Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation

 , , and
, , and 
Abstract
Simple Summary
Abstract
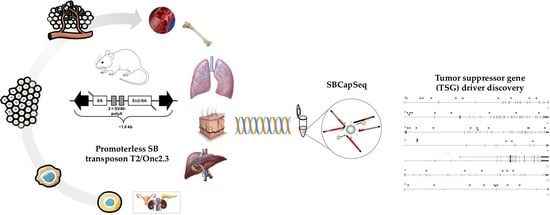
1. Introduction
2. Results
2.1. Harnessing an SB Gene-Trap Allele for Forward Genetic Screens
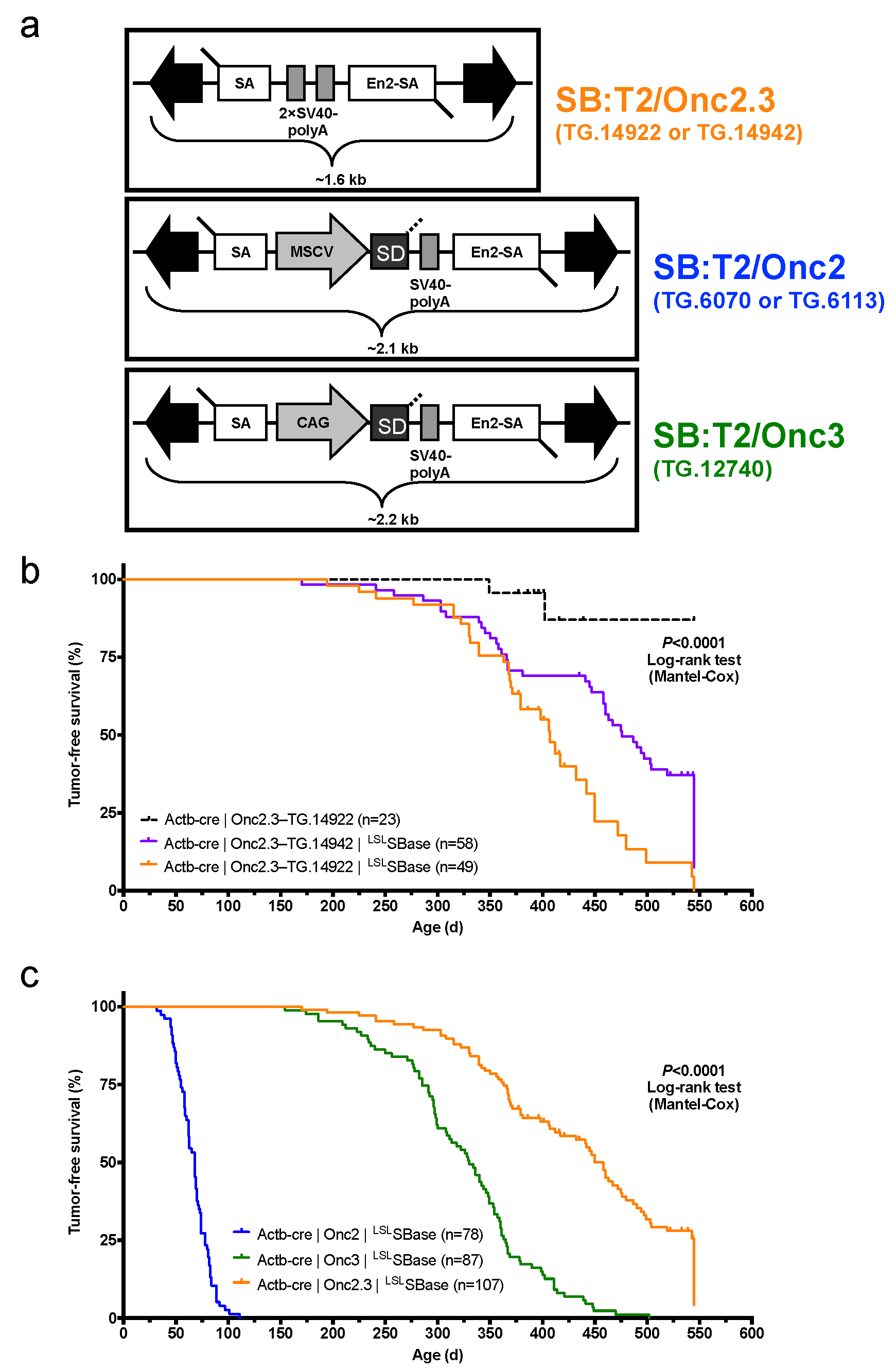
2.1.1. Whole-Body Mutagenesis Using High-Copy, Gene-Trap Only SB Transposons in Wild Type Mice
2.1.2. Systemic, Whole Body Onc2.3 Transposition Extends Latency of Tumor Development
2.2. Solid Tumors Driven by TSG Inactivation in SB-Onc2.3 Mice
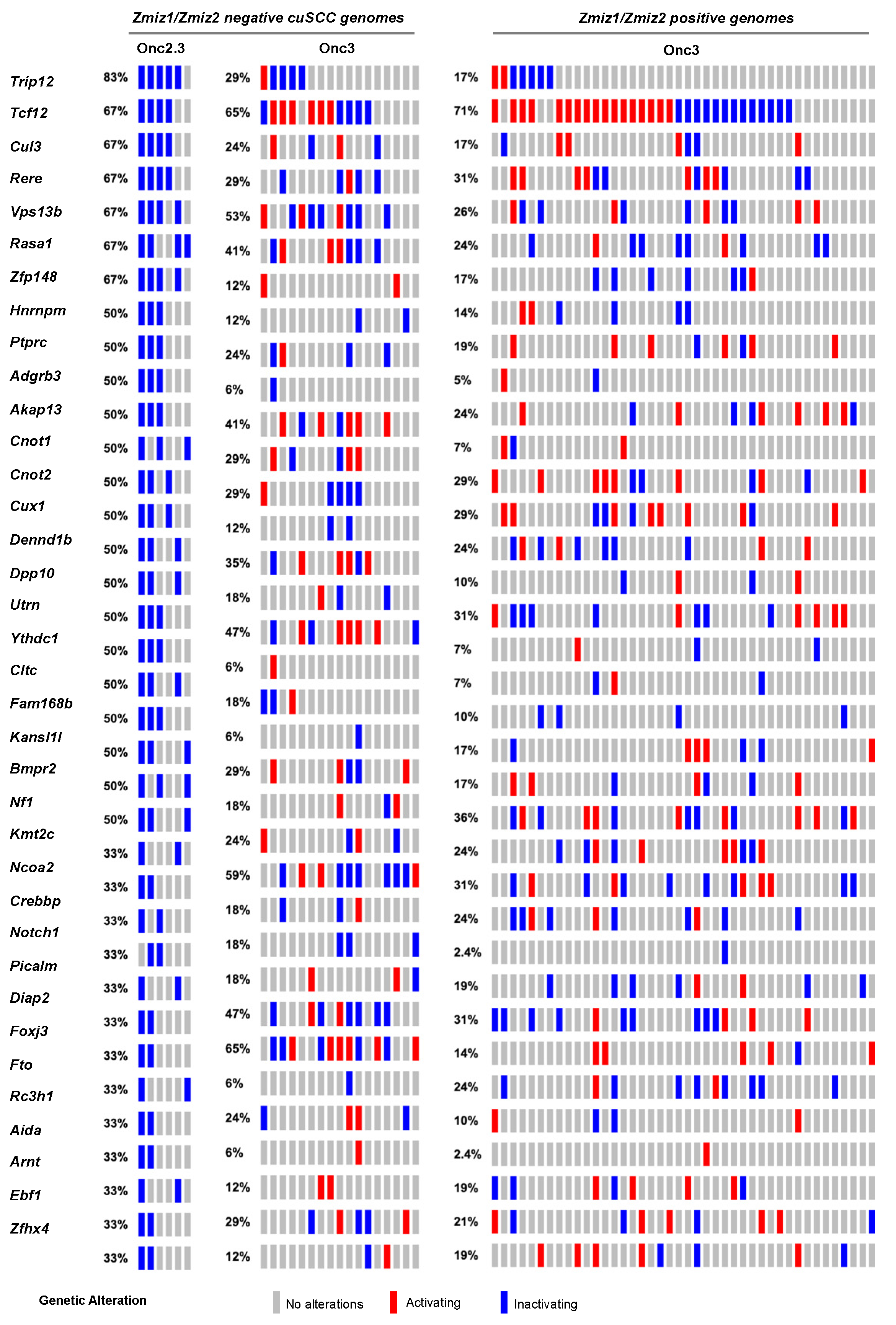
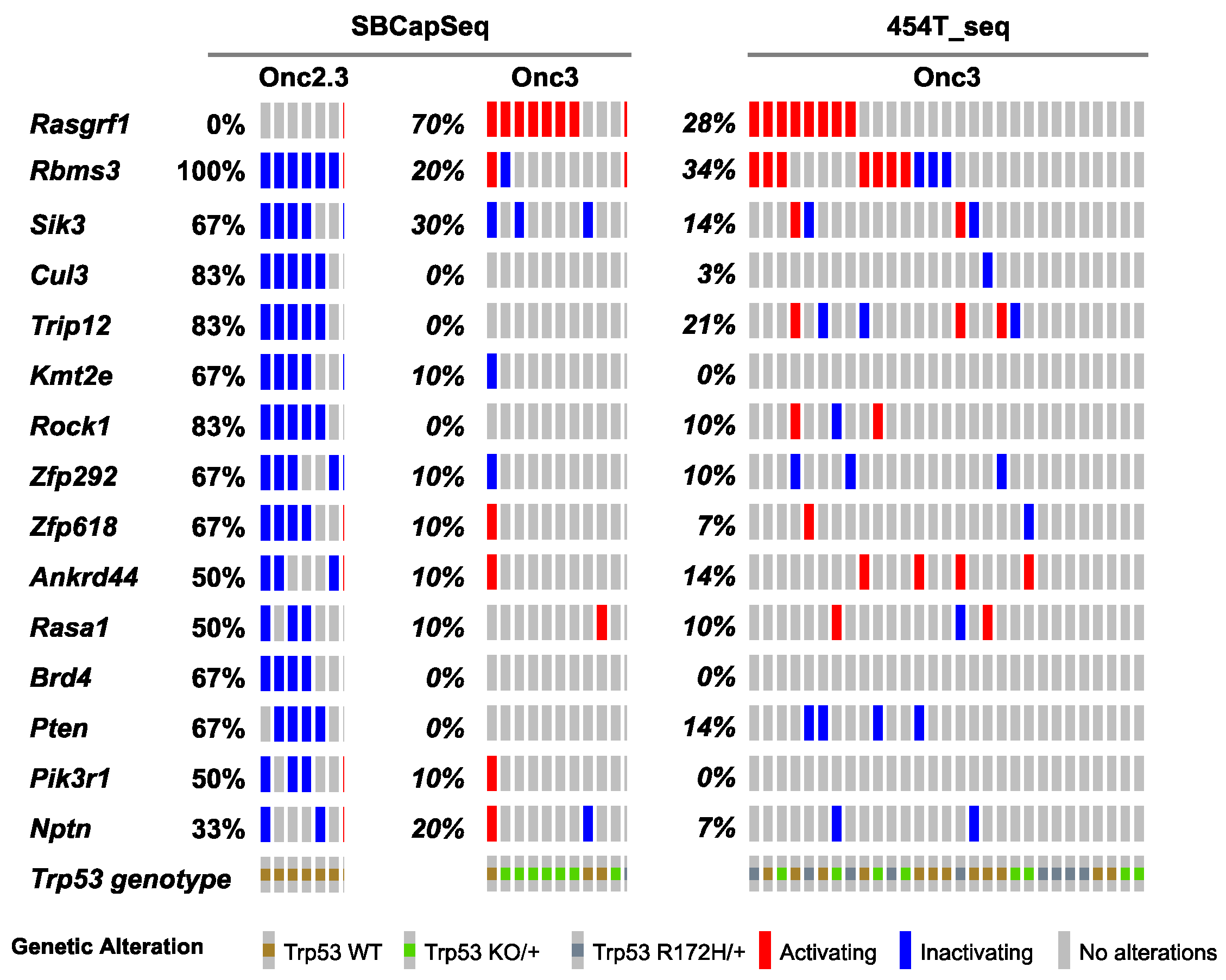
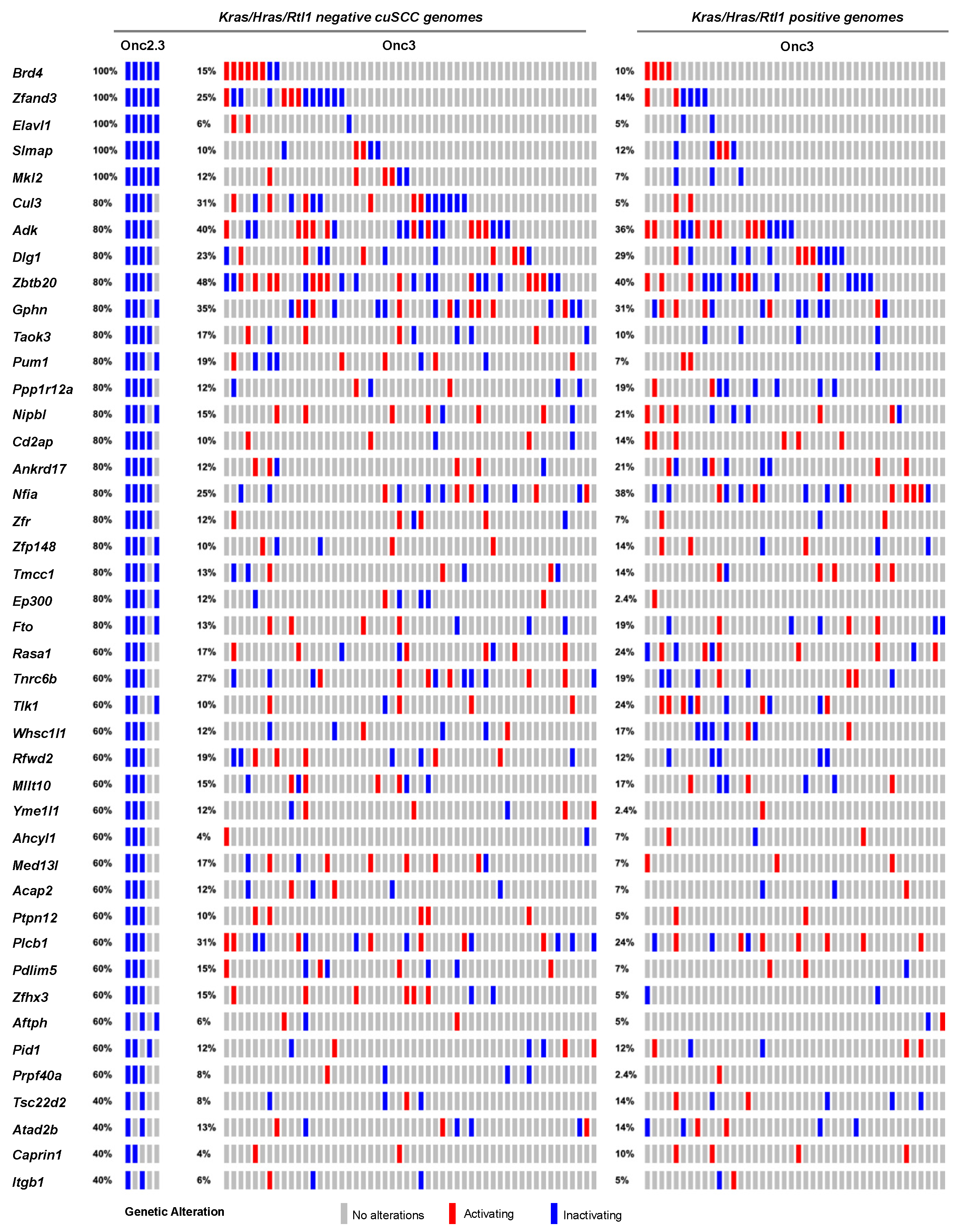
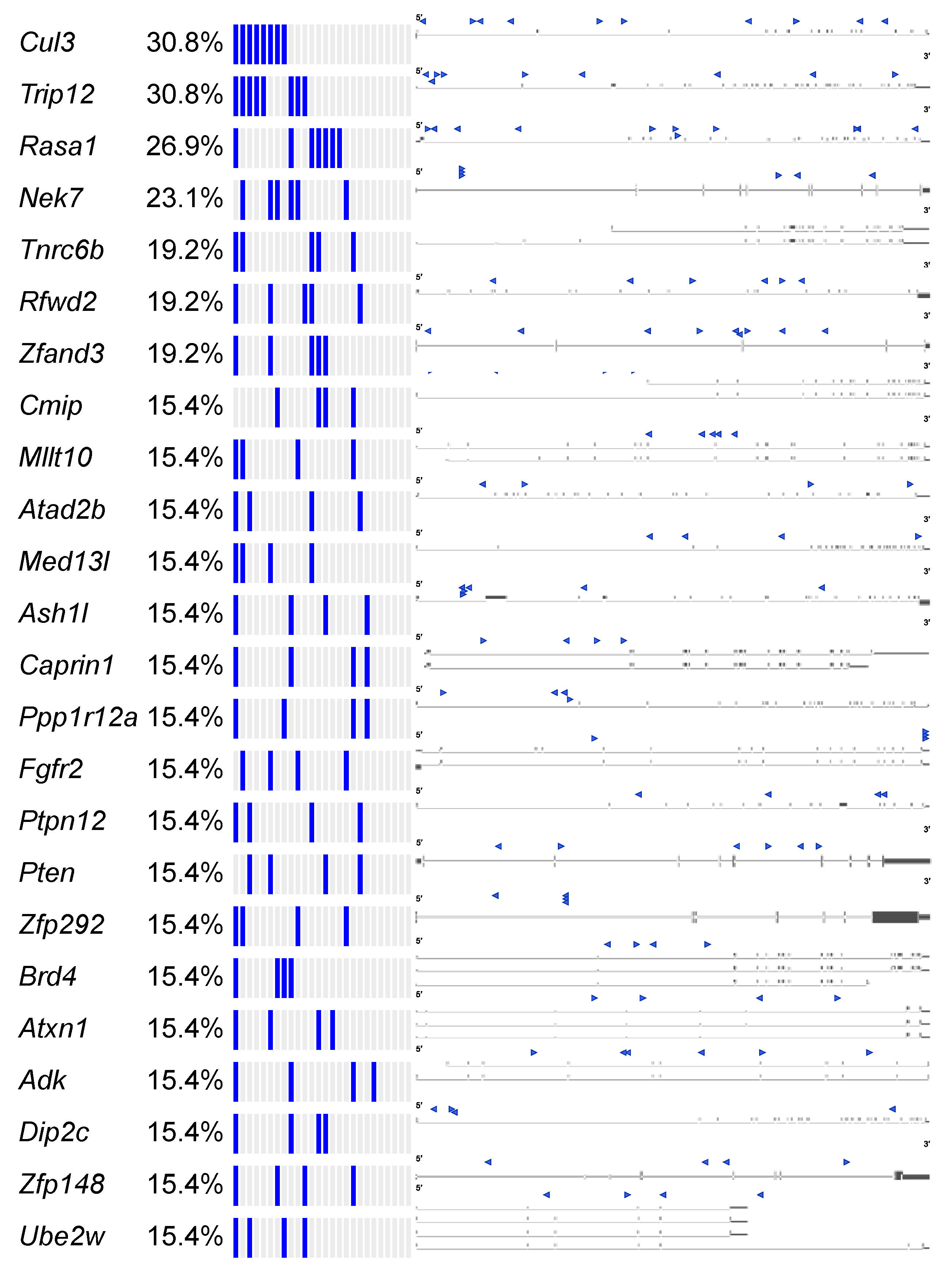
2.2.1. Landscape of cuSCC Driven by Transposon-Mediated Tumor Suppressor Inactivation
2.2.2. Landscape of Lung Cancer Driven by Transposon-Mediated Tumor Suppressor Inactivation
2.2.3. Landscape of Liver Cancer Driven by Transposon-Mediated Tumor Suppressor Inactivation
2.2.4. Landscape of Other Solid Tumors Driven by Transposon-Mediated Tumor Suppressor Inactivation
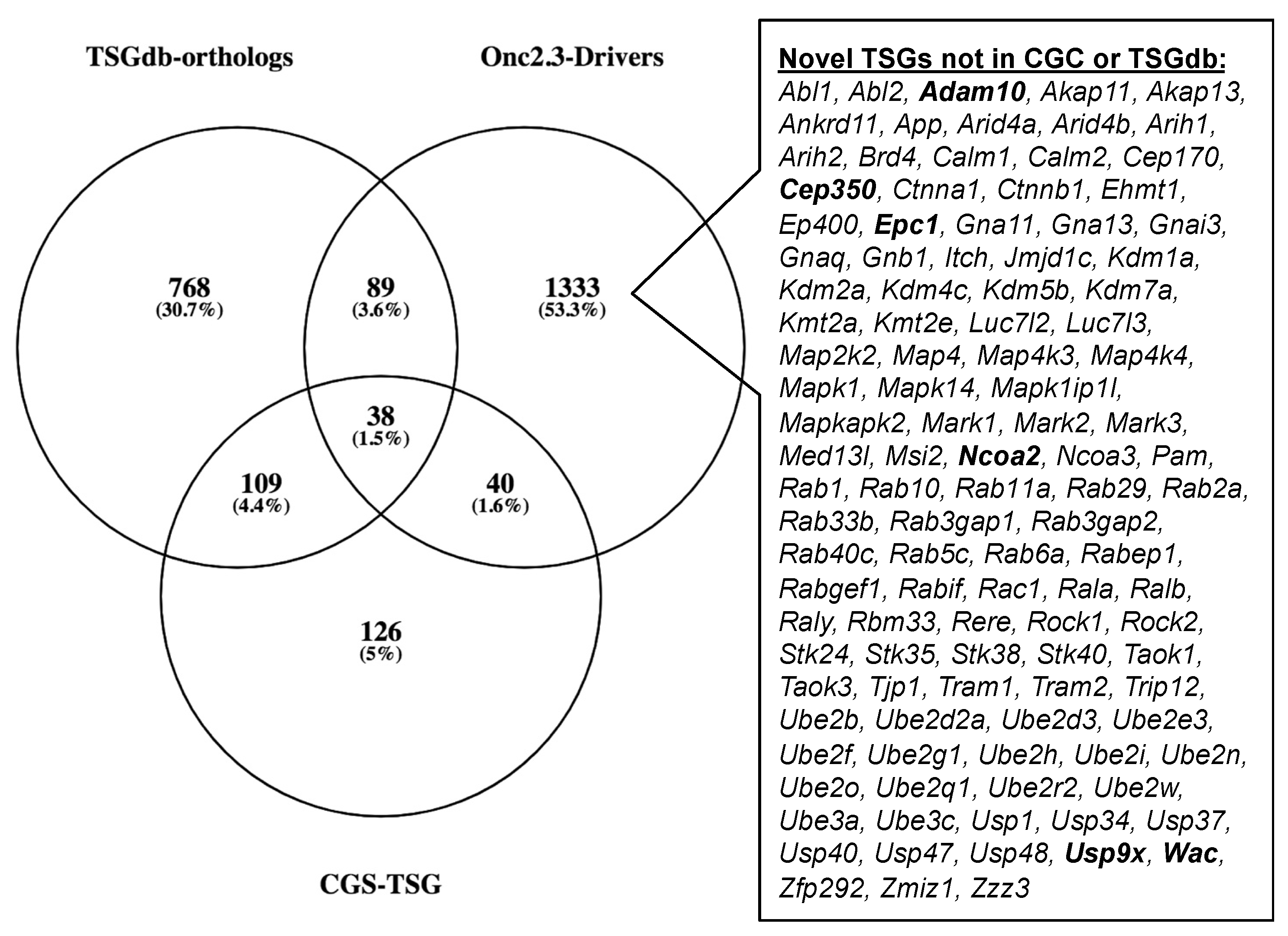
2.2.5. Common Pathways Selected in Response to Whole Genome Tumor Suppressor Gene Inactivation
3. Discussion
4. Materials and Methods
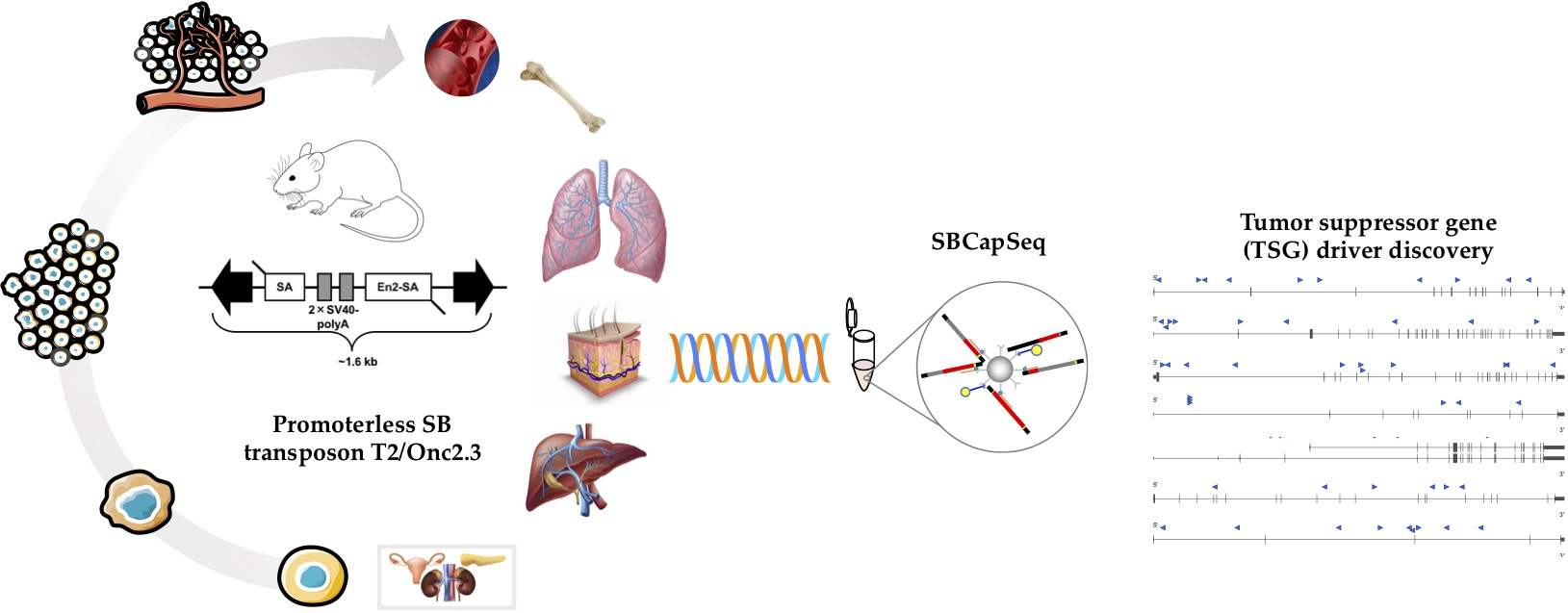
4.1. Generation of pT2/Onc2.3 Vector
 A ACT TGT TTA TTG CAG CTT ATA ATG GTT ACA AAT AAA GCA ATA GCA TCA C-3′ and PCR SuperMix Taq (Invitrogen, Waltham, MA, USA, #10572014), we amplified a 128 bp fragment (5′-ATG CAT GCA TCC CGG GCA TCG ATG CAG TGA AAA AAA TGC TTT ATT TGT GAA ATT TGT GAT GCT ATT GCT TTA TTT GTA ACC ATT ATA AGC TGC AAT AAA CAA GTT
A ACT TGT TTA TTG CAG CTT ATA ATG GTT ACA AAT AAA GCA ATA GCA TCA C-3′ and PCR SuperMix Taq (Invitrogen, Waltham, MA, USA, #10572014), we amplified a 128 bp fragment (5′-ATG CAT GCA TCC CGG GCA TCG ATG CAG TGA AAA AAA TGC TTT ATT TGT GAA ATT TGT GAT GCT ATT GCT TTA TTT GTA ACC ATT ATA AGC TGC AAT AAA CAA GTT  CGA ATT CAT GCA TGC AT-3′) using at thermal cycler program: template denaturation at 94 °C for 90 s followed by 30 cycles of 94 °C for 30 s, 50 °C for 45 s, and 72 °C for 45 s followed by 72 °C for 300 s and cooled to 4 °C. PCR product was visually confirmed by running the PCR product on a 2% agarose TBE gel stained with EtBr viewed under UV light. A small piece of gel containing the linear PCR product was excised, purified using GFX columns (Amersham Biosciences #414581), and used to ligate into pGEM®-T Easy Vector System (Promega, Madison, WI, USA, #A1360) at room temperature for 60 m using the manufacturer’s suggested protocol. Ligation products were electroporated into MAX efficiency DH10B competent cells (Invitrogen #18297010), plated on LB agar plates with ampicillin, and incubated 37 °C for 18 h. Miniprep DNA from selected transformants was sequentially digested with XmaI (NEB) and EcoRI (NEB), at 37 °C for 30 m each, per manufacturer’s suggested protocol. The ligation product was run on a 2% agarose TBE gel stained with EtBr viewed under UV light, and a small piece of gel containing the 108 bp fragment was excised, purified using GFX columns (Amersham Biosciences #414581) prior to ligation. Finally, a ligation reaction using a 1:5 ratio of pT2/Onc2:XmaI-EcoRI linear vector and SV40-polyA: XmaI-EcoRI insert, respectively, was performed using T4 DNA Ligase at room temperature for 30 m, followed by heat inactivation at 65 °C for 20 m. Ligation products were electroporated into MAX efficiency DH10B competent cells (Invitrogen #18297010), plated on LB agar plates with ampicillin, and incubated at 37 °C for 18 h. Miniprep DNA from selected transformants was digested with HindIII (NEB), at 37 °C for 60 m each, per manufacturer’s suggested protocol. Recombinant pT2/Onc2.2 vector was identified by the presence of three fragments at 3.658 kb, 0.8 kb, and 0.6 kb fragments. pT2/Onc2.2 vector DNA was cut with restriction enzyme AscI (NEB #R0558S), at 37 °C for 60 m, to linearize the plasmid. Linear plasmid was visually confirmed on a 1% agarose TAE gel stained with EtBr viewed under UV light. A small piece of agarose gel containing the linear pT2/Onc2.2 vector was excised and purified using GFX columns (Amersham Biosciences #414581). PCR amplification was performed using 100 ng of the purified linearized pT2-Onc2.2 vector template and a pair of oligos T2/Onc2.f1 5′-ATG CGA ATT CAA CGC GCG TTA AGA TAC ATT GA-3′ engineered to contain an EcoRI site (underlined) and T2/Onc2.r1 5′-ATC TAT GGC TCG TAC TCT ATA GGC-3′ designed to amplify the pT2/Onc2 vector excluding the MSCV 5′ LTR minimal promoter and Lun-SD sequences using platinum Hi-Fi DNA Taq polymerase (Invitrogen #11304-028) in 1X Hi-Fi PCR buffer (Invitrogen #52045) supplemented with 2 µL of 50 mM MgSO4 and dNTPs (Roche, Basel, Switzerland, #11-581-295-001) and thermal cycler program: template denaturation at 95 °C for 90 s followed by 30 cycles of 95 °C for 45 s, 55 °C for 60 s, and 72 °C for 270 s followed by 72 °C for 600 s and cooled to 37 °C. Then, 1 unit of restriction enzyme DpnI (NEB #R0176S) was added (to digest the methylated plasmid from vector template grown in bacteria and enrich PCR amplified template during ligation) to each PCR tube and incubated at 37 °C for 30 m, followed by heat inactivation at 80 °C for 20 m, and then cooled to 4 °C. The PCR product was column purified, resuspended in ddH2O, and digested with EcoRI (NEB) at 37 °C for 60 m (one EcoRI site was added by the oligo and the other occurs within the pT2/Onc2.2 vector at position 819). Gel and column purified pT2/Onc2.3 linear vector (4.395 kb) was self-ligated using T4 DNA ligase at room temperature for 30 m, followed by heat inactivation at 65 °C for 20 m. Ligation products were electroporated into MAX efficiency DH10B competent cells, plated on LB agar plates with ampicillin, and incubated at 37 °C for 18 h. Miniprep DNA from selected transformants was digested with HindIII (NEB), at 37 °C for 60 m each, per manufacturer’s suggested protocol. Recombinant pT2/Onc2.3 vector (Figure S1a) was identified by the presence of two fragments at 3.701 kb and 0.694 kb fragments. Cloning maps and sequence files may be downloaded from figshare http://dx.doi.org/10.6084/m9.figshare.12816452 [67]. Several pT2/Onc2.3 vector clones were sequenced using BigDye® direct cycle sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA), using the manufacturer’s suggested protocol, and the following primers: T7, 5′-GTAATACGACTCACTATAGGG-3′; T2/Onc.sp5.1, 5′-GACTGTGCCTTTAAACAGCTTGG-3′; T2/Onc.sp5.2, 5′-TCCTGTGCCAGACTCTGGCGC-3′; and T2/Onc.sp5.3, 5′-GGGTGGTGATATAAACTTGAGGCTG-3′. A single pT2/Onc2.3 clone with full sequence identity to the vector map was re-electroporated into MAX efficiency DH10B competent cells (Invitrogen #18297010), plated on LB agar plates with ampicillin, and incubated at 37 °C for 18 h.
CGA ATT CAT GCA TGC AT-3′) using at thermal cycler program: template denaturation at 94 °C for 90 s followed by 30 cycles of 94 °C for 30 s, 50 °C for 45 s, and 72 °C for 45 s followed by 72 °C for 300 s and cooled to 4 °C. PCR product was visually confirmed by running the PCR product on a 2% agarose TBE gel stained with EtBr viewed under UV light. A small piece of gel containing the linear PCR product was excised, purified using GFX columns (Amersham Biosciences #414581), and used to ligate into pGEM®-T Easy Vector System (Promega, Madison, WI, USA, #A1360) at room temperature for 60 m using the manufacturer’s suggested protocol. Ligation products were electroporated into MAX efficiency DH10B competent cells (Invitrogen #18297010), plated on LB agar plates with ampicillin, and incubated 37 °C for 18 h. Miniprep DNA from selected transformants was sequentially digested with XmaI (NEB) and EcoRI (NEB), at 37 °C for 30 m each, per manufacturer’s suggested protocol. The ligation product was run on a 2% agarose TBE gel stained with EtBr viewed under UV light, and a small piece of gel containing the 108 bp fragment was excised, purified using GFX columns (Amersham Biosciences #414581) prior to ligation. Finally, a ligation reaction using a 1:5 ratio of pT2/Onc2:XmaI-EcoRI linear vector and SV40-polyA: XmaI-EcoRI insert, respectively, was performed using T4 DNA Ligase at room temperature for 30 m, followed by heat inactivation at 65 °C for 20 m. Ligation products were electroporated into MAX efficiency DH10B competent cells (Invitrogen #18297010), plated on LB agar plates with ampicillin, and incubated at 37 °C for 18 h. Miniprep DNA from selected transformants was digested with HindIII (NEB), at 37 °C for 60 m each, per manufacturer’s suggested protocol. Recombinant pT2/Onc2.2 vector was identified by the presence of three fragments at 3.658 kb, 0.8 kb, and 0.6 kb fragments. pT2/Onc2.2 vector DNA was cut with restriction enzyme AscI (NEB #R0558S), at 37 °C for 60 m, to linearize the plasmid. Linear plasmid was visually confirmed on a 1% agarose TAE gel stained with EtBr viewed under UV light. A small piece of agarose gel containing the linear pT2/Onc2.2 vector was excised and purified using GFX columns (Amersham Biosciences #414581). PCR amplification was performed using 100 ng of the purified linearized pT2-Onc2.2 vector template and a pair of oligos T2/Onc2.f1 5′-ATG CGA ATT CAA CGC GCG TTA AGA TAC ATT GA-3′ engineered to contain an EcoRI site (underlined) and T2/Onc2.r1 5′-ATC TAT GGC TCG TAC TCT ATA GGC-3′ designed to amplify the pT2/Onc2 vector excluding the MSCV 5′ LTR minimal promoter and Lun-SD sequences using platinum Hi-Fi DNA Taq polymerase (Invitrogen #11304-028) in 1X Hi-Fi PCR buffer (Invitrogen #52045) supplemented with 2 µL of 50 mM MgSO4 and dNTPs (Roche, Basel, Switzerland, #11-581-295-001) and thermal cycler program: template denaturation at 95 °C for 90 s followed by 30 cycles of 95 °C for 45 s, 55 °C for 60 s, and 72 °C for 270 s followed by 72 °C for 600 s and cooled to 37 °C. Then, 1 unit of restriction enzyme DpnI (NEB #R0176S) was added (to digest the methylated plasmid from vector template grown in bacteria and enrich PCR amplified template during ligation) to each PCR tube and incubated at 37 °C for 30 m, followed by heat inactivation at 80 °C for 20 m, and then cooled to 4 °C. The PCR product was column purified, resuspended in ddH2O, and digested with EcoRI (NEB) at 37 °C for 60 m (one EcoRI site was added by the oligo and the other occurs within the pT2/Onc2.2 vector at position 819). Gel and column purified pT2/Onc2.3 linear vector (4.395 kb) was self-ligated using T4 DNA ligase at room temperature for 30 m, followed by heat inactivation at 65 °C for 20 m. Ligation products were electroporated into MAX efficiency DH10B competent cells, plated on LB agar plates with ampicillin, and incubated at 37 °C for 18 h. Miniprep DNA from selected transformants was digested with HindIII (NEB), at 37 °C for 60 m each, per manufacturer’s suggested protocol. Recombinant pT2/Onc2.3 vector (Figure S1a) was identified by the presence of two fragments at 3.701 kb and 0.694 kb fragments. Cloning maps and sequence files may be downloaded from figshare http://dx.doi.org/10.6084/m9.figshare.12816452 [67]. Several pT2/Onc2.3 vector clones were sequenced using BigDye® direct cycle sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA), using the manufacturer’s suggested protocol, and the following primers: T7, 5′-GTAATACGACTCACTATAGGG-3′; T2/Onc.sp5.1, 5′-GACTGTGCCTTTAAACAGCTTGG-3′; T2/Onc.sp5.2, 5′-TCCTGTGCCAGACTCTGGCGC-3′; and T2/Onc.sp5.3, 5′-GGGTGGTGATATAAACTTGAGGCTG-3′. A single pT2/Onc2.3 clone with full sequence identity to the vector map was re-electroporated into MAX efficiency DH10B competent cells (Invitrogen #18297010), plated on LB agar plates with ampicillin, and incubated at 37 °C for 18 h.4.2. Generation of T2/Onc2.3 Transgenic Mice
4.3. Histological Analysis
4.4. Mapping SB Insertion Sites
4.5. SB Driver Analysis Discovery, Progression and Trunk Drivers
4.6. qRT-PCR
4.7. Software
4.8. URLs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Collier, L.S.; Largaespada, D.A. Transposons for cancer gene discovery: Sleeping Beauty and beyond. Genome Biol. 2007, 8 (Suppl. 1), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer 2010, 10, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.J.; Akagi, K.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 2005, 436, 221–226. [Google Scholar] [CrossRef]
- Dupuy, A.J.; Rogers, L.M.; Kim, J.; Nannapaneni, K.; Starr, T.K.; Liu, P.; Largaespada, D.A.; Scheetz, T.E.; Jenkins, N.A.; Copeland, N.G. A modified Sleeping Beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009, 69, 8150–8156. [Google Scholar] [CrossRef]
- Mann, K.M.; Newberg, J.Y.; Black, M.A.; Jones, D.J.; Amaya-Manzanares, F.; Guzman-Rojas, L.; Kodama, T.; Ward, J.M.; Rust, A.G.; van der Weyden, L.; et al. Analyzing tumor heterogeneity and driver genes in single myeloid leukemia cells with SBCapSeq. Nat. Biotechnol. 2016, 34, 962–972. [Google Scholar] [CrossRef]
- Mann, K.M.; Ward, J.M.; Yew, C.C.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Australian Pancreatic Cancer Genome Initiative; et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941. [Google Scholar] [CrossRef]
- Mann, M.B.; Black, M.A.; Jones, D.J.; Ward, J.M.; Yew, C.C.; Newberg, J.Y.; Dupuy, A.J.; Rust, A.G.; Bosenberg, M.W.; McMahon, M.; et al. Transposon mutagenesis identifies genetic drivers of Braf (V600E) melanoma. Nat. Genet. 2015, 47, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Jenkins, N.A.; Copeland, N.G.; Mann, M.B. Transposon insertional mutagenesis models of cancer. Cold Spring Harb. Protoc. 2014, 3, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Rangel, R.; Lee, S.C.; Hon-Kim Ban, K.; Guzman-Rojas, L.; Mann, M.B.; Newberg, J.Y.; Kodama, T.; McNoe, L.A.; Selvanesan, L.; Ward, J.M.; et al. Transposon mutagenesis identifies genes that cooperate with mutant Pten in breast cancer progression. Proc. Natl. Acad. Sci. USA 2016, 113, E7749–E7758. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Wei, Z.; Koso, H.; Rust, A.G.; Yew, C.C.; Mann, M.B.; Ward, J.M.; Adams, D.J.; Copeland, N.G.; Jenkins, N.A. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat. Genet. 2015, 47, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Jenkins, N.A.; Copeland, N.G.; Mann, K.M. Sleeping Beauty mutagenesis: Exploiting forward genetic screens for cancer gene discovery. Curr. Opin. Genet. Dev. 2014, 24, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Aiderus, A.; Newberg, J.Y.; Guzman-Rojas, L.; Contreras-Sandoval, A.M.; Meshey, A.L.; Jones, D.J.; Amaya-Manzanares, F.; Rangel, R.; Ward, J.M.; et al. Supplementary Tables 1–43 for Transposon mutagenesis identifies cooperating genetic drivers during keratinocyte transformation and cutaneous squamous cell carcinoma progression. Figshare 2020. Available online: https://dx.doi.org/10.35092/yhjc.11441130 (accessed on 30 September 2020).
- Aiderus, A.; Newberg, J.Y.; Guzman-Rojas, L.; Contreras-Sandoval, A.M.; Meshey, A.L.; Jones, D.J.; Amaya-Manzanares, F.; Rangel, R.; Ward, J.M.; Lee, S.-C.; et al. Transposon mutagenesis identifies cooperating genetic drivers during keratinocyte transformation and cutaneous squamous cell carcinoma progression. bioRxiv 2019. Available online: https://dx.doi.org/10.1101/2019.12.24.887968 (accessed on 30 September 2020).
- Newberg, J.Y.; Black, M.A.; Jenkins, N.A.; Copeland, N.G.; Mann, K.M.; Mann, M.B. SB Driver Analysis: A Sleeping Beauty cancer driver analysis framework for identifying and prioritizing experimentally actionable oncogenes and tumor suppressors. Nucleic Acids Res. 2018, 46, e94. [Google Scholar] [CrossRef]
- Izsvak, Z.; Ivics, Z. Sleeping beauty transposition: Biology and applications for molecular therapy. Mol. Ther. 2004, 9, 147–156. [Google Scholar] [CrossRef]
- Izsvak, Z.; Ivics, Z.; Plasterk, R.H. Sleeping Beauty, a wide host-range transposon vector for genetic transformation in vertebrates. J. Mol. Biol. 2000, 302, 93–102. [Google Scholar] [CrossRef]
- Aiderus, A.; Contreras-Sandoval, A.M.; Meshey, A.L.; Newberg, J.; Ward, J.M.; Swing, D.; Copeland, N.G.; Jenkins, N.A.; Mann, K.M.; Mann, M.B. Table 1: SB Allele-Specific Tumor Spectrum by Whole-Body, Transposon-Mediated Mutagenesis in Wild Type Mice. 2020. Available online: https://dx.doi.org/10.6084/m9.figshare.12811685 (accessed on 30 September 2020).
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Johnson, A.; Albacker, L.; Wang, K.; Chmielecki, J.; Frampton, G.; Gay, L.; Elvin, J.A.; Vergilio, J.A.; Ali, S.; et al. Comprehensive Genomic Profiling Facilitates Implementation of the National Comprehensive Cancer Network Guidelines for Lung Cancer Biomarker Testing and Identifies Patients Who May Benefit From Enrollment in Mechanism-Driven Clinical Trials. Oncologist 2016, 21, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; Delrosario, R.; Jen, K.Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015, 517, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Chang, X.; Brait, M.; Fertig, E.; Kagohara, L.T.; Bedi, A.; Marchionni, L.; Agrawal, N.; Ravi, R.; Jones, S.; et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat. Commun. 2015, 6, 8258. [Google Scholar] [CrossRef]
- Liang, Y.N.; Liu, Y.; Meng, Q.; Li, X.; Wang, F.; Yao, G.; Wang, L.; Fu, S.; Tong, D. RBMS3 is a tumor suppressor gene that acts as a favorable prognostic marker in lung squamous cell carcinoma. Med. Oncol. 2015, 32, 459. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Sun, Z.; Habib, G.M.; Lieberman, M.W.; Hannink, M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J. Biol. Chem. 2005, 280, 30091–30099. [Google Scholar] [CrossRef]
- Newberg, J.Y.; Mann, K.M.; Mann, M.B.; Jenkins, N.A.; Copeland, N.G. SBCDDB: Sleeping Beauty Cancer Driver Database for gene discovery in mouse models of human cancers. Nucleic Acids Res. 2018, 46, D1011–D1017. [Google Scholar] [CrossRef]
- Bard-Chapeau, E.A.; Nguyen, A.T.; Rust, A.G.; Sayadi, A.; Lee, P.; Chua, B.Q.; New, L.S.; de Jong, J.; Ward, J.M.; Chin, C.K.; et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat. Genet. 2014, 46, 24–32. [Google Scholar] [CrossRef]
- Keng, V.W.; Sia, D.; Sarver, A.L.; Tschida, B.R.; Fan, D.; Alsinet, C.; Sole, M.; Lee, W.L.; Kuka, T.P.; Moriarity, B.S.; et al. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology 2013, 57, 120–130. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Keng, V.W.; York, B.; Reineke, E.L.; Seo, D.; Fan, D.; Silverstein, K.A.; Schrum, C.T.; Xie, W.R.; Mularoni, L.; et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E1377–E1386. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.M.; Olivier, A.K.; Meyerholz, D.K.; Dupuy, A.J. Adaptive immunity does not strongly suppress spontaneous tumors in a Sleeping Beauty model of cancer. J. Immunol. 2013, 190, 4393–4399. [Google Scholar] [CrossRef] [PubMed]
- Aiderus, A.; Contreras-Sandoval, A.M.; Meshey, A.L.; Newberg, J.Y.; Ward, J.M.; Swing, D.; Copeland, N.G.; Jenkins, N.A.; Mann, K.M.; Mann, M.B. SB-Onc2.3-Pan-TSG Trunk Driver SB insertion maps for “Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation”. Figshare 2020. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Tang, G.; Bindal, N.; Bamford, S.; Dawson, E.; Cole, C.; Kok, C.Y.; Jia, M.; Ewing, R.; Menzies, A.; et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010, 38, D652–D657. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, J.; Zhao, Z. TSGene: A web resource for tumor suppressor genes. Nucleic Acids Res. 2013, 41, D970–D976. [Google Scholar] [CrossRef]
- Perez-Mancera, P.A.; Rust, A.G.; van der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.; Grutzmann, R.; Aust, D.; Rummele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef]
- Collier, L.S.; Carlson, C.M.; Ravimohan, S.; Dupuy, A.J.; Largaespada, D.A. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 2005, 436, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [PubMed]
- de la Rosa, J.; Weber, J.; Friedrich, M.J.; Li, Y.; Rad, L.; Ponstingl, H.; Liang, Q.; de Quiros, S.B.; Noorani, I.; Metzakopian, E.; et al. A single-copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat. Genet. 2017, 49, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; de la Rosa, J.; Grove, C.S.; Schick, M.; Rad, L.; Baranov, O.; Strong, A.; Pfaus, A.; Friedrich, M.J.; Engleitner, T.; et al. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat. Commun. 2019, 10, 1415. [Google Scholar] [CrossRef] [PubMed]
- Katigbak, A.; Cencic, R.; Robert, F.; Senecha, P.; Scuoppo, C.; Pelletier, J. A CRISPR/Cas9 Functional Screen Identifies Rare Tumor Suppressors. Sci. Rep. 2016, 6, 38968. [Google Scholar] [CrossRef]
- Zender, L.; Xue, W.; Zuber, J.; Semighini, C.P.; Krasnitz, A.; Ma, B.; Zender, P.; Kubicka, S.; Luk, J.M.; Schirmacher, P.; et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008, 135, 852–864. [Google Scholar] [CrossRef]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 12601. [Google Scholar] [CrossRef]
- Molina-Sanchez, P.; Lujambio, A. Experimental Models for Preclinical Research in Hepatocellular Carcinoma. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Springer: Cham, Switzerland, 2019; pp. 333–358. [Google Scholar] [CrossRef]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef]
- Cho, S.J.; Yoon, C.; Lee, J.H.; Chang, K.K.; Lin, J.X.; Kim, Y.H.; Kook, M.C.; Aksoy, B.A.; Park, D.J.; Ashktorab, H.; et al. KMT2C Mutations in Diffuse-Type Gastric Adenocarcinoma Promote Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2018, 24, 6556–6569. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Zhao, D.; Lin, T.; Gu, B.; Pal, K.; Wu, S.J.; Alam, H.; Lv, J.; Yun, K.; Gopalakrishnan, V.; et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell 2018, 70, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Augert, A.; Kim, D.W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.B.; Dunn, C.T.; Pillai, S.P.S.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alver, B.H.; San Roman, A.K.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tian, Y.; Zhang, J.; Tong, X.; Huang, H.; Li, S.; Zhao, H.; Tang, Y.; Yuan, C.; Wang, K.; et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E3978–E3986. [Google Scholar] [CrossRef] [PubMed]
- Ichise, T.; Yoshida, N.; Ichise, H. CBP/p300 antagonises EGFR-Ras-Erk signalling and suppresses increased Ras-Erk signalling-induced tumour formation in mice. J. Pathol. 2019, 249, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, M.; Xu, E.S.; Luo, L.; Ma, Y.; Huang, W.; Floyd, W.; Klann, T.S.; Kim, S.Y.; Gersbach, C.A.; et al. Genome-wide CRISPR Screen to Identify Genes that Suppress Transformation in the Presence of Endogenous Kras(G12D). Sci. Rep. 2019, 9, 17220. [Google Scholar] [CrossRef]
- Kas, S.M.; de Ruiter, J.R.; Schipper, K.; Schut, E.; Bombardelli, L.; Wientjens, E.; Drenth, A.P.; de Korte-Grimmerink, R.; Mahakena, S.; Phillips, C.; et al. Transcriptomics and Transposon Mutagenesis Identify Multiple Mechanisms of Resistance to the FGFR Inhibitor AZD4547. Cancer Res. 2018, 78, 5668–5679. [Google Scholar] [CrossRef]
- Hayashi, T.; Desmeules, P.; Smith, R.S.; Drilon, A.; Somwar, R.; Ladanyi, M. RASA1 and NF1 are Preferentially Co-Mutated and Define A Distinct Genetic Subset of Smoking-Associated Non-Small Cell Lung Carcinomas Sensitive to MEK Inhibition. Clin. Cancer Res. 2018, 24, 1436–1447. [Google Scholar] [CrossRef]
- Kodama, T.; Yi, J.; Newberg, J.Y.; Tien, J.C.; Wu, H.; Finegold, M.J.; Kodama, M.; Wei, Z.; Tamura, T.; Takehara, T.; et al. Molecular profiling of nonalcoholic fatty liver disease-associated hepatocellular carcinoma using SB transposon mutagenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E10417–E10426. [Google Scholar] [CrossRef]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Dorr, C.; Janik, C.; Weg, M.; Been, R.A.; Bader, J.; Kang, R.; Ng, B.; Foran, L.; Landman, S.R.; O’Sullivan, M.G.; et al. Transposon Mutagenesis Screen Identifies Potential Lung Cancer Drivers and CUL3 as a Tumor Suppressor. Mol. Cancer Res. 2015, 13, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Larrieu, D.; Brunet, M.; Vargas, C.; Hanoun, N.; Ligat, L.; Dagnon, L.; Lulka, H.; Pommier, R.M.; Selves, J.; Jady, B.E.; et al. The E3 ubiquitin ligase TRIP12 participates in cell cycle progression and chromosome stability. Sci. Rep. 2020, 10, 789. [Google Scholar] [CrossRef] [PubMed]
- Higa, K.C.; DeGregori, J. Decoy fitness peaks, tumor suppression, and aging. Aging Cell 2019, 18, e12938. [Google Scholar] [CrossRef]
- Mann, M.B.; Mann, K.M. SB-Onc2.3 molecular cloning maps and files. Figshare 2020. Available online: https://doi.org/10.6084/m9.figshare.12816452 (accessed on 30 September 2020).
- Mann, M.B.; Mann, K.M. SB-Onc2.3 multiplex PCR genotyping protocol. Figshare 2020. Available online: https://doi.org/10.6084/m9.figshare.12811301 (accessed on 30 September 2020).
- Lewandoski, M.; Meyers, E.N.; Martin, G.R. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb. Symp. Quant. Biol. 1997, 62, 159–168. [Google Scholar]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Aiderus, A.; Contreras-Sandoval, A.M.; Meshey, A.L.; Newberg, J.Y.; Ward, J.M.; Swing, D.; Copeland, N.G.; Jenkins, N.A.; Mann, K.M.; Mann, M.B. Onc2.3 pathology images for “Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation”. Figshare 2020. Available online: https://dx.doi.org/10.6084/m9.figshare.12816305 (accessed on 30 September 2020).
- Mann, M.B.; Mann, K.M.; Contreras-Sandoval, A.M.; Guzman-Rojas, L.; Newberg, J.Y.; Meshey, A.L.; Jones, D.J.; Amaya-Manzanares, F.; Copeland, N.G.; Jenkins, N.A. SBCapSeq Protocol manuscript files for “Quantifying tumor heterogeneity, clonal dynamics, and cancer driver gene evolution from Sleeping Beauty transposon mutagenesis models using SBCapSeq”. Natl. Inst. Health Dataset 2019. Available online: https://dx.doi.org/10.35092/yhjc.11441001 (accessed on 30 September 2020).
- Mann, K.M.; Mann, M.B.; Guzman-Rojas, L.; Amaya-Manzanares, F.; Jones, D.J.; Newberg, J.Y.; Jenkins, N.A.; Copeland, N.G. SBCapSeq Protocol: A method for selective cloning of Sleeping Beauty transposon insertions using liquid capture hybridization and Ion Torrent semiconductor sequencing. Protoc. Exch. 2016. [Google Scholar] [CrossRef]
- March, H.N.; Rust, A.G.; Wright, N.A.; ten Hoeve, J.; de Ridder, J.; Eldridge, M.; van der Weyden, L.; Berns, A.; Gadiot, J.; Uren, A.; et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 2011, 43, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Mann, K.M. SB-Onc2.3 donor chromosome dataset. Figshare 2020. Available online: https://dx.doi.org/10.6084/m9.figshare.12816191 (accessed on 30 December 2020).
- Aiderus, A.; Contreras-Sandoval, A.M.; Meshey, A.L.; Newberg, J.Y.; Ward, J.M.; Swing, D.; Copeland, N.G.; Jenkins, N.A.; Mann, K.M.; Mann, M.B. Supplementary Notes 1–3 for “Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation”. Figshare 2020. Available online: https://dx.doi.org/10.6084/m9.figshare.12811979 (accessed on 30 September 2020).
- Aiderus, A.; Contreras-Sandoval, A.M.; Meshey, A.L.; Newberg, J.Y.; Ward, J.M.; Swing, D.; Copeland, N.G.; Jenkins, N.A.; Mann, K.M.; Mann, M.B. Supplementary Tables 1–24 for “Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation”. Figshare 2020. Available online: https://dx.doi.org/10.6084/m9.figshare.12811772 (accessed on 30 September 2020).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SB Cohort | SB-Onc2.3 | SB-Onc3 | SB-Onc2 | |||
|---|---|---|---|---|---|---|
| Promoter | no promoter | CAG promoter | MSCV promoter | |||
| Reference | This study | Aiderus et al., 2019 | Mann et al., 2016 | |||
| Tumor classification | tumors | mice | tumors | mice | tumors | mice |
| Skin Squamous Cell Carcinoma | 5 | 4 (3%) | 52 | 20 (24%) | 0 | 0 (0%) |
| Skin Keratoacanthoma | 1 | 1 (1%) | 9 | 8 (10%) | 0 | 0 (0%) |
| Hepatocellular Carcinoma | 2 | 2 (2%) | 9 | 9 (11%) | 0 | 0 (0%) |
| Hepatocellular Adenoma | 4 | 3 (2%) | 79 | 42 (50%) | 0 | 0 (0%) |
| Adenoma, Multiple | 25 | 16 (12%) | 39 | 31 (37%) | 0 | 0 (0%) |
| Adenocarcinoma, Multiple | 3 | 3 (2%) | 9 | 8 (10%) | 0 | 0 (0%) |
| Carcinoma, Multiple | 0 | 0 (0%) | 6 | 6 (7%) | 0 | 0 (0%) |
| Sarcoma | 0 | 0 (0%) | 6 | 6 (7%) | 0 | 0 (0%) |
| Papilloma | 1 | 1 (1%) | 4 | 3 (4%) | 0 | 0 (0%) |
| Astrocytoma | 34 | 34 (26%) | 3 | 3 (4%) | 0 | 0 (0%) |
| Hemangiosarcoma | 0 | 0 (0%) | 3 | 3 (4%) | 0 | 0 (0%) |
| Hemangioma | 1 | 1 (1%) | 2 | 2 (2%) | 0 | 0 (0%) |
| Fibrosarcoma | 0 | 0 (0%) | 2 | 2 (2%) | 0 | 0 (0%) |
| Metastasis | 0 | 0 (0%) | 2 | 2 (2%) | 0 | 0 (0%) |
| Mast Cell Tumor | 0 | 0 (0%) | 1 | 1 (1%) | 0 | 0 (0%) |
| Trichoepithelioma | 0 | 0 (0%) | 1 | 1 (1%) | 0 | 0 (0%) |
| Schwannoma | 1 | 1 (2%) | 0 | 0 (0%) | 0 | 0 (0%) |
| Hepatoblastoma | 1 | 1 (1%) | 0 | 0 (0%) | 0 | 0 (0%) |
| Medulloblastoma | 0 | 0 (0%) | 0 | 0 (0%) | 6 | 6 (8%) |
| Lymphoma | 1 | 1 (2%) | 2 | 2 (2%) | 28 | 28 (36%) |
| Leukemia | 0 | 0 (0%) | 0 | 0 (0%) | 78 | 78 (100%) |
| Histiocytic Sarcoma | 0 | 0 (0%) | 8 | 8 (10%) | 0 | 0 (0%) |
| Totals | 79 | 107 | 237 | 87 | 112 | 78 |
| Average tumors per mouse | 0.74 | 2.72 | 1.44 | |||
| Average tumor latency (days) | 450 | 330 | 70 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiderus, A.; Contreras-Sandoval, A.M.; Meshey, A.L.; Newberg, J.Y.; Ward, J.M.; Swing, D.A.; Copeland, N.G.; Jenkins, N.A.; Mann, K.M.; Mann, M.B. Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation. Cancers 2021, 13, 225. https://doi.org/10.3390/cancers13020225
Aiderus A, Contreras-Sandoval AM, Meshey AL, Newberg JY, Ward JM, Swing DA, Copeland NG, Jenkins NA, Mann KM, Mann MB. Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation. Cancers. 2021; 13(2):225. https://doi.org/10.3390/cancers13020225
Chicago/Turabian StyleAiderus, Aziz, Ana M. Contreras-Sandoval, Amanda L. Meshey, Justin Y. Newberg, Jerrold M. Ward, Deborah A. Swing, Neal G. Copeland, Nancy A. Jenkins, Karen M. Mann, and Michael B. Mann. 2021. "Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation" Cancers 13, no. 2: 225. https://doi.org/10.3390/cancers13020225
APA StyleAiderus, A., Contreras-Sandoval, A. M., Meshey, A. L., Newberg, J. Y., Ward, J. M., Swing, D. A., Copeland, N. G., Jenkins, N. A., Mann, K. M., & Mann, M. B. (2021). Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation. Cancers, 13(2), 225. https://doi.org/10.3390/cancers13020225