Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials

 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Biological Rationale of Adoptive Cell Transfer in Hepatocellular Carcinoma
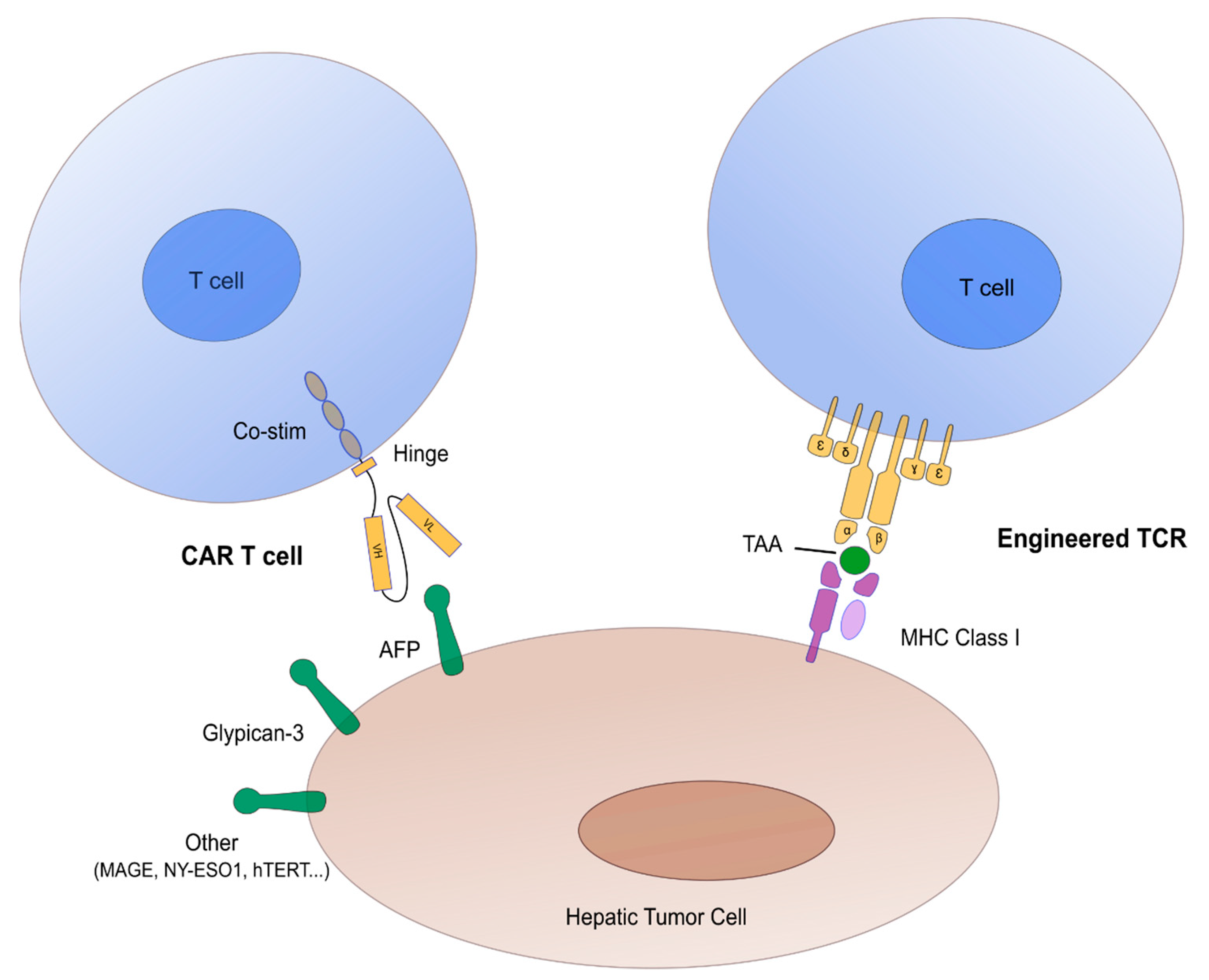
2.1. Concept of CAR/TCR Engineered T Cells
2.2. Targets in Hepatocellular Carcinoma Adoptive Cell Transfer
3. Results of Adoptive Cell Transfer Trials in Hepatocellular Carcinoma
3.1. TCR Engineered T Cells
3.2. CAR-T Cells
4. Ongoing Adoptive Cell Transfer Trials in Hepatocellular Carcinoma
4.1. TCR Engineered T Cells
4.2. CAR-T Cells
4.3. Immune Monitoring of ACT
5. Main Obstacles and Possible Solutions
5.1. A Specific Tumor Micro-Environment (TME)
5.2. Antigen Heterogeneity
5.3. Limited Expansion and Persistence
5.4. Off-Target Toxicity
5.5. Clinical Applicability
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | Adoptive cell transfer |
| AFP | Alpha-fetoprotein |
| CAR | Chimeric antigen receptor |
| CIK | Cytokine-induced killer |
| CTL | Cytotoxic T lymphocyte |
| GPC3 | Glypican-3 |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| IEC | Immune effector cells |
| IFN | Interferon |
| LAK | Lymphokine-activated killer |
| MAGE | Melanoma antigen gene |
| NK | Natural killer |
| TAA | Tumor-associated antigen |
| TCR | T cell receptor |
| TIL | Tumor-infiltrating lymphocyte |
References
- Brar, G.; Greten, T.F.; Graubard, B.I.; McNeel, T.S.; Petrick, J.L.; McGlynn, K.A.; Altekruse, S.F. Hepatocellular Carcinoma Survival by Etiology: A SEER-Medicare Database Analysis. Hepatol. Commun. 2020, 4, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- WHO. Cancer Fact Sheet. In Globocan; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int. J. Cancer 2018, 142, 2471–2477. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; Regalia, E.; Doci, R.; Andreola, S.; Pulvirenti, A.; Bozzetti, F.; Montalto, F.; Ammatuna, M.; Morabito, A.; Gennari, L. Liver Transplantation for the Treatment of Small Hepatocellular Carcinomas in Patients with Cirrhosis. N. Engl. J. Med. 1996, 334, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Maggs, J.R.L.; Suddle, A.R.; Aluvihare, V.; Heneghan, M.A. Systematic review: The role of liver transplantation in the management of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2012, 35, 1113–1134. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up††FootnotesApproved by the ESMO Guidelines Committee: August 2018. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Burroughs, A.; Hochhauser, D.; Meyer, T. Systemic treatment and liver transplantation for hepatocellular carcinoma: Two ends of the therapeutic spectrum. Lancet Oncol. 2004, 5, 409–418. [Google Scholar] [CrossRef]
- Lai, C.-L.; Lok, A.S.-F.; Wu, P.-C.; Chan, G.C.-B.; Lin, H.-J. Doxorubicin versus No Antitumor Therapy in Inoperable Hepatocellular Carcinoma. A Prospective Randomized Trial. Cancer 1988, 62, 479–483. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Lond. Engl. 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Lim, H.Y.; Pracht, M.; et al. REACH-2: A randomized, double-blind, placebo-controlled phase 3 study of ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma (HCC) and elevated baseline alpha-fetoprotein (AFP) following first-line sorafenib. J. Clin. Oncol. 2018, 36, 4003. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Greten, T.F.; Sangro, B. Targets for immunotherapy of liver cancer. J. Hepatol. 2018, 68, 157–166. [Google Scholar] [CrossRef]
- Iñarrairaegui, M.; Melero, I.; Sangro, B. Immunotherapy of Hepatocellular Carcinoma: Facts and Hopes. Clin. Cancer Res. 2018, 24, 1518–1524. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Rochigneux, P.; Nault, J.-C.; Mallet, F.; Chretien, A.-S.; Barget, N.; Garcia, A.J.; Pozo, L.D.; Bourcier, V.; Blaise, L.; Grando-Lemaire, V.; et al. Dynamic of systemic immunity and its impact on tumour recurrence after radiofrequency ablation of hepatocellular carcinoma. OncoImmunology 2019, 8, 1615818. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The Immunology of Hepatocellular Carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Calmels, B.; Mfarrej, B.; Chabannon, C. From clinical proof-of-concept to commercialization of CAR-T cells. Drug Discov. Today 2018, 23, 758–762. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumour activity and long-term fate of chimeric antigen receptor–positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Comoli, P.; Chabannon, C.; Koehl, U.; Lanza, F.; Urbano-Ispizua, A.; Hudecek, M.; Ruggeri, A.; Secondino, S.; Bonini, C.; Pedrazzoli, P. Development of adaptive immune effector therapies in solid tumors. Ann. Oncol. 2019, 30, 1740–1750. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR-T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR-T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR-T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR-T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Singh, I.; Zudaire, E.; Yeh, T.-M.; Allred, A.J.; Olyslager, Y.; Banerjee, A.; Goldberg, J.D.; et al. Update of CARTITUDE-1: A phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2020, 38, 8505. [Google Scholar] [CrossRef]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.-F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C.; et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. 2020, 38, 3794–3804. [Google Scholar] [CrossRef]
- Bôle-Richard, E.; Fredon, M.; Biichlé, S.; Anna, F.; Certoux, J.-M.; Renosi, F.; Tsé, F.; Molimard, C.; Valmary-Degano, S.; Jenvrin, A.; et al. CD28/4-1BB CD123 CAR-T cells in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2020, 34, 3228–3241. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Yáñez, L.; Alarcón, A.; Sánchez-Escamilla, M.; Perales, M.-A. How I treat adverse effects of CAR-T cell therapy. ESMO Open 2020, 4, e000746. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR-T cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Holzinger, A.; Abken, H. CAR-T Cells: A Snapshot on the Growing Options to Design a CAR. HemaSphere 2019, 3, e172. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. Off-the-shelf’ allogeneic CAR-T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Pasquini, M.C.; Hu, Z.-H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef]
- Batlevi, C.L.; Matsuki, E.; Brentjens, R.J.; Younes, A. Novel immunotherapies in lymphoid malignancies. Nat. Rev. Clin. Oncol. 2016, 13, 25–40. [Google Scholar] [CrossRef]
- Wada, Y.; Nakashima, O.; Kutami, R.; Yamamoto, O.; Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 1998, 27, 407–414. [Google Scholar] [CrossRef]
- Unitt, E.; Marshall, A.; Gelson, W.; Rushbrook, S.M.; Davies, S.; Vowler, S.L.; Morris, L.S.; Coleman, N.; Alexander, G.J.M. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J. Hepatol. 2006, 45, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, M.; Bellusci, R.; Cucchetti, A.; Cavrini, G.; De Ruvo, N.; Aden, A.A.; La Barba, G.; Brillanti, S.; Cavallari, A. Low recurrence rate of hepatocellular carcinoma after liver transplantation: Better patient selection or lower immunosuppression? Transplantation 2002, 74, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative Tumour-Specific Antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Bergstrand, C.G.; Csar, B. Demonstration of a New Protein Fraction in Serum from the Human Fetus: Scandinavian Journal of Clinical and Laboratory Investigation: Vol 8, No 2. Available online: https://www.tandfonline.com/doi/abs/10.3109/00365515609049266 (accessed on 26 October 2020).
- He, Y.; Lu, H.; Zhang, L. Chapter Ten—Serum AFP levels in patients suffering from 47 different types of cancers and noncancer diseases. In Progress in Molecular Biology and Translational Science; Glycans and Glycosaminoglycans as Clinical Biomarkers and Therapeutics—Part A; Zhang, L., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 162, pp. 199–212. [Google Scholar]
- Sideras, K.; Bots, S.J.; Biermann, K.; Sprengers, D.; Polak, W.G.; IJzermans, J.N.M.; de Man, R.A.; Pan, Q.; Sleijfer, S.; Bruno, M.J.; et al. Tumour antigen expression in hepatocellular carcinoma in a low-endemic western area. Br. J. Cancer 2015, 112, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Docta, R.Y.; Ferronha, T.; Sanderson, J.P.; Weissensteiner, T.; Pope, G.R.; Bennett, A.D.; Pumphrey, N.J.; Ferjentsik, Z.; Quinn, L.L.; Wiedermann, G.E.; et al. Tuning T-Cell Receptor Affinity to Optimize Clinical Risk-Benefit When Targeting Alpha-Fetoprotein–Positive Liver Cancer. Hepatology 2019, 69, 2061–2075. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Peng, Y.; Wang, L.; Hong, Y.; Jiang, X.; Li, Q.; Liu, H.; Huang, L.; Wu, J.; Celis, E.; et al. Identification of α-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 2018, 68, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Gold, P.J.; El-Khoueiry, A.B.; Abrams, T.A.; Morikawa, H.; Ohishi, N.; Ohtomo, T.; Philip, P.A. First-in-Man Phase I Study of GC33, a Novel Recombinant Humanized Antibody Against Glypican-3, in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2013, 19, 920–928. [Google Scholar] [CrossRef]
- Hanaoka, H.; Nagaya, T.; Sato, K.; Nakamura, Y.; Watanabe, R.; Harada, T.; Gao, W.; Feng, M.; Phung, Y.; Kim, I.; et al. Glypican-3 targeted human heavy chain antibody as a drug carrier for hepatocellular carcinoma therapy. Mol. Pharm. 2015, 12, 2151–2157. [Google Scholar] [CrossRef]
- Shimizu, Y.; Suzuki, T.; Yoshikawa, T.; Endo, I.; Nakatsura, T. Next-Generation Cancer Immunotherapy Targeting Glypican-3. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR-T Cells Suppress the Growth of Tumour Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, L.; Rathi, P.; Marinova, E.; Gao, X.; Wu, M.-F.; Liu, H.; Dotti, G.; Gottschalk, S.; Metelitsa, L.S.; et al. Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumour Activity. Hum. Gene Ther. 2016, 28, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, A.; Croft, P.K.; Vargas, A.C.; Smart, C.E.; Simpson, P.T.; Saunus, J.M.; Lakhani, S.R. Expression of MAGE-A and NY-ESO-1 cancer/testis antigens is enriched in triple-negative invasive breast cancers. Histopathology 2018, 73, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, T.; Matsumine, A.; Kageyama, S.; Asanuma, K.; Matsubara, T.; Nakamura, T.; Iino, T.; Ikeda, H.; Shiku, H.; Sudo, A. Immunohistochemical expression and clinicopathological assessment of the cancer testis antigens NY-ESO-1 and MAGE-A4 in high-grade soft-tissue sarcoma. Oncol. Lett. 2019, 17, 3937–3943. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, Y.; Gong, J.; Rao, L.; Wu, Z.; Nie, T.; Shi, D.; Zhang, L. High expression of MAGE-A9 contributes to stemness and malignancy of human hepatocellular carcinoma. Int. J. Oncol. 2018, 52, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gong, J.; Xiao, C.; Zhu, S.; Hu, Z.; Liang, J.; Li, X.; Yan, X.; Zhang, X.; Li, D.; et al. A comprehensive analysis of the MAGE family as prognostic and diagnostic markers for hepatocellular carcinoma. Genomics 2020, 112, 5101–5114. [Google Scholar] [CrossRef]
- Tahara, K.; Mori, M.; Sadanaga, N.; Sakamoto, Y.; Kitano, S.; Makuuchi, M. Expression of the MAGE gene family in human hepatocellular carcinoma. Cancer 1999, 85, 1234–1240. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Higashi, T.; Nouso, K.; Nakatsukasa, H.; Ishizaki, M.; Kaneyoshi, T.; Toshikuni, N.; Kariyama, K.; Nakayama, E.; Tsuji, T. Expression of MAGE, GAGE and BAGE genes in human liver diseases: Utility as molecular markers for hepatocellular carcinoma. J. Hepatol. 2000, 32, 612–617. [Google Scholar] [CrossRef]
- Marchand, M.; van Baren, N.; Weynants, P.; Brichard, V.; Dréno, B.; Tessier, M.-H.; Rankin, E.; Parmiani, G.; Arienti, F.; Humblet, Y.; et al. Tumour regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int. J. Cancer 1999, 80, 219–230. [Google Scholar] [CrossRef]
- Schooten, E.; Di Maggio, A.; van Bergen en Henegouwen, P.M.P.; Kijanka, M.M. MAGE-A antigens as targets for cancer immunotherapy. Cancer Treat. Rev. 2018, 67, 54–62. [Google Scholar] [CrossRef]
- Wang, H.; Chen, D.; Wang, R.; Quan, W.; Xia, D.; Mei, J.; Xu, J.; Liu, C. NY-ESO-1 expression in solid tumors predicts prognosis: A systematic review and meta-analysis. Medicine 2019, 98, e17990. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nouso, K.; Noguchi, Y.; Higashi, T.; Ono, T.; Jungbluth, A.; Chen, Y.-T.; Old, L.J.; Nakayama, E.; Shiratori, Y. Expression and immunogenicity of NY-ESO-1 in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2006, 21, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Maeda, Y.; Kanbe, T.; Yazama, H.; Takeda, Y.; Sato, R.; Tsukamoto, T.; Sato, E.; Marumoto, A.; Harada, T.; et al. Serum Human Telomerase Reverse Transcriptase Messenger RNA as a Novel Tumour Marker for Hepatocellular Carcinoma. Clin. Cancer Res. 2005, 11, 3205–3209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mizukoshi, E.; Nakamoto, Y.; Marukawa, Y.; Arai, K.; Yamashita, T.; Tsuji, H.; Kuzushima, K.; Takiguchi, M.; Kaneko, S. Cytotoxic T cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology 2006, 43, 1284–1294. [Google Scholar] [CrossRef]
- Sun, B.; Yang, D.; Dai, H.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Gires, O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer 2007, 96, 417–423. [Google Scholar] [CrossRef]
- Marhaba, R.; Klingbeil, P.; Nuebel, T.; Nazarenko, I.; Buechler, M.W.; Zoeller, M. CD44 and EpCAM: Cancer-initiating cell markers. Curr. Mol. Med. 2008, 8, 784–804. [Google Scholar] [CrossRef]
- Munz, M.; Baeuerle, P.A.; Gires, O. The Emerging Role of EpCAM in Cancer and Stem Cell Signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.-X.; et al. EpCAM and α-Fetoprotein Expression Defines Novel Prognostic Subtypes of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef]
- Yang, Y.; McCloskey, J.E.; Yang, H.; Puc, J.; Gallegos, A.A.G.; Vedvyas, Y.; Min, I.M.; von Hofe, E.; Jin, M.M. Abstract 6598: Eradication of EpCAM expressing solid tumors by low-affinity CAR-T cells. Cancer Res. 2020, 80, 6598. [Google Scholar] [CrossRef]
- Kotera, Y.; Fontenot, J.D.; Pecher, G.; Metzgar, R.S.; Finn, O.J. Humoral Immunity against a Tandem Repeat Epitope of Human Mucin MUC-1 in Sera from Breast, Pancreatic, and Colon Cancer Patients. Cancer Res. 1994, 54, 2856–2860. [Google Scholar] [PubMed]
- Finn, O.J.; Jerome, K.R.; Henderson, R.A.; Pecher, G.; Domenech, N.; Magarian-Blander, J.; Barratt-Boyes, S.M. MUC-1 epithelial tumour mucin-based immunity and cancer vaccines. Immunol. Rev. 1995, 145, 61–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR-T Cells Targeting the Tumour MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, K.; Lam, A.K.-Y.; Huang, J.; Qiu, F.; Qiao, B.; Zhang, Y. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med. 2020, 9, 640–652. [Google Scholar] [CrossRef]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T Cells Redirected Against Hepatitis B Virus Surface Proteins Eliminate Infected Hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Krebs, K.; Böttinger, N.; Huang, L.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T Cells Expressing a Chimeric Antigen Receptor That Binds Hepatitis B Virus Envelope Proteins Control Virus Replication in Mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef]
- Koh, S.; Shimasaki, N.; Suwanarusk, R.; Ho, Z.Z.; Chia, A.; Banu, N.; Howland, S.W.; Ong, A.S.M.; Gehring, A.J.; Stauss, H.; et al. A practical approach to immunotherapy of hepatocellular carcinoma using T cells redirected against hepatitis B virus. Mol. Ther. Nucleic Acids 2013, 2, e114. [Google Scholar] [CrossRef] [PubMed]
- Spear, T.T.; Callender, G.G.; Roszkowski, J.J.; Moxley, K.M.; Simms, P.E.; Foley, K.C.; Murray, D.C.; Scurti, G.M.; Li, M.; Thomas, J.T.; et al. TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer Immunol. Immunother. 2016, 65, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Brunetto, M.; Gehring, A.J.; Xue, S.-A.; Schurich, A.; Khakpoor, A.; Zhan, H.; Ciccorossi, P.; Gilmour, K.; Cavallone, D.; et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015, 62, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B. Contents. J. Hepatol. 2020, 73, vii–ix. [Google Scholar] [CrossRef]
- De Azevedo, J.T.C.; Mizukami, A.; Moço, P.D.; Malmegrim, K.C.R. Immunophenotypic Analysis of CAR-T Cells. In Chimeric Antigen Receptor T Cells: Development and Production; Swiech, K., Malmegrim, K.C.R., Picanço-Castro, V., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; pp. 195–201. ISBN 978-1-07-160146-4. [Google Scholar]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric Antigen Receptor-Modified T Cells Derived from Defined CD8 + and CD4 + Subsets Confer Superior Antitumor Reactivity in Vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Li, X.; Guo, X.; Zhu, Y.; Wei, G.; Zhang, Y.; Li, X.; Xu, H.; Cui, J.; Wu, W.; He, J.; et al. Single-Cell Transcriptomic Analysis Reveals BCMA CAR-T Cell Dynamics in a Patient with Refractory Primary Plasma Cell Leukemia. Mol. Ther. 2020, 0. [Google Scholar] [CrossRef] [PubMed]
- Sheih, A.; Voillet, V.; Hanafi, L.-A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal Kinetics and Single-Cell Transcriptional Profiling of CAR-T Cells in Patients Undergoing CD19 CAR-T Immunotherapy. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Xhangolli, I.; Dura, B.; Lee, G.; Kim, D.; Xiao, Y.; Fan, R. Single-Cell Analysis of CAR-T Cell Activation Reveals A Mixed TH1/TH2 Response Independent of Differentiation. Genom. Proteom. Bioinform. 2019, 17, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Barsan, V.; Mackall, C. Prospects and challenges for use of CAR-T cell therapies in solid tumors. Expert Opin. Biol. Ther. 2020, 20, 503–516. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR-T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Vallath, S.; Maher, J. The integrin αvβ6: A novel target for CAR-T-cell immunotherapy? Biochem. Soc. Trans. 2016, 44, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumour Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumour Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR-T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumour Microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumour Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR-T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; Moon, E.; Albelda, S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther. Oncolytics 2016, 3, 16006. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumour heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR-T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Prospects for combined use of oncolytic viruses and CAR-T-cells. J. Immunother. Cancer 2017, 5, 90. [Google Scholar] [CrossRef]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumour durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR-T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR-T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3–Specific CAR-T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumour Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Dolnikov, A.; Yang, S.; Shen, S.; Xu, N.; Chaudhry, K.; Nordon, R.; O’Brien, T. Prolonging CART Cell Persistence Using Conditioning with 5-Azacytidine. Cytotherapy 2016, 18, S98. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of Metastatic Renal Cell Carcinoma with CAIX CAR-engineered T cells: Clinical Evaluation and Management of On-target Toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.-M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR-T cells to ICAM-1 achieves rapid tumour elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR-T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumour Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wen, J.; Yi, H.; Hou, X.; Yin, Y.; Ye, G.; Wu, X.; Jiang, X. Split chimeric antigen receptor-modified T cells targeting glypican-3 suppress hepatocellular carcinoma growth with reduced cytokine release. Ther. Adv. Med. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| NCCT | Phase | Type | Product | Target | Organ | Pop | Sponsor | Primary Endpoint | Start Date |
|---|---|---|---|---|---|---|---|---|---|
| NCT02719782 | I | TCRe | HBV-TCR T cell | HBV Ag | HCC HBV+ | 10 | Lion Company | Safety | 06/2015 |
| NCT03132792 | I | TCRe | AFPᶜ332 T cells | AFP | basket | 45 | Adaptimmune | Safety | 05/2017 |
| NCT03441100 | I | TCRe | IMA202-101 | MAGEA1 | basket | 15 | Immatics | Safety | 05/2019 |
| NCT03899415 | I | TCRe | HBV-TCR T cell | HBV Ag | HCC HBV+ | 10 | Beijing Hospit. | Safety | 06/2019 |
| NCT04368182 | I | TCRe | Autologous C-TCR055 | AFP | HCC | 5 | Zhejiang Univ. | Safety | 06/2020 |
| NCCT | Phase | Type | Product | Target | Organ | Pop | Sponsor | Primary Endpoint | Start Date |
|---|---|---|---|---|---|---|---|---|---|
| NCT03302403 | NA | CAR-T | CAR-CLD18 | CLD18 | HCC/basket | 48 | Kang YU | Safety | 11/2017 |
| NCT03198546 | I | CAR-T | GPC3/TGFβ-CART | GPC3/TGFβ | HCC | 30 | Guangzhou Medical Univ | Safety | 07/2017 |
| NCT03013712 | I-II | CAR-T | EpCAM-CAR-T | EpCAM | basket | 60 | Chengdu Medical Coll | Safety | 01/2018 |
| NCT02905188 | I | CAR-T | GLYCAR | GP3C | HCC | 14 | Baylor Coll of Medicine | Safety | 03/2019 |
| NCT03884751 | I | CAR-T | CAR-GPC3 T Cells | GPC3 | HCC | 15 | Carsgen Therapeutics | Safety | 04/2019 |
| NCT03993743 | I (IAH) | CAR-T | CD147-CART | CD147 | HCC | 34 | Xijing Hospital | Safety | 05/2019 |
| NCT03980288 | I | CAR-T | CAR-GPC3 T Cells | GPC3 | HCC | 36 | Zhejiang Univ | Safety | 07/2019 |
| NCT03638206 | I-II | CAR-T | CAR-T/TCR-T cell | DR5, C-met, EGFR vIII | basket | 73 | Shenzhen BinDeBio | Safety | 08/2020 |
| NCT03941626 | I-II | CAR-T | CAR-T/TCR-T cells | DR5, EGFR vIII | HCC/basket | 50 | Shenzhen BinDeBio | Safety | 09/2019 |
| NCT04121273 | I | CAR-T | NA | GPC3 | HCC | 20 | Nanjing Univ | Safety | 10/2019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochigneux, P.; Chanez, B.; De Rauglaudre, B.; Mitry, E.; Chabannon, C.; Gilabert, M. Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials. Cancers 2021, 13, 271. https://doi.org/10.3390/cancers13020271
Rochigneux P, Chanez B, De Rauglaudre B, Mitry E, Chabannon C, Gilabert M. Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials. Cancers. 2021; 13(2):271. https://doi.org/10.3390/cancers13020271
Chicago/Turabian StyleRochigneux, Philippe, Brice Chanez, Bernadette De Rauglaudre, Emmanuel Mitry, Christian Chabannon, and Marine Gilabert. 2021. "Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials" Cancers 13, no. 2: 271. https://doi.org/10.3390/cancers13020271
APA StyleRochigneux, P., Chanez, B., De Rauglaudre, B., Mitry, E., Chabannon, C., & Gilabert, M. (2021). Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials. Cancers, 13(2), 271. https://doi.org/10.3390/cancers13020271