An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Pancreatic Cancer
2. Pancreatic Tumour Microenvironment (TME)
3. Pancreatic Stellate Cells (PSCs)
4. Interactions between Pancreatic Cancer Cells and Pancreatic Stellate Cells
5. The Endoplasmic Reticulum and ER Stress
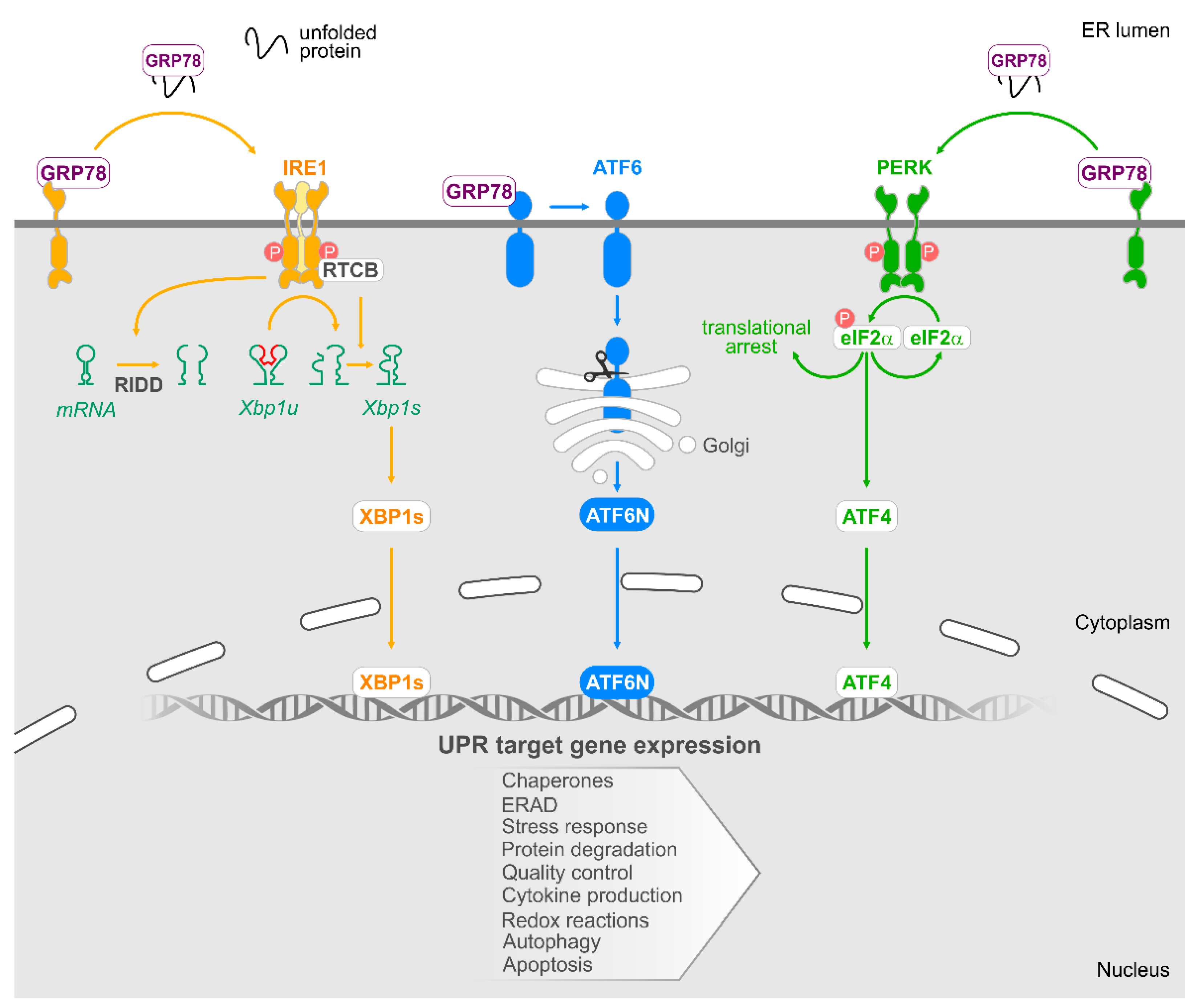
6. The Unfolded Protein Response
6.1. IRE1
6.2. PERK
6.3. ATF6
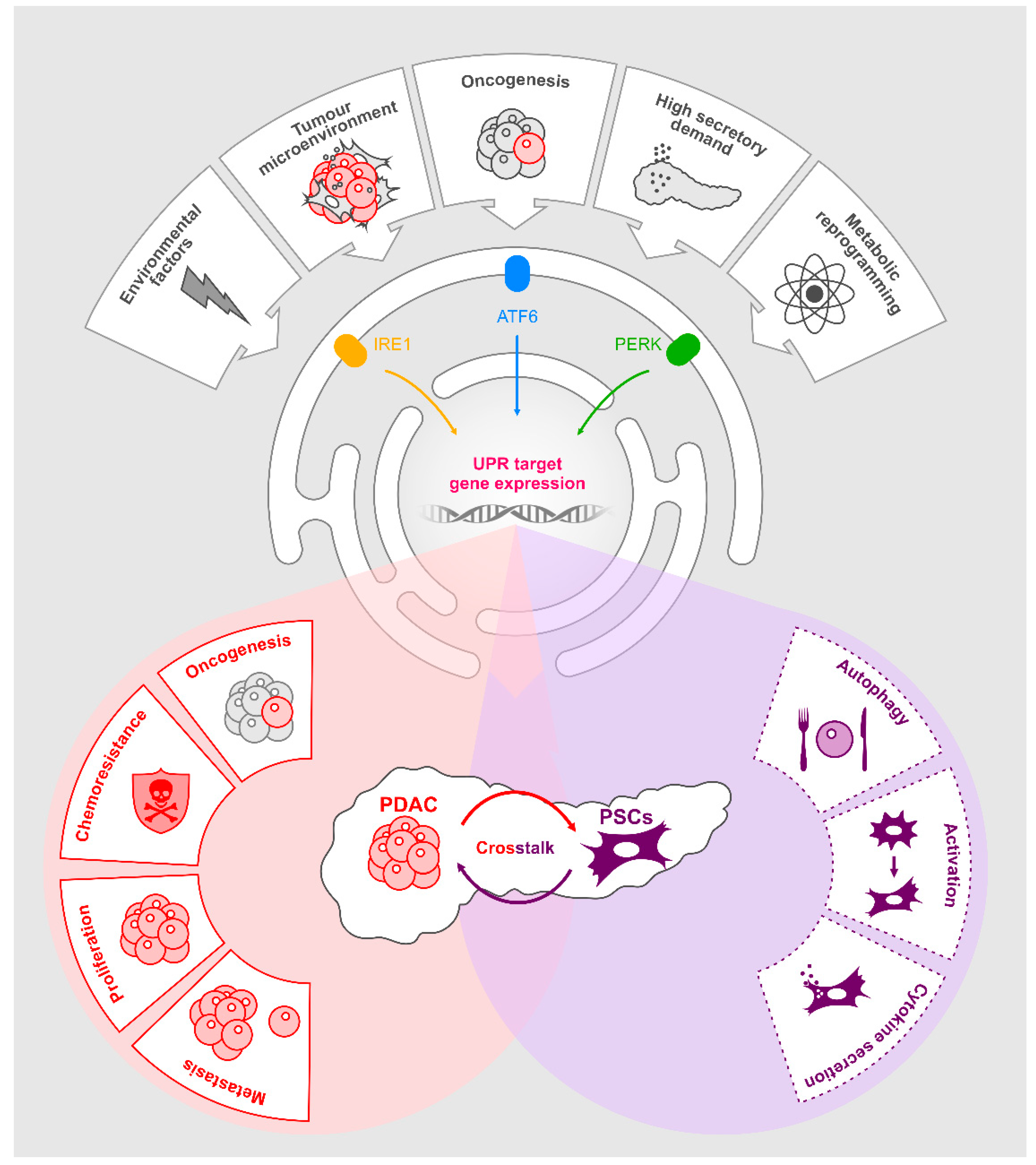
7. UPR Activation in PDAC
8. GRP78
9. IRE1, PERK and ATF6
10. UPR in PDAC Chemoresistance
11. UPR Activation in PSCs
12. UPR-Mediated Regulation of Cytokines and Implications for PDAC
13. Concluding Remarks
Funding
Conflicts of Interest
References
- Kleeff, J.; Korc, M.; Apte, M.; Vecchia, C.L.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the management of pancreatic cancer: Surgery is not enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.C.; Nathan, H.; Cameron, J.L.; Olino, K.; Edil, B.H.; Herman, J.M.; Hirose, K.; Schulick, R.D.; Choti, M.A.; Wolfgang, C.L.; et al. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent. Cancer 2011, 118, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Samuel, N.; Hudson, T.J. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 77–87. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic. Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.-Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.K.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.M.; Herrera, S.G.; et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stalnecker, C.A.; Der, C.J. RAS, wanted dead or alive: Advances in targeting RAS mutant cancers. Sci. Signal. 2020, 13, eaay6013. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Groß, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef]
- Mekapogu, A.R.; Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Multifunctional role of pancreatic stellate cells in pancreatic cancer. Ann. Pancreat. Cancer 2019, 2, 10. [Google Scholar] [CrossRef]
- Bachem, M.G.; Zhou, Z.; Zhou, S.; Siech, M. Role of stellate cells in pancreatic fibrogenesis associated with acute and chronic pancreatitis. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. 3), S92–S96. [Google Scholar] [CrossRef]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangio-genesis in pancreatic cancer. Oncogene 2009, 2, 3513–3525. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Seux, M.; Peuget, S.; Montero, M.P.; Siret, C.; Rigot, V.; Clerc, P.; Gigoux, V.; Pellegrino, E.; Pouyet, L.; N’Guessan, P.; et al. TP53INP1 decreases pancreatic cancer cell migration by regulating SPARC expression. Oncogene 2011, 30, 3049–3061. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral Fibroblast SPARC Expression and Patient Outcome With Resectable Pancreatic Adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic repro-gramming and ROS detoxification. Oncotarget 2016, 7, 36719–36732. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; Lucas, F.A.S.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. ELife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Erratum: Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 540, 150. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef]
- Oldfield, L.E.; Connor, A.A.; Gallinger, S. Molecular Events in the Natural History of Pancreatic Cancer. Trends Cancer 2017, 3, 336–346. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Castellanos, J.A.; VanSaun, M.N.; Dai, X.; Ambrose, M.; Guo, Q.; Xiong, Y.; Merchant, N.B. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016, 7, 65982–65992. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic Stellate Cells: Partners in Crime with Pancreatic Cancer Cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.E.; Bapiro, T.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2017, 67, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 1–16. [Google Scholar] [CrossRef]
- Wehr, A.Y.; Furth, E.E.; Sangar, V.; Blair, I.A.; Yu, K.H. Analysis of the Human Pancreatic Stellate Cell Secreted Proteome. Pancreas 2011, 40, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer: Fig. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Petit, I.; Jin, D.; Rafii, S. The SDF-1–CXCR4 signaling pathway: A molecular hub modulating neo-angiogenesis. Trends Immunol. 2007, 28, 299–307. [Google Scholar] [CrossRef]
- Ding, Y.; Du, Y. Clinicopathological significance and prognostic role of chemokine receptor CXCR4 expression in pancreatic ductal adenocarcinoma, a meta-analysis and literature review. Int. J. Surg. 2019, 65, 32–38. [Google Scholar] [CrossRef]
- Krieg, A.; Riemer, J.C.; Telan, L.A.; Gabbert, H.E.; Knoefel, W.T. CXCR4-A Prognostic and Clinicopathological Biomarker for Pancreatic Ductal Adenocarcinoma: A Meta-Analysis. PLoS ONE 2015, 10, e0130192. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, X.; Wu, K.; Zhao, Y.; Hu, G. Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived fac-tor-1/CXCR4 axis. Pancreatology 2010, 10, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, H.M.; Lv, Y.Q.; Tang, S.M.; Cheng, P. Blockade of SDF-1/CXCR4 reduces adhesion-mediated chemoresistance of multiple myeloma cells via interacting with interleukin. J. Cell. Physiol. 2019, 234, 19702–19714. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.R.; Claude, A.; Fullam, E.F. A Study of Tissue Culture Cells by Electron Microscopy: Methods and Preliminary Observations. J. Exp. Med. 1945, 81, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endo-plasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Kaufman, R.J. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 1996, 271, 18181–18187. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.-X.; Wang, X. A Synthetic Biology Approach Identifies the Mammalian UPR RNA Ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Lipson, K.L.; Fonseca, S.G.; Ishigaki, S.; Nguyen, L.X.; Foss, E.; Bortell, R.; Rossini, A.A.; Urano, F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE. Cell Metab. 2006, 4, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Glimcher, L.H. Fine-Tuning of the Unfolded Protein Response: Assembling the IRE1α Interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Chan, C.-P.; Kok, K.-H.; Tang, H.-M.V.; Wong, C.-M.; Jin, D.-Y. Internal ribosome entry site-mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1833, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Dubrac, A.; Quelen, C.; Desquesnes, A.; Berghe, L.V.D.; Ségura, C.; Filleron, T.; Pyronnet, S.; Prats, H.; Brousset, P.; et al. PERK mediates the IRES-dependent translational activation of mRNAs encoding angiogenic growth factors after ischemic stress. Sci. Signal. 2016, 9, ra44. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Berghe, L.V.D.; Ségura, C.; Mazzolini, L.; Pyronnet, S.; Laurell, H.; Touriol, C. The PERK Branch of the Unfolded Protein Response Promotes DLL4 Expression by Activating an Alternative Translation Mechanism. Cancers 2019, 11, 142. [Google Scholar] [CrossRef]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases that Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, L. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Kong, B.; Cheng, T.; Wu, W.; Regel, I.; Raulefs, S.; Friess, H.; Erkan, M.; Esposito, I.; Kleeff, J.; Michalski, C.W. Hypoxia-induced endoplasmic reticulum stress characterizes a necrotic phenotype of pancreatic cancer. Oncotarget 2015, 6, 32154–32160. [Google Scholar] [CrossRef] [PubMed]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Huang, L.-W.; Lin, C.-Y.; Lee, C.-C.; Liu, T.-Z.; Jeng, C.-J. Overexpression of GRP78 Is Associated With Malignant Transformation in Epithelial Ovarian Tumors. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 381–385. [Google Scholar] [CrossRef]
- Mozos, A.; Roué, G.; López-Guillermo, A.; Jares, P.; Campo, E.; Colomer, D.; Martínez, A. The Expression of the Endoplasmic Reticulum Stress Sensor BiP/GRP78 Predicts Response to Chemotherapy and Determines the Efficacy of Proteasome Inhibitors in Diffuse Large B-Cell Lymphoma. Am. J. Pathol. 2011, 179, 2601–2610. [Google Scholar] [CrossRef]
- Niu, Z.; Wang, M.; Zhou, L.; Yao, L.; Liao, Q.; Zhao, Y.-P. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Sci. Rep. 2015, 5, 16067. [Google Scholar] [CrossRef]
- Thakur, P.C.; Miller-Ocuin, J.L.; Nguyen, K.; Matsuda, R.; Singhi, A.D.; Zeh, H.J.; Bahary, N. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 2018, 16, 190. [Google Scholar] [CrossRef]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Shen, J.; Ha, D.P.; Zhu, G.; Rangel, D.F.; Kobielak, A.; Gill, P.S.; Groshen, S.; Dubeau, L.; Lee, A.S. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant Kras-driven pancreatic tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E4020–E4029. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. GRP 78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Mol. Oncol. 2018, 12, 1498–1512. [Google Scholar] [CrossRef] [PubMed]
- Lev, A.; Lulla, A.R.; Wagner, J.; Ralff, M.D.; Kiehl, J.B.; Zhou, Y.; Benes, C.H.; Prabhu, V.V.; Oster, W.; Astsaturov, I.; et al. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget 2017, 8, 81776–81793. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pan-creatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 1043–1052. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Saito, M.; Kadokura, H.; Miyazaki, J.I.; Tashiro, F.; Imagawa, Y.; Iwawaki, T.; Kohno, K. IRE1-XBP1 pathway regulates oxidative proinsulin folding in pancreatic beta cells. J. Cell Biol. 2018, 217, 1287–1301. [Google Scholar] [CrossRef]
- Gao, Y.; Sartori, D.J.; Li, C.; Yu, Q.-C.; Kushner, J.A.; Simon, M.C.; Diehl, J.A. PERK Is Required in the Adult Pancreas and Is Essential for Maintenance of Glucose Homeostasis. Mol. Cell. Biol. 2012, 32, 5129–5139. [Google Scholar] [CrossRef]
- Iida, K.; Li, Y.; McGrath, B.C.; Frank, A.; Cavener, D.R. PERK eIF2 alpha kinase is required to regulate the viability of the exocrine pancreas in mice. BMC Cell Biol. 2007, 8, 38. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Sheng, X.; Arnoldussen, Y.J.; Storm, M.; Tesikova, M.; Nenseth, H.Z.; Zhao, S.; Fazli, L.; Rennie, P.; Risberg, B.; Wæhre, H.; et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol. Med. 2015, 7, 788–801. [Google Scholar] [CrossRef]
- Li, X.X.; Zhang, H.S.; Xu, Y.M.; Zhang, R.J.; Chen, Y.; Fan, L.; Qin, Y.Q.; Liu, Y.; Li, M.; Fang, J. Knockdown of IRE1alpha inhibits colonic tumorigenesis through decreasing beta-catenin and IRE1alpha targeting suppresses colon cancer cells. Oncogene 2017, 36, 6738–6746. [Google Scholar] [CrossRef]
- Chien, W.; Ding, L.-W.; Sun, Q.-Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Fessart, D.; Delom, F.; Mulot, A.; Vojtesek, B.; Hrstka, R.; Murray, E.; Gray, T.; Hupp, T. Emerging roles for the pro-oncogenic anterior gradient-2 in cancer development. Oncogene 2012, 32, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Dumartin, L.; Alrawashdeh, W.; Trabulo, S.M.; Radon, T.P.; Steiger, K.; Feakins, R.M.; Di Magliano, M.P.; Heeschen, C.; Esposito, I.; Lemoine, N.R.; et al. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene 2016, 36, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.-Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, J.; Nipper, M.; Wang, P. Ductal vs. acinar? Recent insights into identifying cell lineage of pancreatic ductal adenocarcinoma. Ann. Pancreat. Cancer 2019, 2, 11. [Google Scholar] [CrossRef]
- Hess, D.A.; Humphrey, S.E.; Ishibashi, J.; Damsz, B.; Lee, A.; Glimcher, L.H.; Konieczny, S.F. Extensive Pancreas Regeneration Following Acinar-Specific Disruption of Xbp1 in Mice. Gastroenterology 2011, 141, 1463–1472. [Google Scholar] [CrossRef]
- Bulle, A.; Dekervel, J.; Libbrecht, L.; Nittner, D.; Deschuttere, L.; Lambrecht, D.; Van Cutsem, E.; Verslype, C.; Van Pelt, J. Gemcitabine induces Epithelial-to-Mesenchymal Transition in patient-derived pancreatic ductal adenocarcinoma xenografts. Am. J. Transl. Res. 2019, 11, 765–779. [Google Scholar]
- Shah, P.P.; Dupre, T.V.; Siskind, L.J.; Beverly, L.J. Common cytotoxic chemotherapeutics induce epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2017, 8, 22625–22639. [Google Scholar] [CrossRef]
- Wang, R.; Cheng, L.; Xia, J.; Wang, Z.; Wu, Q.; Wang, Z. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1alpha in pan-creatic cancer cells. Curr. Cancer Drug Targets 2014, 14, 407–417. [Google Scholar] [CrossRef]
- Song, Y.; Baba, T.; Li, Y.-Y.; Furukawa, K.; Tanabe, Y.; Matsugo, S.; Sasaki, S.; Mukaida, N. Gemcitabine-induced CXCL8 expression counteracts its actions by inducing tumor neovascularization. Biochem. Biophys. Res. Commun. 2015, 458, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Tyagi, N.; Khan, M.A.; Srivastava, S.K.; Al-Ghadhban, A.; Dugger, K.; Carter, J.E.; Singh, A.P.; Singh, A.P. Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Wang, W.; Zhu, Y.; Chen, Y.; Tian, B. Gemcitabine treatment induces endoplasmic reticular (ER) stress and subsequently upregulates urokinase plas-minogen activator (uPA) to block mitochondrial-dependent apoptosis in Panc-1 cancer stem-like cells (CSCs). PLoS ONE 2017, 12, e0184110. [Google Scholar]
- Tadros, S.B.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Lyudmyla, B.; Lazenby, A.J.; Berim, L.; et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-Y.; Waldron, R.T.; Gong, R.; Ramanujan, V.K.; Pandol, S.J.; Lugea, A. The Unfolded Protein Response Plays a Predominant Homeostatic Role in Response to Mitochondrial Stress in Pancreatic Stellate Cells. PLoS ONE 2016, 11, e0148999. [Google Scholar] [CrossRef]
- Ben-Harosh, Y.; Anosov, M.; Salem, H.; Yatchenko, Y.; Birk, R. Pancreatic stellate cell activation is regulated by fatty acids and ER stress. Exp. Cell Res. 2017, 359, 76–85. [Google Scholar] [CrossRef]
- Buchholz, M.; Kestler, H.A.; Holzmann, K.; Ellenrieder, V.; Schneiderhan, W.; Siech, M.; Adler, G.; Bachem, M.G.; Gress, T.M. Transcriptome analysis of human hepatic and pancreatic stellate cells: Organ-specific variations of a common transcriptional phenotype. J. Mol. Med. 2005, 83, 795–805. [Google Scholar] [CrossRef]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef]
- Kim, R.S.; Hasegawa, D.; Goossens, N.; Tsuchida, T.; Athwal, V.; Sun, X.; Robinson, C.L.; Bhattacharya, D.; Chou, H.-I.; Zhang, D.Y.; et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci. Rep. 2016, 6, 39342. [Google Scholar] [CrossRef]
- Galarreta, M.R.; Navarro, A.; Ansorena, E.; Garzón, A.G.; Mòdol, T.; López-Zabalza, M.J.; Martínez-Irujo, J.J.; Iraburu, M.J. Unfolded protein response induced by Brefeldin A increases collagen type I levels in hepatic stellate cells through an IRE1alpha, p38 MAPK and Smad-dependent pathway. Biochim. Biophys. Acta 2016, 1863, 2115–2123. [Google Scholar] [CrossRef]
- Smith, J.A. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front. Immunol. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Razidlo, G.L.; Burton, K.M.; McNiven, M.A. Interleukin-6 promotes pancreatic cancer cell migration by rapidly activating the small GTPase CDC. J. Biol. Chem. 2018, 293, 11143–11153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; Di Magliano, M.P. Interleukin-6 Is Required for Pancreatic Cancer Progression by Promoting MAPK Signaling Activation and Oxidative Stress Resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Abbruzzese, J.L.; Huang, S.; Fidler, I.J.; Xiong, Q.; Xie, K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin. Cancer Res. 1999, 5, 3711–3721. [Google Scholar]
- Guthrie, L.N.; Abiraman, K.; Plyler, E.S.; Sprenkle, N.T.; Gibson, S.A.; McFarland, B.C.; Rajbhandari, R.; Rowse, A.L.; Benveniste, E.N.; Meares, G.P. Attenuation of PKR-like ER Kinase (PERK) Signaling Selectively Controls Endoplasmic Reticulum Stress-induced Inflammation Without Compromising Immunological Responses. J. Biol. Chem. 2016, 291, 15830–15840. [Google Scholar] [CrossRef]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflam-mation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ische-mia-reperfusion injury. Am. J. Transpl. 2014, 14, 1552–1561. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed]
- Rodvold, J.J.; Chiu, K.T.; Hiramatsu, N.; Nussbacher, J.K.; Galimberti, V.; Mahadevan, N.R.; Willert, K.; Lin, J.H.; Zanetti, M. Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci. Signal. 2017, 10, eaah7177. [Google Scholar] [CrossRef] [PubMed]
- Sprenkle, N.T.; Lahiri, A.; Simpkins, J.W.; Meares, G.P. Endoplasmic reticulum stress is transmissible in vitro between cells of the central nervous system. J. Neurochem. 2019, 148, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Van Der Harg, J.M.; Van Heest, J.C.; Bangel, F.N.; Patiwael, S.; Van Weering, J.R.; Scheper, W. The UPR reduces glucose metabolism via IRE1 signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 655–665. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, H.; Jiang, C.; Fang, L.; Chen, C.; Zhang, X.D.; Jiang, Z.-W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Talty, A.; Deegan, S.; Ljujic, M.; Mnich, K.; Naicker, S.D.; Quandt, D.; Zeng, Q.; Patterson, J.B.; Gorman, A.M.; Griffin, M.D.; et al. Inhibition of IRE1alpha RNase activity reduces NLRP3 inflammasome assembly and processing of pro-IL1beta. Cell Death Dis. 2019, 10, 622. [Google Scholar] [CrossRef]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, C.M.; Talty, A.; Logue, S.E.; Mnich, K.; Gorman, A.M.; Samali, A. An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer. Cancers 2021, 13, 261. https://doi.org/10.3390/cancers13020261
Robinson CM, Talty A, Logue SE, Mnich K, Gorman AM, Samali A. An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer. Cancers. 2021; 13(2):261. https://doi.org/10.3390/cancers13020261
Chicago/Turabian StyleRobinson, Claire M., Aaron Talty, Susan E. Logue, Katarzyna Mnich, Adrienne M. Gorman, and Afshin Samali. 2021. "An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer" Cancers 13, no. 2: 261. https://doi.org/10.3390/cancers13020261
APA StyleRobinson, C. M., Talty, A., Logue, S. E., Mnich, K., Gorman, A. M., & Samali, A. (2021). An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer. Cancers, 13(2), 261. https://doi.org/10.3390/cancers13020261