Simple Summary
Myeloproliferative neoplasms (MPN) are clonal hematologic malignancies with dysregulated myeloid blood cell production driven by JAK2, calreticulin, and MPL gene mutations. Technological advances have revealed a heterogeneous genomic landscape with additional mutations mainly in epigenetic regulators and splicing factors, which are of diagnostic and prognostic value and may inform treatment decisions. Thus, genetic testing has become an integral part of the state-of-the-art work-up for MPN. The finding that JAK2, CALR, and MPL mutations activate JAK2 signaling has promoted the development of targeted JAK2 inhibitor therapies. However, their disease-modifying potential remains limited and investigations of additional molecular vulnerabilities in MPN are imperative to advance the development of new therapeutic options. Here, we summarize the current insights into the genetic basis of MPN, its use as diagnostic and prognostic tool in clinical settings, and recent advances in targeted therapies for MPN.
Abstract
Somatic mutations in JAK2, calreticulin, and MPL genes drive myeloproliferative neoplasms (MPN), and recent technological advances have revealed a heterogeneous genomic landscape with additional mutations in MPN. These mainly affect genes involved in epigenetic regulation and splicing and are of diagnostic and prognostic value, predicting the risk of progression and informing decisions on therapeutic management. Thus, genetic testing has become an integral part of the current state-of-the-art laboratory work-up for MPN patients and has been implemented in current guidelines for disease classification, tools for prognostic risk assessment, and recommendations for therapy. The finding that JAK2, CALR, and MPL driver mutations activate JAK2 signaling has provided a rational basis for the development of targeted JAK2 inhibitor therapies and has fueled their translation into clinical practice. However, the disease-modifying potential of JAK2 inhibitors remains limited and is further impeded by loss of therapeutic responses in a substantial proportion of patients over time. Therefore, the investigation of additional molecular vulnerabilities involved in MPN pathogenesis is imperative to advance the development of new therapeutic options. Combination of novel compounds with JAK2 inhibitors are of specific interest to enhance therapeutic efficacy of molecularly targeted treatment approaches. Here, we summarize the current insights into the genetic basis of MPN, its use as a diagnostic and prognostic tool in clinical settings, and the most recent advances in targeted therapies for MPN.
1. Introduction
Myeloproliferative neoplasms (MPN) are clonal hematologic malignancies, which are characterized by an excessive output of mature myeloid blood cells and arise from mutant hematopoietic stem/progenitor cells [1]. MPN were first recognized as an entity by William Dameshek in 1951, who proposed that dysregulated myeloid blood cell proliferation was driven by a yet undefined stimulus [2]. In polycythemia vera (PV), this primarily becomes manifest as erythrocytosis, while essential thrombocythemia (ET) is characterized by thrombocytosis, and primary myelofibrosis (PMF) shows increased megakaryocytes along with progressive fibrosis of the bone marrow. Thrombohemorrhagic complications account in large parts for morbidity and mortality of MPN. Of note, all three forms may progress to acute myeloid leukemia mostly with high risk genetic features and dismal prognosis [1]. It was not until 2005 that technological advances in DNA sequencing led to the discovery of the JAK2V617F mutation [3,4,5,6], the most frequent driver mutation in MPN, followed by the identification of additional driver mutations in exon 12 of JAK2 [7], the thrombopoietin receptor MPL [8,9] and calreticulin (CALR) genes [10,11]. With the increased accessibility of next-generation sequencing (NGS) approaches to large patient cohorts, we have recently gained more detailed insights into the heterogeneous genetic landscape of MPN and how additional mutations, together and in interplay with the so-called driver mutations, play significant roles in determining the clinical phenotype and course of MPN [12,13,14]. This has impacted on the way we diagnose MPN, as we increasingly rely on genetic/genomic information in addition to typical bone marrow morphology findings [15,16,17,18]. It has also enabled the development of small molecule tyrosine kinase inhibitors targeting JAK2 with clinical benefits for MPN patients [19,20,21,22,23,24,25]. In this review, we discuss the current knowledge regarding the mutational landscape of MPN and its diagnostic and prognostic value, as well as novel therapeutic options rationally designed based on the recent molecular insights and potential future targets for MPN therapy.
2. Genetic Landscape of Myeloproliferative Neoplasms
2.1. Somatic Driver Mutations
Myeloproliferative neoplasms arise from hematopoietic stem/progenitor cells with somatic gene mutations enabling clonal expansion. Limiting dilution experiments have demonstrated that a single hematopoietic stem/progenitor cell harboring the JAK2V617F mutation is sufficient to drive the emergence of an MPN phenotype in mouse models [26]. Thus, JAK2V617F and analogously mutations in CALR or MPL genes, which per se can drive outgrowth of a mutant cell clone and induce an MPN phenotype, are considered as driver mutations. They have been incorporated as a major criterion into the World Health Organization (WHO) diagnostic criteria of MPN and represent standard laboratory testing for patients with suspected MPN [15].
The valine to phenylalanine substitution at position 617 of the JAK2 protein is located in the pseudokinase domain of JAK2 and was the first driver mutation to be discovered in 2005 (Figure 1) [3,4,5,6]. JAK2 is an intracellular tyrosine kinase essential for intracellular signaling downstream of hematopoietic cytokine receptors including thrombopoietin (TPO), erythropoietin (EPO), and granulocyte-colony stimulating factor (G-CSF) receptors [27,28]. The JAK2V617F mutation impedes the inhibitory effect of the pseudokinase on the kinase domain and results in constitutive activation of JAK2 and its downstream effectors, including the signal transducer and activator of transcription (STAT) proteins, the phosphatidylinositol 3-kinase (PI3K), and the mitogen-activated protein kinase (MAPK) pathways promoting proliferation and survival of myeloid cells [29,30]. JAK2V617F is the most frequent MPN driver mutation detected in 95% of PV and 50–60% of ET and PMF patients [1,31]. It is still not completely understood how JAK2V617F gives rise to the differential clinical phenotypes of PV, ET, and MF. JAK2V617F mutant allele frequency and type and sequence of additional mutations in hematopoietic stem/progenitor cell sub-clones seem to influence the specific disease manifestations [32,33,34,35]. JAK2 exon 12 mutations are identified in ~4% of PV patients and lead to a similar constitutive activation of JAK-STAT signaling. While two thirds of JAK2 exon 12 mutated patients present with isolated erythrocytosis in the absence of concomitant leuko- or thrombocytosis, the clinical course is analogous to JAK2V617F mutant PV [36].
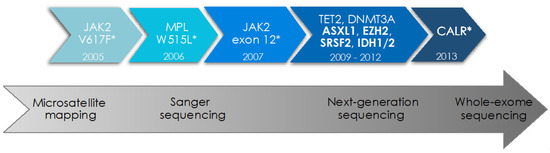
Figure 1.
Identification of driver mutations and additional somatic mutations in myeloproliferative neoplasms by modern sequencing technologies. The advent of modern sequencing technologies over the last two decades has led to the characterization of the genetic landscape in myeloproliferative neoplasms (MPN), including driver mutations (*) in JAK2, MPL, and CALR genes as well as concomitant somatic mutations in cancer genes frequently mutated in myeloid malignancies. Mutations in the genes highlighted in bold are considered of “high molecular risk” (HMR) given their adverse prognostic impact. The year of discovery of specific mutations is indicated as well as the development of sequencing methodologies over time.
Calreticulin (CALR) is a chaperone protein in the endoplasmic reticulum (ER) involved in calcium homeostasis and protein folding. Mutations of the calreticulin gene are detected in 30–40% of ET and PMF patients [10,11]. They were first identified in 2013 with a 52 bp deletion (type 1 mutation) or 5 bp insertion (type 2 mutation) in exon 9 of CALR being most prevalent, while more than 35 mutations were described overall. It has been shown that the various CALR mutations all result in one-base pair frameshift leading to a novel C-terminus of the protein. Mutant CALR induces aberrant activation of MPL by binding to the receptor both in the ER and on the cell surface [37,38,39]. Of note, CALR mutations associate with a favorable prognosis and a lower risk for leukemic transformation compared to JAK2V617F mutated MPN, which impacts on therapeutic management [40,41,42].
MPL gene mutations affecting the thrombopoietin receptor MPL are less frequent, and account for only 5–8% of ET and PMF patients. The most common mutation at position 515 is located in the juxtamembrane protein region and induces constitutive activation of MPL and, consecutively, JAK2 signaling [8]. Several missense mutations, including W515L and W515K, and, rarely, other mutations have also been reported and analogously induce MPL activation [9]. Thus, it has become clear that driver mutations in JAK2, CALR, and MPL converge on the activation of JAK2 signaling as a common feature [43].
2.2. Triple-Negative MPN
For 10–15% of PMF and ET patients, neither JAK2V617F nor a CALR or MPL driver mutation are identified. These patients, who are termed “triple-negative” driver mutations, may be found later in the clinical course or upon genetic testing with higher sensitivity. Non-canonical mutations in JAK2 (e.g., V625F, F556V, R683G, and E627A) or MPL (e.g., S505N, S204P, T119I, and Y591D/N) were found in some triple-negative patients and shown to result in constitutive activation of the JAK-STAT signaling [44,45,46]. Some of these mutations are germline rather than somatically acquired genetic alterations, therefore representing a familial, non-clonal erythrocytosis or thrombocytosis. In the remaining proportion of ET and PMF patients, a driver mutation cannot be identified, suggesting an unknown genetic alteration underlying MPN pathogenesis or that they might not actually have a malignancy.
2.3. Concomitant Gene Mutations in Myeloid Cancer Genes
The advent of modern sequencing technologies including next generation sequencing (NGS) has enabled a detailed characterization of the genetic/genomic landscape of MPN in recent years. These comprehensive investigations of genetic alterations underlying MPN have revealed that additional somatic mutations co-occur with the driver mutations in JAK2, CALR, and MPL genes in more than half of MPN patients [11,33,44]. They mostly affect genes commonly mutated in myeloid malignancies include acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). They primarily affect epigenetic regulators, factors of the mRNA splicing machinery, and signaling molecules, while a set of specific mutations is indicative of imminent leukemic transformation. The overall number of somatic mutations is higher in PMF as compared to PV and ET and has been shown to confer an adverse prognostic effect [11,33,44].
Epigenetic regulators. Mutations in epigenetic regulators include TET2 mutations in 10–15%, DNMT3A and ASXL1 mutations in 5–15%, EZH2 in 3–10%, and IDH1/2 mutations in 1–2% of MPN patients. TET2 mutations impair TET2 enzymatic function, which catalyzes the conversion of 5-methylcytosine (5-mc) to 5-hydroxymethylcytosine (5-hmc) and initiates DNA demethylation and upregulated transcription. It has been shown in murine models that TET2 mutations promote the expansion of the hematopoietic stem cell pool [47,48,49], but their prognostic significance regarding transformation and survival is unclear [42,43]. The order of appearance of TET2 and JAK2 mutations impacts on the clinical phenotype, with “JAK2 first” patients typically presenting with PV, while “TET2 first” patients would rather present with ET/PMF [32]. TET2 function is modulated by the conversion of isocitrate to alpha-ketoglutarate catalyzed by isocitrate dehydrogenases IDH1 and IDH2. IDH1/2 mutations interfere with this process and lead to the accumulation of the oncometabolite 2-hydroxyglutarate impeding TET2 function [50]. IDH1/2 mutations confer adverse prognostic effects in MPN and are enriched in patients progressing to secondary AML [51].
Analysis of global DNA methylation patterns in MPN patients identified differential methylation as compared to healthy controls highlighting the important role of perturbed DNA methylation in MPN [52]. DNA methyltransferases as DNMT3A mediate de novo DNA methylation and DNMT3A mutations were found to be prevalent across myeloid malignancies, primarily in AML and MPN [53]. The R882H mutation is most frequent and mediates DNMT3A loss of function leading to expansion of the hematopoietic stem cell pool and increased gene transcription and thrombocytosis in murine models [54,55]. DNMT3A mutations are considered early events in MPN evolution and, similarly to TET2 mutations, the order of acquisition affects the MPN phenotypes with a propensity for ET upon DNMT3A followed by JAK2 mutations and for PV upon acquisition of DNMT3A after JAK2 mutations [56].
Mutations in EZH2 and ASXL1 interfere with the function of the polycomb repressive complex 2 (PRC2), which mediates histone modification via histone H3 di- and tri-methylation at lysine 27 (H3K27me2/3). ASXL1 mutations frequently occur in PMF patients (10–35%) and are rather rare in ET or PV (2–5%). As in other myeloid malignancies, they confer an adverse prognosis with inferior survival [57,58,59]. EZH2, which represents the PRC2 catalytic component, is mostly mutated in MF patients (2–10%) and is associated with an unfavorable prognosis [60,61]. The JAK2V617F mutation also interferes with epigenetic processes, in addition to mediating constitutive activation of hematopoietic cytokine signaling. It has been shown that both wild-type and mutant JAK2 translocate to the nucleus and phosphorylate arginine methyltransferase PRMT5, which highlights the importance of histone methylation changes in MPN [62,63,64].
Splicing factors. Mutations in RNA splicing factor genes, including SRSF2, SF3B1, U2AF1, and ZRSR2, which are characteristic of myelodysplastic syndromes, are also prevalent in PMF (5–15%), but rare in ET and PV (1–5%). They lead to perturbation of mRNA splicing [65]. For SRSF2 mutations, an unfavorable prognosis with lower overall survival has been observed. Genetic alterations in SRSF2 are enriched in secondary AML, similarly to IDH1/2 and TP53 mutations [66].
3. Genetic Testing in Clinical Settings in MPN
Modern sequencing efforts over the last decade have revealed the heterogeneous genetic landscape of MPN in large parts. These discoveries into the genetic basis of MPN have led to a detailed insight into disease development, clonal evolution, and progression of MPN and have revealed specific molecular features of clinical sub-entities such as PV, ET, and MF. This genetic knowledge is increasingly integrated into clinical practice including the implementation into standard diagnostic criteria and modern prognostication schemes (Table 1, Figure 2) [19,67].

Table 1.
Risk stratification schemes for myelofibrosis involving clinical and genetic factors.
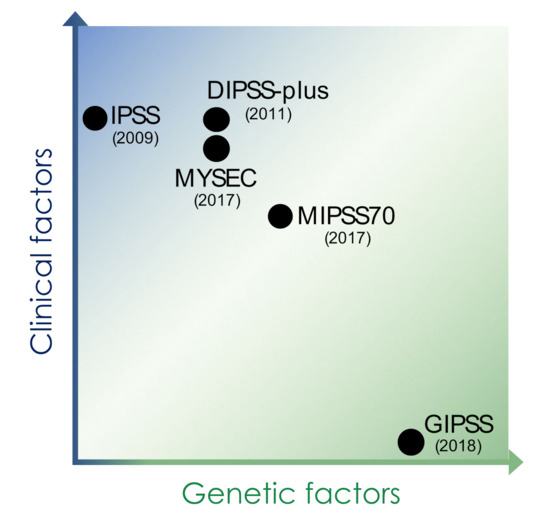
Figure 2.
Prognostic scoring systems for myelofibrosis. Early prognostication schemes for myelofibrosis relied on clinical factors including age, hematologic parameters including cytopenias and blast count, and symptoms, while more recent scores have increasingly implemented genetic factors such as information on cytogenetic features, driver mutations, and concomitant somatic mutations of prognostic impact. IPSS: international prognostic scoring system, DIPSS: dynamic international prognostic system, MYSEC: myelofibrosis secondary to PV and ET-prognostic model, MIPPS70: mutation-enhanced international prognostic score system (validated up to 70 years of age), GIPSS: genetically inspired prognostic scoring system.
3.1. Genetic Testing for MPN Diagnosis
Based on the extensive genetic characterization of MPN over the last years, driver mutations can be detected by routine genetic testing in 98% of PV patients and 85–90% of ET and PMF patients. The presence of a JAK2, CALR, or MPL driver mutation is not specific, but highly suggestive of an MPN and thus represents a valuable tool in support of diagnosing PV, ET, or PMF [68]. Accordingly, JAK2, CALR, or MPL driver mutations have been implemented as a major criterion into the standard diagnostic criteria by the World Health Organization (WHO) for PV, ET, and PMF [15]. The ease and high reproducibility of genetic testing has supported this process, while other, more laborious and delicate assays such as the evaluation of endogenous colony formation have been largely abandoned [15]. The extensive availability of next generation sequencing platforms at tertiary care centers has further supported the testing for additional gene mutations prevalent in MPN. This is particularly helpful to demonstrate clonality of hematopoiesis in triple negative MPN patients, and such testing is recommended by the WHO 2016 guidelines for ASXL1, EZH2, IDH1/2, SRSF2, TET2, and SF3B1 in the absence of the classical driver mutations [19]. Despite the fulminant development of genetic/genomic tools for routine diagnostics, conventional cyto- and histomorphologic assessment remains valuable given the long-standing experience with these readouts and their availability in less-equipped settings.
3.2. Genetic Testing for Prognostication and Treatment Decisions
Correlative studies have revealed associations of specific gene mutations with clinical presentation, progression dynamics and outcome, and this knowledge has been increasingly utilized for prognostication of individual patients’ courses. It has become clear that CALR mutations are associated with lower risk courses in terms of thrombosis and leukemic transformation, with the longest overall survival in CALR mutated and ASXL1 unmutated patients [69,70]. In contrast, it has consistently been shown that the presence of the so-called “high molecular risk” (HMR) mutations affecting ASXL1, EZH2, IDH1/2 and SRSF2 relates to adverse prognosis [12,66].
While prognostic modeling for PMF relied on the international prognostic scoring system (IPSS) in 2009 [71], which is solely based on age and hematological parameters for risk stratification, more recent schemas have implemented molecular factors to refine prognostication (Table 1, Figure 2). Thus, the dynamic IPSS-plus score (DIPSSplus) involves the patient’s cytogenetic features, while the MYSEC score specifically validated for post-ET and post-PV MF considers the presence or absence of CALR mutations [13,72]. The mutation enhanced IPSS-70 (MIPSS70), which was specifically developed for assisting the decision making for allogeneic stem cell transplantation in MF patients up to 70 years of age, involves CALR mutation status as well as the presence and number of HMR mutations including ASXL1, EZH2, IDH1/2, and SRSF2 mutations [20]. It was further refined by adding cytogenetic information including unfavorable and very high risk karyotypes as well as U2AF1 mutations in the MIPSS70-plus 2.0 [73]. The genetically inspired IPSS (GIPSS) so far represents the only prognostication scheme purely relying on genetic factors including driver mutations, HMR mutations, and cytogenetic information [65]. Given the significance of molecular factors for the prognosis of MF patients, genetic features have a growing impact also on therapeutic decisions. Currently, the European Leukemia Net (ELN) and the European Society on Blood and Marrow Transplantation (EBMT) advise to evaluate MF patients for allogeneic hematopoietic stem cell transplantation if their IPSS or DIPSS scores reach intermediate-2 or high risk or if they show intermediate-1 risk disease with molecular high risk features such as ASXL1 mutations or adverse cytogenetics [74].
For PV and ET patients, thrombotic and hemorrhagic events represent the major cause of morbidity and mortality, but fibrotic or leukemic transformation may also occur. Current prediction tools, such as the international prognostic score for thrombosis in ET (IPSET-thrombosis), focus on the risk for thrombo-hemorrhagic complications and rely on clinically derived variables including age, previous thrombosis, and presence of cardiovascular risk factors and the JAK2V617F mutation [75]. Although additional somatic mutations in myeloid cancer genes are less frequent in ET and PV than in MF, recent studies have shown that genetic information could also support outcome prediction in ET and PV. This has led to the development of MIPSS scores for ET and PV, which implement the information on spliceosome mutations, particularly SRSF2, SF3B1, and U2AF1 mutations, in addition to the established clinical factors age, leukocyte counts, and history of thrombosis [76].
Currently, further detailed prognostication tools are being developed, which aim for personalized predictions based on large scale genomic analysis [44] (https://cancer.sanger.ac.uk/mpn-multistage, accessed on 31 July 2021). These developments will hopefully soon reach clinical practice to support specifically tailored therapeutic approaches for individual patients.
4. Molecular Therapies in MPN
4.1. Treatment Recommendation
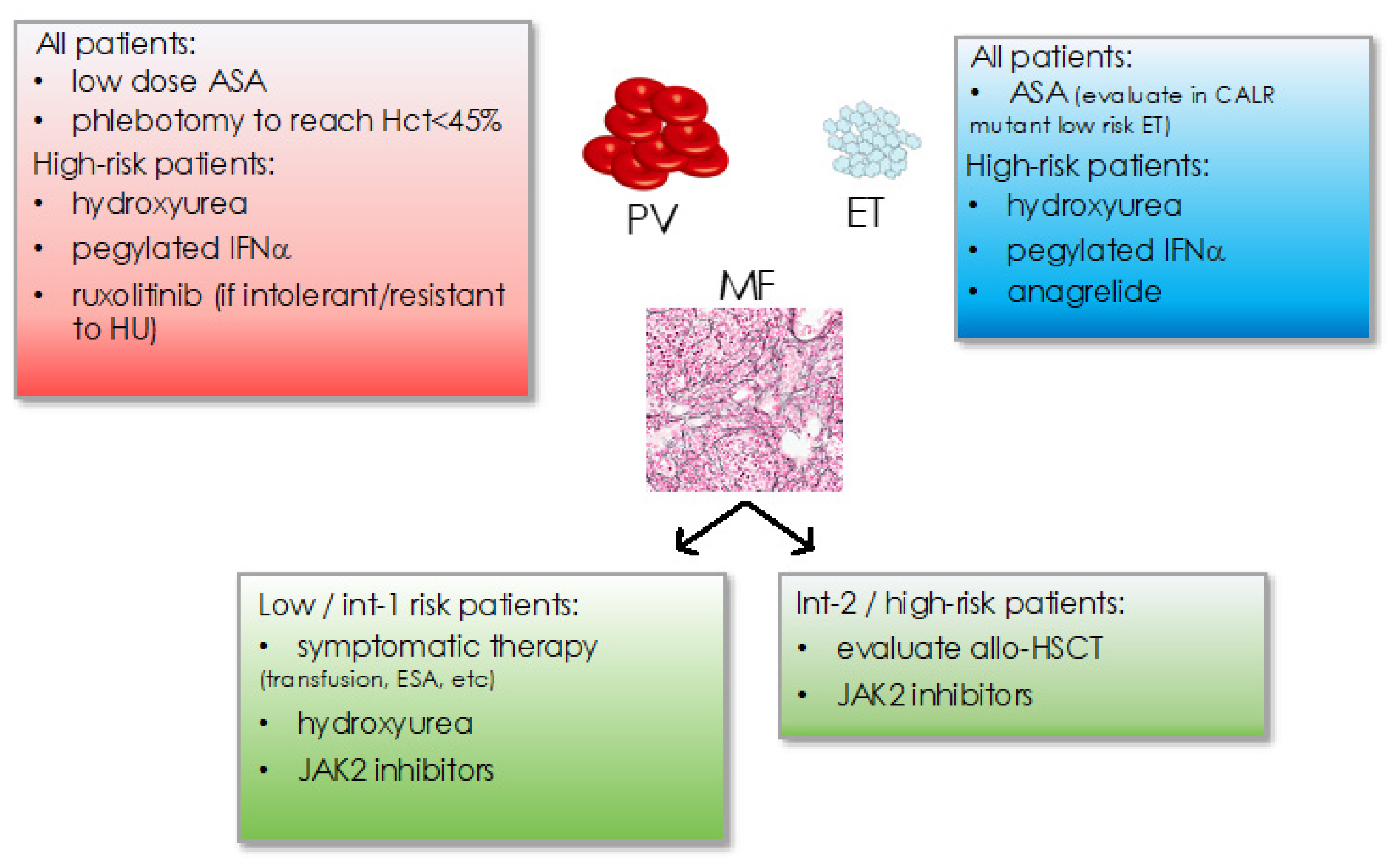
Classic treatment of MPN has traditionally focused on platelet inhibition as well as cytoreductive measures including phlebotomies to achieve hematocrit <45% in PV or hydroxyurea, anagrelide, or pegylated interferon-alpha (Figure 3) [19,77]. Cytoreductive treatment is recommended in patients aged >60 years or with a history of thrombosis or excessive thrombocytosis to counteract an increased risk for thrombo-hemorrhagic complications. Management of symptoms such as cytopenias (e.g., by transfusions, erythropoietin substitution, etc.) or splenomegaly is essential in myelofibrosis. Intermediate-2 to high risk myelofibrosis or the presence of high risk molecular features mandates the evaluation of allogeneic hematopoietic stem cell transplantation as the only curative treatment concept so far.
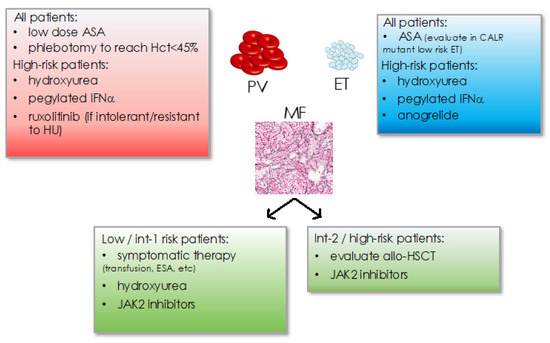
Figure 3.
Current treatment recommendations in MPN for PV, ET, and MF subtypes. Hct: hematocrit, ASA: acetyl-salicylic acid, IFNα: interferon-alpha, int: intemediate, ESA: erythropoiesis stimulating agents, allo-HSCT: allogeneic hematopoietic stem cell transplantation.
4.2. JAK2 Inhibitors
The increasing insight into the molecular pathogenesis of MPN has revealed rational targets for therapy. The discovery of the JAK2V617F oncogene and the finding that CALR and MPL mutations similarly activate JAK2 signaling has posed the JAK2 tyrosine kinase as an important therapeutic target [78]. The JAK1/2 inhibitor ruxolitinib approved in 2012 represents a current standard of care for the treatment of symptomatic intermediate to high risk MF and for PV patients intolerant or refractory to hydroxyurea [21,22,23]. Fedratinib, a JAK2/FLT3 inhibitor with analogous type 1 mode of JAK2 binding, has recently been approved for MF patients [24]. Neurologic toxicities, which became evident in the JAKARTA trials including Wernicke’s encephalopathy were judged as rare and manageable by preventive measures such as thiamine supplementation and monitoring [79]. Additional JAK2 inhibitors are currently in advanced clinical trials and include momelotinib and pacritinib. They show promising profiles for anemic and thrombocytopenic MF patients and are thus eagerly awaited [80,81,82]. JAK2 inhibitors such as ruxolitinib provide clinical benefits in terms of splenomegaly reduction and symptom control and have demonstrated a survival benefit [21,22,83]. However, their disease-modifying effects are modest, with a limited potential for mutant clone reduction and continuing clonal evolution [84,85]. Refined JAK2 inhibitors truly reducing the MPN clone and halting clonal progression are highly desirable. Investigative efforts are ongoing, as, e.g., for JAK inhibitors with a type II mode of binding similar to BCR-ABL inhibitors [86,87]. In addition, mutant calreticulin, which is exposed at the cell surface in association with MPL, could be addressed as an therapeutic target, directly relating to JAK2-STAT signaling in CALR mutant patients [38,88,89].
4.3. Resistance to JAK Inhibitors
The limited clinical efficacy of JAK2 inhibitors as ruxolitinib seen in treatment-naïve patients, represents an active area of research. It has been shown that the MAPK pathway involving MEK and ERK kinases, remains activated despite JAK2 inhibitor therapy and requires to be targeted to increase therapeutic efficacy [90,91]. Several mechanisms have been reported including compensatory PDGF-PDGFR signaling or nuclear translocation of the splicing factor YBX1 ultimately resulting in persistent activation of the MAPK pathway [90,92].
In addition, clinical responses to the treatment with the JAK2 inhibitor ruxolitinib are lost in a substantial proportion of patients, as reflected by loss of the spleen response in approximately half of the patients with initial therapeutic benefit over five years [83]. Secondary mutations inducing genetic resistance as seen in BCR-ABL driven malignancies exposed to BCR-ABL inhibitors occur upon exposure to JAK2 inhibitors in vitro, but have not been observed in MPN patients [93,94]. In contrast, adaptive changes of JAK2 signaling have been described, which are reversible. It has been shown that JAK2 is able to form heterodimers with JAK1 or TYK2 upon JAK2 inhibitor treatment, which leads to reactivation of JAK2 signaling and re-occurrence of symptoms [87,95]. Of note, MPN cells would re-sensitize to JAK inhibitor therapy upon cessation and re-exposure, a phenomenon which has also been observed in patients [96]. Additional mechanisms of resistance have also been described such as pro-survival signals emerging from hematopoietic and/or stromal cells of the bone marrow microenvironment. Aberrant pro-inflammatory cytokine signals from both JAK2 mutant and non-mutant bone marrow cells have also been implicated as mediators of resistance to JAK2 inhibition [97,98].
4.4. Novel Therapies in MPN
Given the limitations of the current targeted therapies with JAK2 inhibitors, there is an important need for improved therapeutic approaches with enhanced clinical efficacy. Targeting JAK2 currently remains a central aspect of MPN treatment given the essential role of aberrant JAK2 signaling in PV, ET, and MF. Dual treatment approaches combining JAK2 inhibitors with additional therapeutic principles are thus being explored as promising options. Clinical studies are currently evaluating approaches of JAK2 inhibition in combination with inhibition of MEK-ERK or PI3K signaling, interference with apoptosis regulation by Bcl-2/Bcl-xL inhibition such as navitoclax, targeting methylation changes by the hypomethylating agents azacytidine or decitabine, as well as by histone deacetylase inhibitors panobinostat or givinostat [90,99,100,101,102]. As epigenetic mechanisms enhance the inflammatory milieu in MPN via activation of NF-kB signaling, BET inhibition has gained important interest as a therapeutic approach in MPN. A phase 2 study with the BET inhibitor CPI-0610 as monotherapy or in combination with ruxolitinib is currently ongoing in MF patients [103,104]. Of note, immune modulatory effects by interferon alpha represent a longstanding treatment concept in MPN, which has recently gained substantial interest as novel pegylated forms of interferon alpha with improved tolerability profiles have become available [105]. Pegylated interferon alpha has evident potential to decrease MPN clone size and has been approved for the treatment of PV [105], while data in MF is accumulating [106,107]. Pegylated interferon alpha is also a promising approach in combination with JAK2 inhibition by ruxolitinib, as evidenced by recent/ongoing clinical studies leading to decreased mutant allele burden and lowered symptom burden [108,109].
Several additional innovative treatment approaches are under clinical investigation, such as telomerase inhibition interfering with telomere function, MDM2 inhibition impacting on TP53 tumor suppressor function, and others [110]. These efforts will hopefully soon add valid options to the molecularly targeted treatment approaches for MPN patients.
5. Perspectives
The advent of modern sequencing technologies has driven the progress in the molecular understanding of MPN pathogenesis. These insights into the genetic/genomic landscape of MPN has translated in recent years to the routine clinical work-up of MPN patients integrating molecular markers into the diagnostic criteria and providing molecularly enriched prognostication schemes as a basis for decisions on clinical management. It is a hopeful perspective that additional, novel omics technologies addressing, e.g., the methylome and/or spliceosome of MPN will also be available for molecular diagnostics of MPN in the future and will inform further advanced, molecular-based therapeutic approaches for MPN patients.
Author Contributions
Conceptualization and writing including original draft preparation, review and editing were by S.S. and S.C.M. Both authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge research support to S.C.M. by the Swiss National Science Foundation (PZ00P3_161145, PCEFP3_181357), the Swiss Cancer League/Swiss Cancer Research (KFS-3858-02-2016), the Swiss BRIDGE Foundation (PSB-4066-06-2016), the Cancer League Basel (KLbB-4784-02-2019), the Swiss Group for Clinical Cancer Research SAKK, the Foundation for the Fight against Cancer, the Nora van Meeuwen-Häfliger Foundation, the Foundation Peter-Anton and Anna-Katharina Miescher and the Swiss Society of Hematology.
Conflicts of Interest
S.C.M. has consulted for and received honoraria from Celgene/BMS and Novartis. S.S. has no conflicts of interest to disclose.
References
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Dameshek, W. Editorial: Some Speculations on the Myeloproliferative Syndromes. Blood 1951, 6, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired Mutation of the Tyrosine Kinase JAK2 in Human Myeloproliferative Disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A Unique Clonal JAK2 Mutation Leading to Constitutive Signalling Causes Polycythaemia Vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 Mutations in Myeloproliferative and Other Myeloid Disorders: A Study of 1182 Patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and Prognosis in Primary Myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A Clinical-Molecular Prognostic Model to Predict Survival in Patients with Post Polycythemia Vera and Post Essential Thrombocythemia Myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Godfrey, A.L.; Nangalia, J. Genomic Heterogeneity in Myeloproliferative Neoplasms and Applications to Clinical Practice. Blood Rev. 2020, 42, 100708. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, R.; Gómez-Casares, M.T. Recent Advances in the Use of Molecular Analyses to Inform the Diagnosis and Prognosis of Patients with Polycythaemia Vera. Int. J. Mol. Sci. 2021, 22, 5042. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Kralovics, R. Progress in Elucidation of Molecular Pathophysiology of Myeloproliferative Neoplasms and Its Application to Therapeutic Decisions. Int. J. Hematol. 2020, 111, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, K. Progress in Elucidation of Molecular Pathophysiology and Its Application in Therapeutic Decision-Making for Myeloproliferative Neoplasms. Int. J. Hematol. 2020, 111, 180–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.J.; Hehlmann, R.; et al. Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms: Revised Management Recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talpaz, M.; Kiladjian, J.-J. Fedratinib, a Newly Approved Treatment for Patients with Myeloproliferative Neoplasm-Associated Myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef]
- Schwaab, J.; Naumann, N.; Luebke, J.; Jawhar, M.; Somervaille, T.C.P.; Williams, M.S.; Frewin, R.; Jost, P.J.; Lichtenegger, F.S.; La Rosée, P.; et al. Response to Tyrosine Kinase Inhibitors in Myeloid Neoplasms Associated with PCM1-JAK2, BCR-JAK2 and ETV6-ABL1 Fusion Genes. Am. J. Hematol. 2020, 95, 824–833. [Google Scholar] [CrossRef]
- Lundberg, P.; Takizawa, H.; Kubovcakova, L.; Guo, G.; Hao-Shen, H.; Dirnhofer, S.; Orkin, S.H.; Manz, M.G.; Skoda, R.C. Myeloproliferative Neoplasms Can Be Initiated from a Single Hematopoietic Stem Cell Expressing JAK2-V617F. J. Exp. Med. 2014, 211, 2213–2230. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, H.; Cumano, A.; Müller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 Deficiency Defines an Essential Developmental Checkpoint in Definitive Hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.C.; Levine, R.L. Molecular Pathways: Molecular Basis for Sensitivity and Resistance to JAK Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F Expression Causes a Lethal Myeloproliferative Neoplasm with Differential Effects on Hematopoietic Stem and Progenitor Cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Bandaranayake, R.M.; Ungureanu, D.; Shan, Y.; Shaw, D.E.; Silvennoinen, O.; Hubbard, S.R. Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 2012, 19, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic Basis and Molecular Pathophysiology of Classical Myeloproliferative Neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal Evolution and Clinical Correlates of Somatic Mutations in Myeloproliferative Neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical Profile of Homozygous JAK2 617V>F Mutation in Patients with Polycythemia Vera or Essential Thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective Identification of High-Risk Polycythemia Vera Patients Based on JAK2V617F Allele Burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.-J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and Clinical Features of the Myeloproliferative Neoplasm Associated with JAK2 Exon 12 Mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [Green Version]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin Receptor Activation by Myeloproliferative Neoplasm Associated Calreticulin Mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-Terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Marty, C.; Pecquet, C.; Nivarthi, H.; El-Khoury, M.; Chachoua, I.; Tulliez, M.; Villeval, J.-L.; Raslova, H.; Kralovics, R.; Constantinescu, S.N.; et al. Calreticulin Mutants in Mice Induce an MPL-Dependent Thrombocytosis with Frequent Progression to Myelofibrosis. Blood 2016, 127, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR Mutation Status Defines Subtypes of Essential Thrombocythemia with Substantially Different Clinical Course and Outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-Mutated or Triple-Negative Myelofibrosis: Clinical, Cytogenetic and Molecular Comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Belachew, A.A.; Wassie, E.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A. Type 1 vs. Type 2 Calreticulin Mutations in Primary Myelofibrosis: Differences in Phenotype and Prognostic Impact. Leukemia 2014, 28, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated Genomic Analysis Illustrates the Central Role of JAK-STAT Pathway Activation in Myeloproliferative Neoplasm Pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-Exome Sequencing Identifies Novel MPL and JAK2 Mutations in Triple-Negative Myeloproliferative Neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of Atypical Thrombopoietin Receptor (MPL) Mutations in Triple-Negative Essential Thrombocythemia Patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation InTET2in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 Mutations and Their Clinical Correlates in Polycythemia Vera, Essential Thrombocythemia and Myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Jimma, T.; Sulai, N.H.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; McClure, R.F.; Pardanani, A. IDH Mutations in Primary Myelofibrosis Predict Leukemic Transformation and Shortened Survival: Clinical Evidence for Leukemogenic Collaboration with JAK2V617F. Leukemia 2012, 26, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Pérez, C.; Pascual, M.; Ignacio Martín-Subero, J.; Bellosillo, B.; Segura, V.; Delabesse, E.; Álvarez, S.; Larrayoz, M.J.; Rifón, J.; Cruz Cigudosa, J.; et al. Aberrant DNA Methylation Profile of Chronic and Transformed Classic Philadelphia-Negative Myeloproliferative Neoplasms. Haematologica 2013, 98, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Pardanani, A.; Rampal, R.; Lasho, T.L.; Levine, R.L.; Tefferi, A. DNMT3A Mutational Analysis in Primary Myelofibrosis, Chronic Myelomonocytic Leukemia and Advanced Phases of Myeloproliferative Neoplasms. Leukemia 2011, 25, 1219–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a Loss Cooperate to Induce Myelofibrosis through Activated Enhancer-Driven Inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A Mutations Occur Early or Late in Patients with Myeloproliferative Neoplasms and Mutation Order Influences Phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [Green Version]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.-J.; Birnbaum, D. Mutations in ASXL1 Are Associated with Poor Prognosis across the Spectrum of Malignant Myeloid Diseases. J. Hematol. Oncol. 2012, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Pardanani, A.; Patel, J.; Wadleigh, M.; Lasho, T.; Heguy, A.; Beran, M.; Gilliland, D.G.; Levine, R.L.; Tefferi, A. Concomitant Analysis of EZH2 and ASXL1 Mutations in Myelofibrosis, Chronic Myelomonocytic Leukemia and Blast-Phase Myeloproliferative Neoplasms. Leukemia 2011, 25, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrtue Nielsen, H.; Lykkegaard Andersen, C.; Westman, M.; Sommer Kristensen, L.; Asmar, F.; Arvid Kruse, T.; Thomassen, M.; Stauffer Larsen, T.; Skov, V.; Lotte Hansen, L.; et al. Epigenetic Changes in Myelofibrosis: Distinct Methylation Changes in the Myeloid Compartments and in Cases with ASXL1 Mutations. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 Mutational Status Predicts Poor Survival in Myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene-Regulatory Networks Leading to Therapeutic Efficacy in JAK2 V617F -Mutant MPN. Cancer Discov. 2020, 1742–1758. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D. JAK2 Phosphorylates Histone H3Y41 and Excludes HP1 α from Chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hautin, M.; Mornet, C.; Chauveau, A.; Bernard, D.; Corcos, L.; Lippert, E. Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues. Cancers 2020, 12, 2216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic Analysis of Patients with Leukemic Transformation of Myeloproliferative Neoplasms Shows Recurrent SRSF2 Mutations That Are Associated with Adverse Outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Pietra, D.; Pane, F.; Pancrazzi, A.; Cazzola, M.; Vannucchi, A.M.; Tura, S.; Barosi, G. Recommendations for Molecular Testing in Classical Ph1-Neg Myeloproliferative Disorders–A Consensus Project of the Italian Society of Hematology. Leuk. Res. 2017, 58, 63–72. [Google Scholar] [CrossRef]
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The Number of Prognostically Detrimental Mutations and Prognosis in Primary Myelofibrosis: An International Study of 797 Patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 Mutations-Based Molecular Prognostication in Primary Myelofibrosis: An International Study of 570 Patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New Prognostic Scoring System for Primary Myelofibrosis Based on a Study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information From Karyotype, Platelet Count, and Transfusion Status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically Inspired Prognostic Scoring System for Primary Myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Kröger, N.M.; Deeg, J.H.; Olavarria, E.; Niederwieser, D.; Bacigalupo, A.; Barbui, T.; Rambaldi, A.; Mesa, R.; Tefferi, A.; Griesshammer, M.; et al. Indication and Management of Allogeneic Stem Cell Transplantation in Primary Myelofibrosis: A Consensus Process by an EBMT/ELN International Working Group. Leukemia 2015, 29, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and Validation of an International Prognostic Score of Thrombosis in World Health Organization–Essential Thrombocythemia (IPSET-Thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-Enhanced International Prognostic Systems for Essential Thrombocythaemia and Polycythaemia Vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and Functional Analysis of Leukemic Transformation of Myeloproliferative Neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.-J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus Kinase-2 Inhibitor Fedratinib in Patients with Myelofibrosis Previously Treated with Ruxolitinib (JAKARTA-2): A Single-Arm, Open-Label, Non-Randomised, Phase 2, Multicentre Study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.; Egyed, M.; Szoke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus Best Available Therapy for the Treatment of Myelofibrosis Irrespective of Baseline Cytopenias (PERSIST-1): An International, Randomised, Phase 3 Trial. Lancet. Haematol. 2017, 4, e225–e236. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus Best Available Therapy in Patients with Myelofibrosis Previously Treated with Ruxolitinib (SIMPLIFY 2): A Randomised, Open-Label, Phase 3 Trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.-J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-Term Survival in Patients Treated with Ruxolitinib for Myelofibrosis: COMFORT-I and -II Pooled Analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal Evolution and Outcomes in Myelofibrosis after Ruxolitinib Discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-Cell Analysis Based Dissection of Clonality in Myelofibrosis. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Andraos, R.; Qian, Z.; Bonenfant, D.; Rubert, J.; Vangrevelinghe, E.; Scheufler, C.; Marque, F.; Régnier, C.H.; De Pover, A.; Ryckelynck, H.; et al. Modulation of Activation-Loop Phosphorylation by JAK Inhibitors Is Binding Mode Dependent. Cancer Discov. 2012, 2, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, S.C.; Keller, M.D.; Chiu, S.; Koppikar, P.; Guryanova, O.A.; Rapaport, F.; Xu, K.; Manova, K.; Pankov, D.; O’Reilly, R.J.; et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2015, 28, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; LaCava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.V.; et al. Targeting the CALR Interactome in Myeloproliferative Neoplasms. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Araki, M.; Imai, M.; Mori, Y.; Horino, M.; Ogata, S.; Yoshikawa, S.; Taguchi, T.; Masubuchi, N.; Mabuchi, Y.; et al. Therapeutic Potential of an Antibody Targeting the Cleaved Form of Mutant Calreticulin in Myeloproliferative Neoplasms. Blood 2020, 136, 9–10. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting Compensatory MEK/ERK Activation Increases JAK Inhibitor Efficacy in Myeloproliferative Neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef]
- Williams, D.A. Pairing JAK with MEK for Improved Therapeutic Efficiency in Myeloproliferative Disorders. J. Clin. Investig. 2019, 130, 1519–1521. [Google Scholar] [CrossRef] [Green Version]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing Factor YBX1 Mediates Persistence of JAK2-Mutated Neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Reddy, M.M.; Schade, G.O.M.; Ray, A.; Chowdary, T.K.; Griffin, J.D.; Sattler, M. Kinase Domain Mutations Confer Resistance to Novel Inhibitors Targeting JAK2V617F in Myeloproliferative Neoplasms. Leukemia 2012, 26, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Weigert, O.; Lane, A.A.; Bird, L.; Kopp, N.; Chapuy, B.; van Bodegom, D.; Toms, A.V.; Marubayashi, S.; Christie, A.L.; McKeown, M.; et al. Genetic Resistance to JAK2 Enzymatic Inhibitors Is Overcome by HSP90 Inhibition. J. Exp. Med. 2012, 209, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK-STAT Activation as a Mechanism of Persistence to JAK2 Inhibitor Therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Schalling, M.; Gisslinger, B.; Skrabs, C.; Müllauer, L.; Kralovics, R. Restoration of Response to Ruxolitinib upon Brief Withdrawal in Two Patients with Myelofibrosis. Am. J. Hematol. 2014, 89, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Manshouri, T.; Estrov, Z.; Quintás-Cardama, A.; Burger, J.; Zhang, Y.; Livun, A.; Knez, L.; Harris, D.; Creighton, C.J.; Kantarjian, H.M.; et al. Bone Marrow Stroma–Secreted Cytokines Protect JAK2V617F-Mutated Cells from the Effects of a JAK2 Inhibitor. Cancer Res. 2011, 71, 3831–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT Pathway Activation in Malignant and Nonmalignant Cells Contributes to MPN Pathogenesis and Therapeutic Response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartalucci, N.; Tozzi, L.; Bogani, C.; Martinelli, S.; Rotunno, G.; Villeval, J.-L.; Vannucchi, A.M. Co-Targeting the PI3K/MTOR and JAK2 Signalling Pathways Produces Synergistic Activity against Myeloproliferative Neoplasms. J. Cell. Mol. Med. 2013, 17, 1385–1396. [Google Scholar] [CrossRef]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.-K.; Banks, K.-M.; Vidacs, E.; Virely, C.; Sia, K.C.S.; Bracken, L.S.; et al. Combined Targeting of JAK2 and Bcl-2/Bcl-XL to Cure Mutant JAK2-Driven Malignancies and Overcome Acquired Resistance to JAK2 Inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, J.; Marcellino, B.K.; Lu, M.; Kremyanskaya, M.; Fabris, F.; Sandy, L.; Mehrotra, M.; Houldsworth, J.; Najfeld, V.; El Jamal, S.; et al. A Phase I Study of Panobinostat and Ruxolitinib in Patients with Primary Myelofibrosis (PMF) and Post- polycythemia Vera/Essential Thrombocythemia Myelofibrosis (Post- PV/ET MF). Leuk. Res. 2020, 88, 106272. [Google Scholar] [CrossRef]
- Masarova, L.; Verstovsek, S.; Hidalgo-Lopez, J.E.; Pemmaraju, N.; Bose, P.; Estrov, Z.; Jabbour, E.J.; Ravandi-Kashani, F.; Takahashi, K.; Cortes, J.E.; et al. A Phase 2 Study of Ruxolitinib in Combination with Azacitidine in Patients with Myelofibrosis. Blood 2018, 132, 1664–1674. [Google Scholar] [CrossRef]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2018, 33, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Kremyanskaya, M.; Hoffman, R.; Bose, P.; Talpaz, M.; Harrison, C.N.; Gupta, V.; Leber, B.; Sirhan, S.; Kabir, S.; et al. MANIFEST, a Phase 2 Study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), As Monotherapy or “Add-on” to Ruxolitinib, in Patients with Refractory or Intolerant Advanced Myelofibrosis. Blood 2019, 134, 670. [Google Scholar] [CrossRef]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon Alfa-2b versus Standard Therapy for Polycythaemia Vera (PROUD-PV and CONTINUATION-PV): A Randomised, Non-Inferiority, Phase 3 Trial and Its Extension Study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Yacoub, A.; Mascarenhas, J.; Kosiorek, H.; Prchal, J.T.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. Pegylated Interferon Alfa-2a for Polycythemia Vera or Essential Thrombocythemia Resistant or Intolerant to Hydroxyurea. Blood 2019, 134, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Ianotto, J.C.; Chauveau, A.; Boyer-Perrard, F.; Gyan, E.; Laribi, K.; Cony-Makhoul, P.; Demory, J.L.; de Renzis, B.; Dosquet, C.; Rey, J.; et al. Benefits and Pitfalls of Pegylated Interferon-A2a Therapy in Patients with Myeloproliferative Neoplasm-Associated Myelofibrosis: A French Intergroup of Myeloproliferative Neoplasms (FIM) Study. Haematologica 2018, 103, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, A.L.; Mikkelsen, S.U.; Knudsen, T.A.; Bjørn, M.E.; Andersen, C.L.; Bjerrum, O.W.; Brochmann, N.; Patel, D.A.; Gjerdrum, L.M.R.; El Fassi, D.; et al. Ruxolitinib and Interferon-A2 Combination Therapy for Patients with Polycythemia Vera or Myelofibrosis: A Phase II Study. Haematologica 2020, 105, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Kiladjian, J.-J.; Soret-Dulphy, J.; Resche-Rigon, M.; Boyer-Perrard, F.; Barraco, F.; Rolland-Neyret, V.; Capron, C.; Chevret, S.; Giraudier, S.; Cassinat, B. Ruxopeg, a Multi-Center Bayesian Phase 1/2 Adaptive Randomized Trial of the Combination of Ruxolitinib and Pegylated Interferon Alpha 2a in Patients with Myeloproliferative Neoplasm (MPN)-Associated Myelofibrosis. Blood 2018, 132, 581. [Google Scholar] [CrossRef]
- Brkic, S.; Meyer, S.C. Challenges and Perspectives for Therapeutic Targeting of Myeloproliferative Neoplasms. HemaSphere 2021, 5, e516. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).