Adenosinergic Pathway: A Hope in the Immunotherapy of Glioblastoma


Abstract
Simple Summary
Abstract
1. Introduction
2. Glioblastoma Immunotherapy
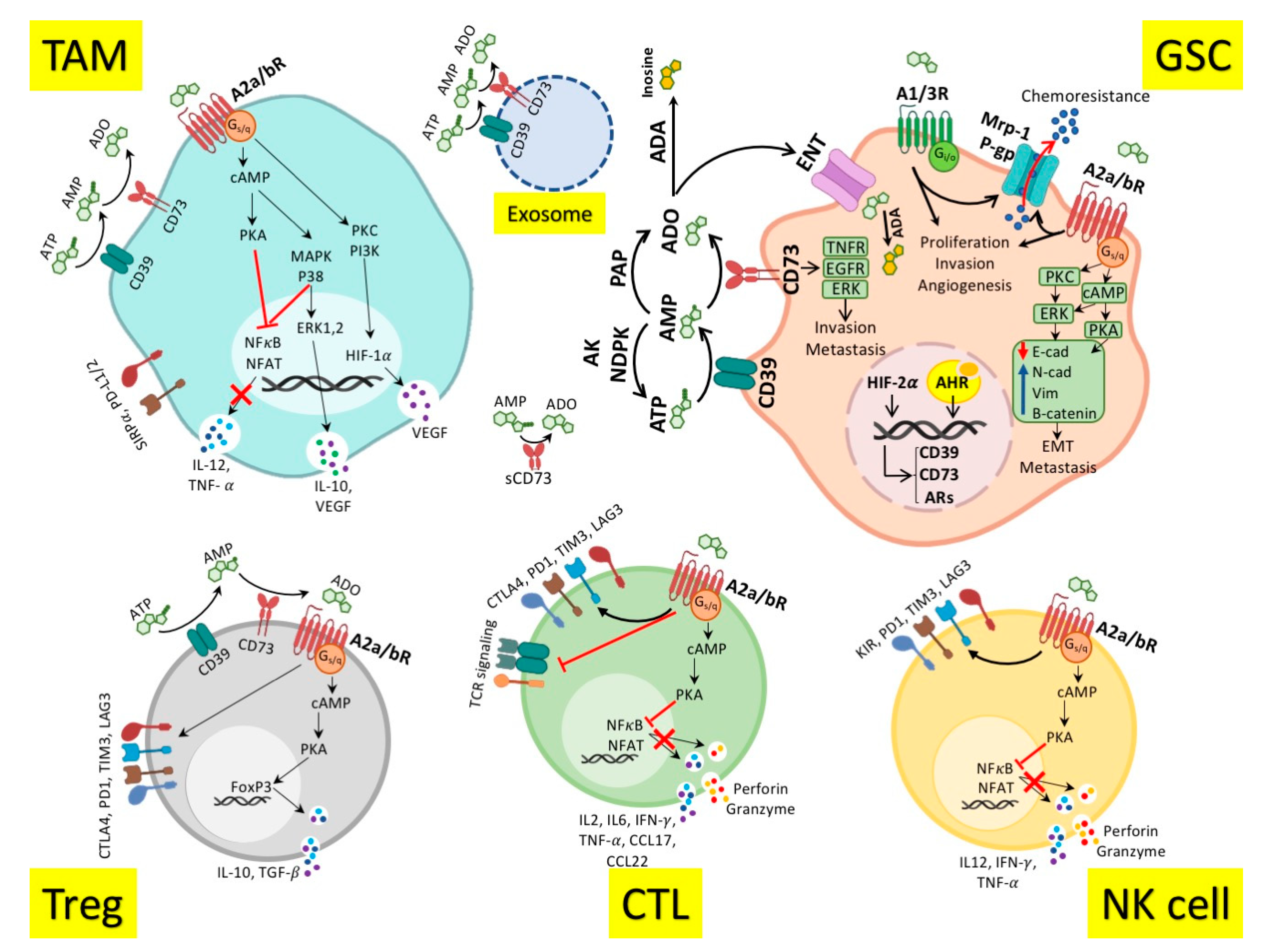
3. Role of Adenosinergic Pathway in Antitumor Immune Response
4. Adenosinergic Pathway in the Glioblastoma Immunopathogenesis
5. Targeting Adenosinergic Pathway in Glioblastoma Immunotherapy

{kind=link}
| Target | Pro-Tumor and Immunosuppressive Roles | Diagnostic/Prognostic Roles | Therapeutic Potentials |
|---|---|---|---|
| CD39 |
|
| |
| CD73 |
|
| |
| A1R |
|
|
|
| A2aR |
|
|
|
| A2bR |
|
|
|
| A3R |
|
|
|
| ADA |
|
| |
| PAP |
|
|
6. The Role of Adenosinergic Pathway in Glioblastoma Prognosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jovčevska, I.; Kočevar, N.; Komel, R. Glioma and glioblastoma-how much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zhang, X.; Chen, Z.; Liu, N. Establishment and validation of an immune-based prognostic score model in glioblastoma. Int. Immunopharmacol. 2020, 85, 106636. [Google Scholar] [CrossRef] [PubMed]
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Aldape, K.; Colman, H.; Figrarella-Branger, D.; Fuller, G.N.; Giannini, C.; Holland, E.C.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Komori, T. cIMPACT-NOW update 5: Recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020, 139, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Perry, A.; Reifenberger, G.; Stupp, R. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Zong, H.; Verhaak, R.G.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef]
- Iacob, G.; Dinca, E.B. Current data and strategy in glioblastoma multiforme. J. Med. Life 2009, 2, 386. [Google Scholar]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neuro-Oncol. 2020. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, M.J.; Jung, H.H.; Chang, W.S.; Choi, H.S.; Rachmilevitch, I.; Zadicario, E.; Chang, J.W. One-Year Outcome of Multiple Blood–Brain Barrier Disruptions with Temozolomide for the Treatment of Glioblastoma. Front. Oncol. 2020, 10, 1663. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Fabian, D.; Guillermo Prieto Eibl, M.D.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of glioblastoma (GBM) with the addition of tumor-treating fields (TTF): A review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Hajifathali, A.; Parkhideh, S.; Kazemi, M.H.; Chegeni, R.; Roshandel, E.; Gholizadeh, M. Immune checkpoints in hematologic malignancies: What made the immune cells and clinicians exhausted! J. Cell. Physiol. 2020. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Reardon, D.; Omuro, A.; Brandes, A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.; Lim, M. OS10. 3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro-Oncology 2017, 19, iii21. [Google Scholar] [CrossRef]
- Phase, B.-M.S.A. CheckMate−498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo (nivolumab) Plus Radiation in Patients with Newly Diagnosed MGMT-Unmethylated Glioblastoma Multiforme. BMS Newsroom, 5 September 2019. [Google Scholar]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, G.J.; Raber, J. CNS side effects of immune checkpoint inhibitors: Preclinical models, genetics and multimodality therapy. Immunotherapy 2017, 9, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Omuro, A.; Vlahovic, G.; Reardon, D.; Sahebjam, S.; Cloughesy, T.; Baehring, J.; Butowski, N.; Potter, V.; Zwirtes, R. 325ONivolumab (nivo) in combination with radiotherapy (RT)±temozolomide (TMZ): Updated safety results from CheckMate 143 in pts with methylated or unmethylated newly diagnosed glioblastoma (GBM). Ann. Oncol. 2017, 28, v109–v121. [Google Scholar] [CrossRef]
- Sampson, J.H.; Vlahovic, G.; Sahebjam, S.; Omuro, A.M.P.; Baehring, J.M.; Hafler, D.A.; Voloschin, A.D.; Paliwal, P.; Grosso, J.; Coric, V. Preliminary safety and activity of nivolumab and its combination with ipilimumab in recurrent glioblastoma (GBM): CHECKMATE-143. Am. Soc. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Liu, E.K.; Sulman, E.P.; Wen, P.Y.; Kurz, S.C. Novel Therapies for Glioblastoma. Curr. Neurol. Neurosci. Rep. 2020, 20, 19. [Google Scholar] [CrossRef]
- Neyns, B.; Sadones, J.; Joosens, E.; Bouttens, F.; Verbeke, L.; Baurain, J.-F.; D’Hondt, L.; Strauven, T.; Chaskis, C.; In’t Veld, P. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009, 20, 1596–1603. [Google Scholar] [CrossRef]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Falls, H.D.; Mitten, M.J.; DeVries, P.J.; Benatuil, L.; Hsieh, C.-M.; Meulbroek, J.A.; Panchal, S.C. Characterization of ABBV-221, a tumor-selective EGFR-targeting antibody drug conjugate. Mol. Cancer Ther. 2018, 17, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A. Her2-specific chimeric antigen receptor–modified virus-specific t cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): Results of a double-blind randomized phase II trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Belal, A.; Qiu, J.; Mogensen, K.; Schilero, C.; Hutson, A. SurVaxM with standard therapy in newly diagnosed glioblastoma: Phase II trial update. J. Clin. Oncol. 2019, 37, 2016. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 1–9. [Google Scholar]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511+ Toca FC. Neuro-Oncology 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Landolfi, J.; Hogan, D.J.; Bloomfield, S.; Carter, B.; Chen, C.C.; Elder, J.B.; Kalkanis, S.N.; Kesari, S.; Lai, A. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef]
- Cockle, J.V.; Rajani, K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.M.; Shim, K.; Peterson, T.; Parney, I.F.; Short, S. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro-Oncology 2015, 18, 518–527. [Google Scholar] [CrossRef]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef]
- Brenner, A.J.; Peters, K.B.; Vredenburgh, J.; Bokstein, F.; Blumenthal, D.T.; Yust-Katz, S.; Peretz, I.; Oberman, B.; Freedman, L.S.; Ellingson, B.M. Safety and efficacy of VB-111, an anticancer gene therapy, in patients with recurrent glioblastoma: Results of a phase I/II study. Neuro-Oncology 2020, 22, 694–704. [Google Scholar] [CrossRef]
- Chiocca, E.A.; John, S.Y.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, eaaw5680. [Google Scholar] [CrossRef]
- Westphal, M.; Ylä-Herttuala, S.; Martin, J.; Warnke, P.; Menei, P.; Eckland, D.; Kinley, J.; Kay, R.; Ram, Z.; Group, A.S. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 823–833. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018, 36, 1419. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Tomaszowski, K.-H.; Marisetty, A.; Kong, L.-Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5, e134386. [Google Scholar] [CrossRef]
- Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The adenosine A3 receptor regulates differentiation of glioblastoma stem-like cells to endothelial cells under hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef]
- Niechi, I.; Uribe-Ojeda, A.; Erices, J.I.; Torres, Á.; Uribe, D.; Rocha, J.D.; Silva, P.; Richter, H.G.; San Martín, R.; Quezada, C. Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness. Cells 2019, 8, 1353. [Google Scholar] [CrossRef]
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; López, M.N.; Melo, R.; Oyarzún, C.; San Martín, R. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373. [Google Scholar] [CrossRef]
- Wang, J.; Matosevic, S. NT5E/CD73 as correlative factor of patient survival and natural killer cell infiltration in glioblastoma. J. Clin. Med. 2019, 8, 1526. [Google Scholar] [CrossRef]
- Daniele, S.; Zappelli, E.; Natali, L.; Martini, C.; Trincavelli, M.L. Modulation of A 1 and A 2B adenosine receptor activity: A new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis. 2014, 5, e1539. [Google Scholar] [CrossRef]
- Quezada, C.; Garrido, W.; Oyarzún, C.; Fernández, K.; Segura, R.; Melo, R.; Casanello, P.; Sobrevia, L.; San Martín, R. 5′-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J. Cell. Physiol. 2013, 228, 602–608. [Google Scholar] [CrossRef]
- Xu, S.; Shao, Q.-Q.; Sun, J.-T.; Yang, N.; Xie, Q.; Wang, D.-H.; Huang, Q.-B.; Huang, B.; Wang, X.-Y.; Li, X.-G. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro-Oncology 2013, 15, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.; Schuh, R.; Michels, L.; Gelsleichter, N.; Beckenkamp, L.; Iser, I.; Lenz, G.; De Oliveira, F.; Venturin, G.; Greggio, S. Nasal administration of cationic nanoemulsions as CD73-siRNA delivery system for glioblastoma treatment: A new therapeutical approach. Mol. Neurobiol. 2020, 57, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.; Schuh, R.; Michels, L.; Iser, I.; Beckenkamp, L.; Roliano, G.; Lenz, G.; Scholl, J.; Sévigny, J.; Wink, M. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol. Immunother. CII 2020, 69, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, S.; Pathak, K. Physicochemical and physiological considerations for efficient nose-to-brain targeting. Expert Opin. Drug Deliv. 2012, 9, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.; Schuh, R.; Michels, L.; Gelsleichter, N.; Beckenkamp, L.; Lenz, G.; de Oliveira, F.; Wink, M.; Stefani, M.; Battastini, A. CD73 as a target to improve temozolomide chemotherapy effect in glioblastoma preclinical model. Cancer Chemother. Pharmacol. 2020, 85, 1177–1182. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Wikstrand, C.J.; McLendon, R.E.; Friedman, A.H.; Bigner, D.D. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997, 57, 4130–4140. [Google Scholar]
- González-Tablas, M.; Arandia, D.; Jara-Acevedo, M.; Otero, Á.; Vital, A.-L.; Prieto, C.; González-Garcia, N.; Nieto-Librero, A.B.; Tao, H.; Pascual, D. Heterogeneous EGFR, CDK4, MDM4, and PDGFRA Gene Expression Profiles in Primary GBM: No Association with Patient Survival. Cancers 2020, 12, 231. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722. [Google Scholar] [CrossRef]
- Chuntova, P.; Downey, K.M.; Hegde, B.; Almeida, N.D.; Okada, H. Genetically engineered T-cells for malignant glioma: Overcoming the barriers to effective immunotherapy. Front. Immunol. 2019, 9, 3062. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, S.; Han, D.; Tang, B.; Ma, J. Delivery and Biosafety of Oncolytic Virotherapy. Front. Oncol. 2020, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.; Peterson, T.E.; Balgeman, A.; Chen, S.; Zhang, L.; Renner, D.N.; Johnson, A.J.; Parney, I.F. Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro-Oncology 2015, 17, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance mechanisms and barriers to successful immunotherapy for treating glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, C.; Wang, B.; Zhang, H.; Li, C.; Qin, G.; Cao, L.; Gao, Q.; Ping, Y.; Zhang, K. Regulatory T cells promote glioma cell stemness through TGF-β–NF-κB–IL6–STAT3 signaling. Eur. PMC 2020. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.-S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar]
- Maxwell, R.; Luksik, A.S.; Garzon-Muvdi, T.; Hung, A.L.; Kim, E.S.; Wu, A.; Xia, Y.; Belcaid, Z.; Gorelick, N.; Choi, J. Contrasting impact of corticosteroids on anti-PD-1 immunotherapy efficacy for tumor histologies located within or outside the central nervous system. Oncoimmunology 2018, 7, e1500108. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.-K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M. Anti–PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, ra180–ra370. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A. Targeting CD39 in cancer reveals an extracellular ATP-and inflammasome-driven tumor immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, G.; Zimmerman, T.P.; Hiemstra, K.; Winston, M.; Chu, L.-C. Adenosine inhibition of lymphocyte-mediated cytolysis: Possible role of cyclic adenosine monophosphate. Science 1975, 187, 957–959. [Google Scholar] [CrossRef]
- Ghalamfarsa, G.; Kazemi, M.H.; Raoofi Mohseni, S.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a potential opportunity for cancer immunotherapy. Expert Opin. Ther. Targets 2019, 23, 127–142. [Google Scholar]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef]
- Street, S.E.; Kramer, N.J.; Walsh, P.L.; Taylor-Blake, B.; Yadav, M.C.; King, I.F.; Vihko, P.; Wightman, R.M.; Millán, J.L.; Zylka, M.J. Tissue-nonspecific alkaline phosphatase acts redundantly with PAP and NT5E to generate adenosine in the dorsal spinal cord. J. Neurosci. 2013, 33, 11314–11322. [Google Scholar] [CrossRef]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investig. 2013, 123, 3552–3563. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Et Biophys. Acta (Bba)-Biomembr. 2011, 1808, 1290–1308. [Google Scholar]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Hatfield, S.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A receptor stimulation inhibits TCR-induced Notch1 activation in CD8+ T-Cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Rodrigo, B.N.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef]
- Hazenberg, M.D.; Haverkate, N.J.; van Lier, Y.F.; Spits, H.; Krabbendam, L.; Bemelman, W.A.; Buskens, C.J.; Blom, B.; Shikhagaie, M.M. Human ectoenzyme-expressing ILC3: Immunosuppressive innate cells that are depleted in graft-versus-host disease. Blood Adv. 2019, 3, 3650–3660. [Google Scholar] [CrossRef]
- Yago, T.; Tsukamoto, H.; Liu, Z.; Wang, Y.; Thompson, L.F.; McEver, R.P. Multi-inhibitory effects of A2A adenosine receptor signaling on neutrophil adhesion under flow. J. Immunol. 2015, 195, 3880–3889. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Jacobson, K.A. Purinergic signaling in mast cell degranulation and asthma. Front. Pharmacol. 2017, 8, 947. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Köhler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232. [Google Scholar] [CrossRef]
- Bowser, J.L.; Lee, J.W.; Yuan, X.; Eltzschig, H.K. The hypoxia-adenosine link during inflammation. J. Appl. Physiol. 2017, 123, 1303–1320. [Google Scholar] [CrossRef] [PubMed]
- Torres, Á.; Erices, J.I.; Sanchez, F.; Ehrenfeld, P.; Turchi, L.; Virolle, T.; Uribe, D.; Niechi, I.; Spichiger, C.; Rocha, J.D. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A3 Adenosine Receptor activation under hypoxia. Cancer Lett. 2019, 446, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef]
- Schneider, E.; Rissiek, A.; Winzer, R.; Puig, B.; Rissiek, B.; Haag, F.; Mittrücker, H.-W.; Magnus, T.; Tolosa, E. Generation and Function of Non-cell-bound CD73 in Inflammation. Front. Immunol. 2019, 10, 1729. [Google Scholar] [CrossRef]
- Pietrobono, D.; Giacomelli, C.; Marchetti, L.; Martini, C.; Trincavelli, M.L. High Adenosine Extracellular Levels Induce Glioblastoma Aggressive Traits Modulating the Mesenchymal Stromal Cell Secretome. Int. J. Mol. Sci. 2020, 21, 7706. [Google Scholar] [CrossRef]
- Gholami, M.D.; Falak, R.; Heidari, S.; Khoshmirsafa, M.; Kazemi, M.H.; Zarnani, A.-H.; Safari, E.; Tajik, N.; Kardar, G.A. A truncated snail1 transcription factor alters the expression of essential EMT markers and suppresses tumor cell migration in a human lung cancer cell line. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 158–169. [Google Scholar] [CrossRef]
- Gao, Z.-W.; Wang, H.-P.; Lin, F.; Wang, X.; Long, M.; Zhang, H.-Z.; Dong, K. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer 2017, 17, 1–8. [Google Scholar] [CrossRef]
- Zhou, L.; Jia, S.; Chen, Y.; Wang, W.; Wu, Z.; Yu, W.; Zhang, M.; Ding, G.; Cao, L. The distinct role of CD73 in the progression of pancreatic cancer. J. Mol. Med. 2019, 97, 803–815. [Google Scholar] [CrossRef]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A2B adenosine receptor signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef]
- González-Tablas, M.; Otero, Á.; Arandia, D.; Pascual, D.; Ruiz, L.; Sousa, P.; García, A.; Roa, J.C.; Villaseñor, J.J.M.; Torres, L. Tumor cell and immune cell profiles in primary human glioblastoma: Impact on patient outcome. Brain Pathol. 2020, e12927. [Google Scholar] [CrossRef]
- Zhou, W.; Bao, S. Reciprocal supportive interplay between glioblastoma and tumor-associated macrophages. Cancers 2014, 6, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When immune cells turn bad—tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.H.; da Silveira, E.F.; de Carvalho, T.R.; Oliveira, P.S.; Pacheco, S.; do Couto, C.T.; Beira, F.T.; Stefanello, F.M.; Spanevello, R.M.; Braganhol, E. Glioma sensitive or chemoresistant to temozolomide differentially modulate macrophage protumor activities. Biochim. Et Biophys. Acta (Bba)-Gen. Subj. 2017, 1861, 2652–2662. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Fu, J.; Wang, W.; Hofman, F.M.; Chen, T.C.; Chen, L. Distribution of cancer stem cells in two human brain gliomas. Oncol. Lett. 2019, 17, 2123–2130. [Google Scholar] [CrossRef]
- Jang, J.-W.; Song, Y.; Kim, S.-H.; Kim, J.; Seo, H.R. Potential mechanisms of CD133 in cancer stem cells. Life Sci. 2017, 184, 25–29. [Google Scholar] [CrossRef]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-β signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.; Leite, S.; Sauvageot, N.; Sarkisjan, D. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; John, S.Y. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shen, G.; Shi, Z.; Shen, F.; Zheng, X.; Wen, L.; Yang, X. Brain tumour stem cells and neural stem cells: Still explored by the same approach? J. Int. Med. Res. 2008, 36, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Torres, Á.; Arriagada, V.; Erices, J.I.; Toro, M.D.l.Á.; Rocha, J.D.; Niechi, I.; Carrasco, C.; Oyarzún, C.; Quezada, C. FK506 Attenuates the MRP1-Mediated Chemoresistant Phenotype in Glioblastoma Stem-Like Cells. Int. J. Mol. Sci. 2018, 19, 2697. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Asp. Med. 2017, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Sacchetto, V.; Fogli, E.; Merighi, S.; Varani, K.; Baraldi, P.G.; Tabrizi, M.A.; Leung, E.; Maclennan, S.; Borea, P.A. Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem. Pharmacol. 2010, 79, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Borea, P.A. Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem. Pharmacol. 2006, 72, 19–31. [Google Scholar] [CrossRef]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Baraldi, P.G.; Borea, P.A. Hypoxia inhibits paclitaxel-induced apoptosis through adenosine-mediated phosphorylation of bad in glioblastoma cells. Mol. Pharmacol. 2007, 72, 162–172. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.; Rao, A.; Braganhol, E.; Whiteside, T.L. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neuro-Oncol. Adv. 2020, 2, vdaa056. [Google Scholar] [CrossRef]
- Scholl, J.N.; de Fraga Dias, A.; Pizzato, P.R.; Lopes, D.V.; Moritz, C.E.J.; Jandrey, E.H.F.; Souto, G.D.; Colombo, M.; Rohden, F.; Sévigny, J. Characterization and antiproliferative activity of glioma-derived extracellular vesicles. Nanomedicine 2020, 15, 1001–1018. [Google Scholar] [CrossRef]
- Zhou, Y.; Tong, L.; Chu, X.; Deng, F.; Tang, J.; Tang, Y.; Dai, Y. The adenosine A1 receptor antagonist DPCPX inhibits tumor progression via the ERK/JNK pathway in renal cell carcinoma. Cell. Physiol. Biochem. 2017, 43, 733–742. [Google Scholar] [CrossRef]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biol. Targets Ther. 2009, 3, 349. [Google Scholar]
- Azambuja, J.; Gelsleichter, N.; Beckenkamp, L.; Iser, I.; Fernandes, M.; Figueiró, F.; Battastini, A.; Scholl, J.; de Oliveira, F.; Spanevello, R. CD73 downregulation decreases in vitro and in vivo glioblastoma growth. Mol. Neurobiol. 2019, 56, 3260–3279. [Google Scholar] [CrossRef] [PubMed]
- Tekade, R.K.; Tekade, M.; Kesharwani, P.; D’Emanuele, A. RNAi-combined nano-chemotherapeutics to tackle resistant tumors. Drug Discov. Today 2016, 21, 1761–1774. [Google Scholar]
- Malhotra, M.; Toulouse, A.; Godinho, B.M.; Mc Carthy, D.J.; Cryan, J.F.; O’Driscoll, C.M. RNAi therapeutics for brain cancer: Current advancements in RNAi delivery strategies. Mol. Biosyst. 2015, 11, 2635–2657. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, S.; Toro, M.d.l.Á.; Villarreal, C.; Melo, R.; Fernández, R.; Ayuso Sacido, A.; Uribe, D.; San Martín, R.; Quezada, C. Decreased Equilibrative Nucleoside Transporter 1 (ENT1) Activity Contributes to the High Extracellular Adenosine Levels in Mesenchymal Glioblastoma Stem-Like Cells. Cells 2020, 9, 1914. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S. First-in-human study of AMG 820, a monoclonal anti-colony-stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]

| Immunotherapy | Advantage | Disadvantage | Refs. |
|---|---|---|---|
| Immune checkpoint inhibitors (PD-1, CTLA-4, LAG-3, TIM-3, IDO, CD27) |
|
| [19,20,21,22,25,26] |
| Bevacizumab (anti-VEGF) |
|
| [27,28,29] |
| Cetuximab (anti-EGFR) |
|
| [29,30] |
|
|
| [31,32] |
| Anti-CSF-1R mAb |
|
| [33,34] |
| CAR T cell against IL13Rα2, EGFRvIII, Her-2, EphA2 |
|
| [9,35,36,37] |
| BiTE (against EGFR) |
|
| [38] |
| Tumor vaccines using specific peptides (Rindopepimut, survivin) or tumor lysate |
|
| [39,40,41,42,43] |
| DC Vaccines (ICT-107:pulsed with six peptides)(DCVax: pulsed with tumor lysate) |
|
| [44,45] |
|
|
| [46,47,48,49,50,51] |
| Oncolytic virotherapy (Adenovirus, polio-rhinovirus chimera, herpes simplex virus) |
|
| [52,53,54] |
| Adenosinergic pathway (ARs, CD39, CD73, ADA) |
|
| [55,56,57,58,59,60,61,62,63,64,65,66,67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, K.; Mao, C.; Chen, L.; Wang, L.; Liu, Y.; Yuan, J. Adenosinergic Pathway: A Hope in the Immunotherapy of Glioblastoma. Cancers 2021, 13, 229. https://doi.org/10.3390/cancers13020229
Jin K, Mao C, Chen L, Wang L, Liu Y, Yuan J. Adenosinergic Pathway: A Hope in the Immunotherapy of Glioblastoma. Cancers. 2021; 13(2):229. https://doi.org/10.3390/cancers13020229
Chicago/Turabian StyleJin, Ketao, Chunsen Mao, Lin Chen, Lude Wang, Yuyao Liu, and Jianlie Yuan. 2021. "Adenosinergic Pathway: A Hope in the Immunotherapy of Glioblastoma" Cancers 13, no. 2: 229. https://doi.org/10.3390/cancers13020229
APA StyleJin, K., Mao, C., Chen, L., Wang, L., Liu, Y., & Yuan, J. (2021). Adenosinergic Pathway: A Hope in the Immunotherapy of Glioblastoma. Cancers, 13(2), 229. https://doi.org/10.3390/cancers13020229