Consequences Persists after Viral Clearance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Chung, R.T.; Baumert, T.F. Curing chronic hepatitis c—The arc of a medical triumph. N. Engl. J. Med. 2014, 370, 1576–1578. [Google Scholar] [CrossRef]
- Rinaldi, L.; Nevola, R.; Franci, G.; Perrella, A.; Corvino, G.; Marrone, A.; Berretta, M.; Morone, M.V.; Galdiero, M.; Giordano, M.; et al. Risk of hepatocellular carcinoma after hcv clearance by direct-acting antivirals treatment predictive factors and role of epigenetics. Cancers 2020, 12, 1351. [Google Scholar] [CrossRef]
- Ioannou, G.N. Hcc surveillance after svr in patients with f3/f4 fibrosis. J. Hepatol. 2021, 74, 458–465. [Google Scholar] [CrossRef]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with hcv-related hcc undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dustin, L.B.; Bartolini, B.; Capobianchi, M.R.; Pistello, M. Hepatitis c virus: Life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin. Microbiol. Infect. 2016, 22, 826–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Rong, L.; Li, Y.P. Flaviviridae viruses and oxidative stress: Implications for viral pathogenesis. Oxidative Med. Cell. Longev. 2019, 2019, 1409582. [Google Scholar] [CrossRef] [Green Version]
- Maki, A.; Kono, H.; Gupta, M.; Asakawa, M.; Suzuki, T.; Matsuda, M.; Fujii, H.; Rusyn, I. Predictive power of biomarkers of oxidative stress and inflammation in patients with hepatitis c virus-associated hepatocellular carcinoma. Ann. Surg. Oncol. 2007, 14, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Ploen, D.; Hildt, E. Hcv and oxidative stress: Implications for hcv life cycle and hcv-associated pathogenesis. Oxidative Med. Cell. Longev. 2016, 2016, 9012580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.Y.K.; Ou, J.J. Autophagy in hcv replication and protein trafficking. Int. J. Mol. Sci. 2021, 22, 1089. [Google Scholar] [CrossRef]
- Chan, S.T.; Ou, J.J. Hepatitis c virus-induced autophagy and host innate immune response. Viruses 2017, 9, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis c virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the atm-nbs1/mre11/rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol. 2010, 185, 6985–6998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.T.; Park, E.M.; Lim, Y.S.; Hwang, S.B. Nonstructural protein 5a impairs DNA damage repair: Implications for hepatitis c virus-mediated hepatocarcinogenesis. J. Virol. 2018, 92, e00178-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA damage response by rna viruses. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijetunga, N.A.; Pascual, M.; Tozour, J.; Delahaye, F.; Alani, M.; Adeyeye, M.; Wolkoff, A.W.; Verma, A.; Greally, J.M. A pre-neoplastic epigenetic field defect in hcv-infected liver at transcription factor binding sites and polycomb targets. Oncogene 2017, 36, 2030–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongrui, L.; Na, H.; Zongfang, L.; Fanpu, J.; Shiwen, J. Epigenetic mechanism involved in the hbv/hcv-related hepatocellular carcinoma tumorigenesis. Curr. Pharm. Des. 2014, 20, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, N.; Juhling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.; et al. Hcv-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology 2019, 156, 2313–2329.e2317. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Alison Holmes, J.; Dai, C.Y.; Huang, C.F.; Chung, R.T.; et al. Hepatitis c virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Shinjo, K.; Shimizu, Y.; Sano, T.; Yamao, K.; Gao, W.; Fujii, M.; Osada, H.; Sekido, Y.; Murakami, S.; et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 2014, 146, 562–572. [Google Scholar] [CrossRef]
- Shukla, R.; Upton, K.R.; Muñoz-Lopez, M.; Gerhardt, D.J.; Fisher, M.E.; Nguyen, T.; Brennan, P.M.; Baillie, J.K.; Collino, A.; Ghisletti, S.; et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 2013, 153, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by line-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Schauer, S.N.; Carreira, P.E.; Shukla, R.; Gerhardt, D.J.; Gerdes, P.; Sanchez-Luque, F.J.; Nicoli, P.; Kindlova, M.; Ghisletti, S.; Santos, A.D.; et al. L1 retrotransposition is a common feature of mammalian hepatocarcinogenesis. Genome Res. 2018, 28, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Hlady, R.A.; Joyce, B.T.; Robertson, K.D.; He, C.; Nannini, D.R.; Kibbe, W.A.; Achenbach, C.J.; Murphy, R.L.; Roberts, L.R.; et al. DNA methylation of individual repetitive elements in hepatitis c virus infection-induced hepatocellular carcinoma. Clin. Epigenetics 2019, 11, 145. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., 3rd; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Angus, A.G.; Loquet, A.; Stack, S.J.; Dalrymple, D.; Gatherer, D.; Penin, F.; Patel, A.H. Conserved glycine 33 residue in flexible domain i of hepatitis c virus core protein is critical for virus infectivity. J. Virol. 2012, 86, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Ozerov, A.A.; Tunitskaya, V.L.; Valuev-Elliston, V.T.; Wahid, A.; Pirisi, M.; Simmonds, P.; Ivanov, A.V.; Novikov, M.S.; Patel, A.H. Exploration of acetanilide derivatives of 1-(omega-phenoxyalkyl)uracils as novel inhibitors of hepatitis c virus replication. Sci. Rep. 2016, 6, 29487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flasch, D.A.; Macia, A.; Sanchez, L.; Ljungman, M.; Heras, S.R.; Garcia-Perez, J.L.; Wilson, T.E.; Moran, J.V. Genome-wide de novo l1 retrotransposition connects endonuclease activity with replication. Cell 2019, 177, 837–851.e828. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.V.; Holmes, S.E.; Naas, T.P.; DeBerardinis, R.J.; Boeke, J.D.; Kazazian, H.H. High frequency retrotransposition in cultured mammalian cells. Cell 1996, 87, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Klawitter, S.; Fuchs, N.V.; Upton, K.R.; Munoz-Lopez, M.; Shukla, R.; Wang, J.; Garcia-Canadas, M.; Lopez-Ruiz, C.; Gerhardt, D.J.; Sebe, A.; et al. Reprogramming triggers endogenous l1 and alu retrotransposition in human induced pluripotent stem cells. Nat. Commun. 2016, 7, 10286. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, A.; O’Donnell, R.; Drew, Y.; Curtin, N.J.; Sharma Saha, S. Characterisation of ovarian cancer cell line nih-ovcar3 and implications of genomic, transcriptomic, proteomic and functional DNA damage response biomarkers for therapeutic targeting. Cancers 2020, 12, 1939. [Google Scholar] [CrossRef]
- Wu, C.E.; Esfandiari, A.; Ho, Y.H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by mdm2 and wip1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Gilbert, N.; Ooi, S.L.; Lawler, J.F.; Ostertag, E.M.; Kazazian, H.H.; Boeke, J.D.; Moran, J.V. Human l1 retrotransposition: Cis preference versus trans complementation. Mol. Cell. Biol. 2001, 21, 1429–1439. [Google Scholar] [CrossRef] [Green Version]
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse transcription of r2bm rna is primed by a nick at the chromosomal target site: A mechanism for non-ltr retrotransposition. Cell 1993, 72, 595–605. [Google Scholar] [CrossRef]
- Feng, Q.; Moran, J.V.; Kazazian, H.H., Jr.; Boeke, J.D. Human l1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 1996, 87, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Morrish, T.A.; Garcia-Perez, J.L.; Stamato, T.D.; Taccioli, G.E.; Sekiguchi, J.; Moran, J.V. Endonuclease-independent line-1 retrotransposition at mammalian telomeres. Nature 2007, 446, 208–212. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human line-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Marchetto, M.C.; Muotri, A.R.; Mu, Y.; Carson, C.T.; Macia, A.; Moran, J.V.; Gage, F.H. Ataxia telangiectasia mutated (atm) modulates long interspersed element-1 (l1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20382–20387. [Google Scholar] [CrossRef] [Green Version]
- Mita, P.; Sun, X.; Fenyo, D.; Kahler, D.J.; Li, D.; Agmon, N.; Wudzinska, A.; Keegan, S.; Bader, J.S.; Yun, C.; et al. Brca1 and s phase DNA repair pathways restrict line-1 retrotransposition in human cells. Nat. Struct. Mol. Biol. 2020, 27, 179–191. [Google Scholar] [CrossRef]
- Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N.J. Targeting the atr-chk1 axis in cancer therapy. Cancers 2017, 9, 41. [Google Scholar] [CrossRef]
- Schobel, A.; Nguyen-Dinh, V.; Schumann, G.G.; Herker, E. Hepatitis c virus infection restricts human line-1 retrotransposition in hepatoma cells. PLoS Pathog. 2021, 17, e1009496. [Google Scholar] [CrossRef]
- Nakayama, R.; Ueno, Y.; Ueda, K.; Honda, T. Latent infection with kaposi’s sarcoma-associated herpesvirus enhances retrotransposition of long interspersed element-1. Oncogene 2019, 38, 4340–4351. [Google Scholar] [CrossRef] [PubMed]
- Kawano, K.; Doucet, A.J.; Ueno, M.; Kariya, R.; An, W.; Marzetta, F.; Kuroki, M.; Turelli, P.; Sukegawa, S.; Okada, S.; et al. Hiv-1 vpr and p21 restrict line-1 mobility. Nucleic Acids Res. 2018, 46, 8454–8470. [Google Scholar] [CrossRef] [Green Version]
- Iijima, K.; Okudaira, N.; Tamura, M.; Doi, A.; Saito, Y.; Shimura, M.; Goto, M.; Matsunaga, A.; Kawamura, Y.I.; Otsubo, T.; et al. Viral protein r of human immunodeficiency virus type-1 induces retrotransposition of long interspersed element-1. Retrovirology 2013, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; Song, H.; Xu, Y.; Garrison, K.E.; Buzdin, A.A.; Anwar, N.; Hunter, D.V.; Mujib, S.; Mihajlovic, V.; Martin, E.; et al. Line-1 retrotransposable element DNA accumulates in hiv-1-infected cells. J. Virol. 2013, 87, 13307–13320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Carbone, C.J.; Katlinskaya, Y.V.; Zheng, H.; Zheng, K.; Luo, M.; Wang, P.J.; Greenberg, R.A.; Fuchs, S.Y. Type i interferon controls propagation of long interspersed element-1. J. Biol. Chem. 2015, 290, 10191–10199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Huang, Q.; Boeke, J.D. Effect of reverse transcriptase inhibitors on line-1 and ty1 reverse transcriptase activities and on line-1 retrotransposition. BMC Biochem. 2011, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; Garrison, K.E.; Wong, J.C.; Duan, E.H.; Nixon, D.F.; Ostrowski, M.A. Nucleoside analogue reverse transcriptase inhibitors differentially inhibit human line-1 retrotransposition. PLoS ONE 2008, 3, e1547. [Google Scholar] [CrossRef]
- Maeda, K.; Das, D.; Kobayakawa, T.; Tamamura, H.; Takeuchi, H. Discovery and development of anti-hiv therapeutic agents: Progress towards improved hiv medication. Curr. Top. Med. Chem. 2019, 19, 1621–1649. [Google Scholar] [CrossRef] [PubMed]
- Mavilia, M.G.; Wu, G.Y. Hbv-hcv coinfection: Viral interactions, management, and viral reactivation. J. Clin. Transl. Hepatol. 2018, 6, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Platt, L.; Easterbrook, P.; Gower, E.; McDonald, B.; Sabin, K.; McGowan, C.; Yanny, I.; Razavi, H.; Vickerman, P. Prevalence and burden of hcv co-infection in people living with hiv: A global systematic review and meta-analysis. Lancet Infect. Dis. 2016, 16, 797–808. [Google Scholar] [CrossRef]
- Global Burden Of Hepatitis, C.W.G. Global burden of disease (gbd) for hepatitis c. J. Clin. Pharmacol. 2004, 44, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Aydin, Y.; Widmer, K.E.; Nayak, L. Hepatocellular carcinoma mechanisms associated with chronic hcv infection and the impact of direct-acting antiviral treatment. J. Hepatocell. Carcinoma 2020, 7, 45–76. [Google Scholar] [CrossRef] [Green Version]
- Roche, B.; Coilly, A.; Duclos-Vallee, J.C.; Samuel, D. The impact of treatment of hepatitis c with daas on the occurrence of hcc. Liver Int. 2018, 38, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, I.M.; Lim, J.K.; Fried, M.W. American gastroenterological association institute clinical practice update-expert review: Care of patients who have achieved a sustained virologic response after antiviral therapy for chronic hepatitis c infection. Gastroenterology 2017, 152, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Eldafashi, N.; Darlay, R.; Shukla, R.; McCain, M.V.; Watson, R.; Liu, Y.L.; McStraw, N.; Fathy, M.; Fawzy, M.A.; Zaki, M.Y.W.; et al. A pdcd1 role in the genetic predisposition to nafld-hcc? Cancers 2021, 13, 1412. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the pnpla3 rs738409 c >g polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, J.; Mei, T.T.; Guo, H.Q.; Wei, X.H.; Zhang, W.Y.; Liu, Y.L.; Liang, S.; Fan, Z.P.; Ma, L.X.; et al. Association of tm6sf2 rs58542926 t/c gene polymorphism with hepatocellular carcinoma: A meta-analysis. BMC Cancer 2019, 19, 1128. [Google Scholar] [CrossRef]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef]
- Ponomaryova, A.A.; Rykova, E.Y.; Gervas, P.A.; Cherdyntseva, N.V.; Mamedov, I.Z.; Azhikina, T.L. Aberrant methylation of line-1 transposable elements: A search for cancer biomarkers. Cells 2020, 9, 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Shin, T.J.; Kim, W.H.; Cho, J.Y. Methylation of line-1 in cell-free DNA serves as a liquid biopsy biomarker for human breast cancers and dog mammary tumors. Sci. Rep. 2019, 9, 175. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudhindar, P.D.; Wainwright, D.; Saha, S.; Howarth, R.; McCain, M.; Bury, Y.; Saha, S.S.; McPherson, S.; Reeves, H.; Patel, A.H.; et al. HCV Activates Somatic L1 Retrotransposition—A Potential Hepatocarcinogenesis Pathway. Cancers 2021, 13, 5079. https://doi.org/10.3390/cancers13205079
Sudhindar PD, Wainwright D, Saha S, Howarth R, McCain M, Bury Y, Saha SS, McPherson S, Reeves H, Patel AH, et al. HCV Activates Somatic L1 Retrotransposition—A Potential Hepatocarcinogenesis Pathway. Cancers. 2021; 13(20):5079. https://doi.org/10.3390/cancers13205079
Chicago/Turabian StyleSudhindar, Praveen D., Daniel Wainwright, Santu Saha, Rachel Howarth, Misti McCain, Yvonne Bury, Sweta S. Saha, Stuart McPherson, Helen Reeves, Arvind H. Patel, and et al. 2021. "HCV Activates Somatic L1 Retrotransposition—A Potential Hepatocarcinogenesis Pathway" Cancers 13, no. 20: 5079. https://doi.org/10.3390/cancers13205079
APA StyleSudhindar, P. D., Wainwright, D., Saha, S., Howarth, R., McCain, M., Bury, Y., Saha, S. S., McPherson, S., Reeves, H., Patel, A. H., Faulkner, G. J., Lunec, J., & Shukla, R. (2021). HCV Activates Somatic L1 Retrotransposition—A Potential Hepatocarcinogenesis Pathway. Cancers, 13(20), 5079. https://doi.org/10.3390/cancers13205079