Simple Summary
Protein phosphorylation is a key regulatory mechanism that controls a wide variety of cellular responses. This process is catalysed by the members of the protein kinase superfamily that are classified into two main families based on their ability to phosphorylate either tyrosine or serine and threonine residues in their substrates. Massive research efforts have been invested in dissecting the functions of tyrosine kinases, revealing their importance in the initiation and progression of human malignancies. Based on these investigations, numerous tyrosine kinase inhibitors have been included in clinical protocols and proved to be effective in targeted therapies for various haematological malignancies. In this review, we provide insights into the role of tyrosine kinases in leukaemia and discuss their targeting for therapeutic purposes with the currently available inhibitory compounds.
Abstract
Protein kinases constitute a large group of enzymes catalysing protein phosphorylation and controlling multiple signalling events. The human protein kinase superfamily consists of 518 members and represents a complicated system with intricate internal and external interactions. Protein kinases are classified into two main families based on the ability to phosphorylate either tyrosine or serine and threonine residues. Among the 90 tyrosine kinase genes, 58 are receptor types classified into 20 groups and 32 are of the nonreceptor types distributed into 10 groups. Tyrosine kinases execute their biological functions by controlling a variety of cellular responses, such as cell division, metabolism, migration, cell–cell and cell matrix adhesion, cell survival and apoptosis. Over the last 30 years, a major focus of research has been directed towards cancer-associated tyrosine kinases owing to their critical contributions to the development and aggressiveness of human malignancies through the pathological effects on cell behaviour. Leukaemia represents a heterogeneous group of haematological malignancies, characterised by an uncontrolled proliferation of undifferentiated hematopoietic cells or leukaemia blasts, mostly derived from bone marrow. They are usually classified as chronic or acute, depending on the rates of their progression, as well as myeloid or lymphoblastic, according to the type of blood cells involved. Overall, these malignancies are relatively common amongst both children and adults. In malignant haematopoiesis, multiple tyrosine kinases of both receptor and nonreceptor types, including AXL receptor tyrosine kinase (AXL), Discoidin domain receptor 1 (DDR1), Vascular endothelial growth factor receptor (VEGFR), Fibroblast growth factor receptor (FGFR), Mesenchymal–epithelial transition factor (MET), proto-oncogene c-Src (SRC), Spleen tyrosine kinase (SYK) and pro-oncogenic Abelson tyrosine-protein kinase 1 (ABL1) mutants, are implicated in the pathogenesis and drug resistance of practically all types of leukaemia. The role of ABL1 kinase mutants and their therapeutic inhibitors have been extensively analysed in scientific literature, and therefore, in this review, we provide insights into the impact and mechanism of action of other tyrosine kinases involved in the development and progression of human leukaemia and discuss the currently available and emerging treatment options based on targeting these molecules.
1. Introduction
Protein kinases are a large family of enzymes that catalyse the phosphorylation of protein molecules. While kinases act as enzymes that transfer a phosphate group to a target molecule, phosphatases are enzymes with an opposite function to remove a phosphate group from a protein. These two enzymatic processes play a key role in the cell by regulating numerous signalling pathways in response to an external stimulus [1]. The human protein kinase gene family, which consists of 518 members, along with 106 pseudogenes, is the second largest enzyme family and the fifth-largest gene family in humans [2]. The deregulation of kinase activity results in the dramatic change of different cellular responses, such as cell cycle regulation, growth, differentiation, proliferation, survival, apoptosis, migration and several others [3]. As protein phosphorylation is one of the most common and important post-translational modifications, protein kinases are involved in multiple key regulatory pathways in the cell; deregulation or loss of their activities are often associated with the development of various types of cancers [4,5]. Further, genome-wide studies have provided key insights into the role of genetically inherited variants of specific kinases in several stages of cancer, including initiation, promotion, progression and recurrence [5,6]. Additionally, chromosomal mapping has provided evidence indicating that, out of the 518 protein kinase genes, 244 map to either disease loci or cancer amplicons, further highlighting the therapeutic potential of protein kinase inhibitors and their targets [2]. Several studies have linked protein kinases and deactivated phosphatases to multiple human malignancies owing to their genetic mutations and, also, chromosomal reshuffling patterns [7,8,9,10,11]. Since the abnormal regulation of protein kinases is strongly associated with human malignancies, including chronic and acute leukaemia, considerable efforts have been expended to dissect the functions of protein kinase-controlled signal transduction pathways in cancer [2,12]. Within leukemic malignancies, acute leukaemia are characterised by a rapid progression and are subdivided into two types based on their origins: Acute myeloid leukaemia (AML) and Acute lymphoblastic leukaemia (ALL), with about 80–85% of cases of the B-ALL and 15–20% of cases of the T-ALL [13,14] nature. While AML accounts for about 90% of all acute leukaemia in adults, ALL is the most common subtype found in childhood [15,16]. On the other hand, in chronic leukaemia, Chronic lymphocytic leukaemia (CLL) represents a heterogeneous disease with a variety of clinical outcomes that is characterised by the proliferation and accumulation of mature CD5+B cells in the blood, bone marrow and lymphoid tissues [17]. Chronic myelogenous leukaemia (CML) originates in a hematopoietic stem cell and is a chronic myeloproliferative disease that is characterised by an abnormal fusion gene (BCR-ABL1) on the Philadelphia chromosome in neoplastic cells [18]. Amongst the protein kinases, tyrosine kinases (TKs) [Discoidin domain receptor (DDRs), Erythropoietin-producing human haepatocellular (Eph) receptors, proto-oncogene c-Src (SRC), Spleen tyrosine kinase (SYK), Fms-like tyrosine kinase 3 (FLT3), Janus kinase (JAK), etc.] play a major role in haematopoiesis, and the deregulation of TK signalling has long been associated with the development of haematological malignancies [11,19,20]. The examples of TKs involved in leukaemia are highlighted in Figure 1 [21]. While TK mutations in AML account for FLT3 (30%), KIT (5%) and Janus kinase 2 (JAK2) (2%), the deregulated activity of ABL1 (within the BCR-ABL1 fusion, resulting from the t(9;22) (q34;q11) chromosomal translocation) drives the initiation and maintenance of CML [18,22,23]. Zeta chain of T-cell receptor-associated protein kinase 70 (ZAP-70), B lymphocyte kinase (BLK), JAK2, tyrosine-protein kinase (LYN) and neurotrophic Receptor Tyrosine Kinase 3 (NTRK3) are found to be the most commonly mutated kinases in CLL [24]. The abelson tyrosine-protein kinase 1 (ABL1) mutants and their therapeutic inhibitors have been extensively reported to play a critical role in leukaemia and discussed in detail in recent reviews [25,26,27].
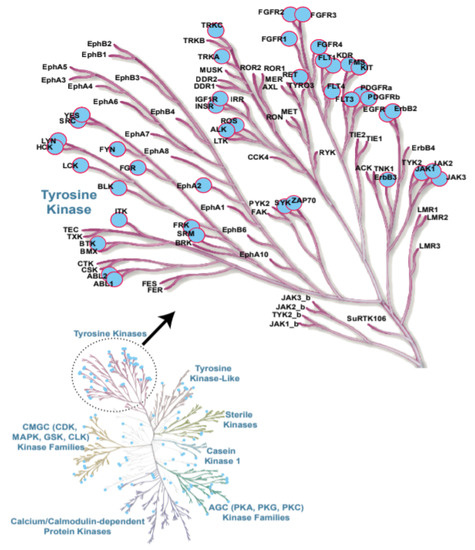
Figure 1.
Protein kinases contributing to leukaemia are highlighted in blue. Examples of tyrosine kinases (TKs) involved in leukaemia are shown in the insert [21].
This review is mainly focused on the impact and targeting of TKs not related to ABL1 but having a relevant role in the development and progression of different types of leukaemia. Emerging treatment options based on targeting these molecules and the currently available modulators of their activities are also being discussed.
2. Classification of TKs
The human genome contains 90 unique TK genes, out of which, 58 are receptor types, distributed into 20 subfamilies, and 32 are of nonreceptor types grouped into 10 subfamilies [2,28] (Table 1). Over the last 30 years, a major focus of research has been linked to multiple TKs owing to their importance in the development of different malignancies, including leukaemia, as well as nonmalignant disorders. TKs are important mediators of signal transduction, controlling cell proliferation, differentiation, migration, metabolism and programmed cell death, and are actively involved in all stages of neoplastic development and progression. The finding of the protein-tyrosine kinase (PTK) activities to be linked with viral transforming proteins and the SRC oncogene that has transforming nonreceptor tyrosine kinase (NRTK) activity led the way to the understanding of the role and significance of TKs in cancer [29,30]. Receptor tyrosine kinases (RTKs), a subclass of TKs, are key regulators of cellular processes and are essential regulators of signal transduction pathways, controlling a wide range of complex biological functions [31]. Due to their role as growth factor receptors and their ability to initiate complex network of signalling pathways in a wide variety of cell types, their abnormal activities, including gain-of-function mutations or receptor/ligand overexpression, have been associated with multiple cancers and, in particular, leukaemia [31,32]. In general, the activation of RTKs occurs through the formation of intermolecular dimerization in the presence of ligands, which, in turn, leads to the activation of their kinase function and phosphorylation of tyrosine residues in their cytoplasmic portions. In oncogenic conditions, gain-of-function mutations in RTKs often lead to their constitutive activation, even in the absence of a matching ligand.

Table 1.
Receptor and nonreceptor Classes of tyrosine kinase (TK) families.
The overexpression of RTKs also frequently occurs in cancer as a consequence of genomic amplifications of RTK genes, which results in the increased accumulation of receptor molecules on the cell membrane and, ultimately, abnormally high level of signalling [31] (Figure 2a). Thus, deregulated RTKs have been reported to be responsible for the enhanced survival and apoptotic resistance in CLL [33,34,35,36,37]. NRTKs are cytosolic TK enzymes that lack a transmembrane domain. NRTKs exhibit significant structural variability due to the presence of a kinase domain, some protein–protein interacting domains (SH2, SH3 and PH domains) and additional signalling [10]. Therefore, the activation of NRTKs are regulated by other factors on the cell surface or in the cytoplasm integrating heterologous protein–protein interactions, thus enabling transphosphorylation (Figure 2b). NRTKs play crucial roles in the regulation of the immune system, cell growth, proliferation, differentiation, adhesion, migration and apoptosis. Mutations in NRTKs result in the formation of oncogenes [including ABL1, proto-oncogene tyrosine protein kinase c-Fes (FES), SRC, etc.] and leads to aberrant signalling. This has been reported in many forms of haematological malignancies, leading to their prolonged viability and overall survival [11].
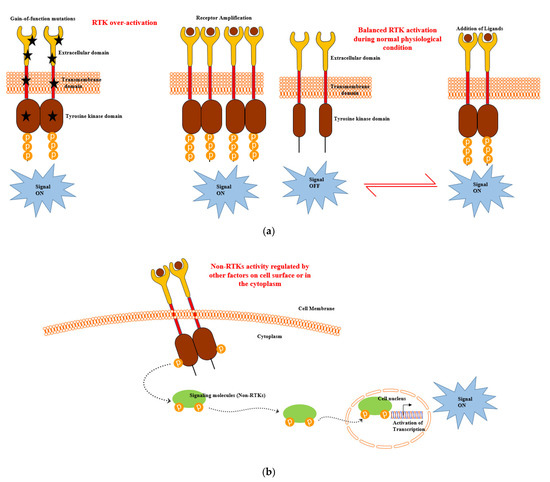
Figure 2.
(a) Receptor tyrosine kinase (RTK) and (b) nonreceptor tyrosine kinase (NRTK) activation during leukaemia development and progression [10,31].
In the following sections, we discuss the roles of individual TKs involved in the pathogenesis of leukaemia, including their roles in the onset and progression of different types of leukemic diseases and their targeting for therapeutic purposes.
2.1. FLT3
FLT3 belongs to the class III receptor TK family, which also includes platelet-derived growth factor receptor (PDGFR), macrophage colony-stimulating factor receptor (FMS) and stem cell factor receptor (KIT). FLT3 is important for the normal development of hematopoietic stem cells and the immune system [38,39]. In acute leukaemia, the expression of FLT3 is observed in 92% of AMLs and 27% of T-ALLs, and the overexpression of the FLT3 protein is usually found in 70–100% leukemic blast cells of AML patients. Mutations in the FLT3 gene have been reported in many cases of AML [40,41]. The activating mutations in FLT3 can be found either in the internal tandem duplications (ITDs) of the juxta membrane domain or in the activation loop mutations (ALM) of the kinase domain (TKD) [42]. While the former interferes with the normal negative regulatory role of FLT3 gene, the latter locks the receptor into an active conformation, resulting in a constitutively open ATP-binding pocket. In fact, FLT3 ITDs have been reported in 25–30% of patients with AML [43]. In a study conducted by Yamamoto et al. [44], the authors examined the D835 and ITD mutations of the FLT3 gene in a total of 589 patients with haematological malignancies. The results of the study revealed several kinds of D835 missense mutations in 30 of the 429 (7.0%) AML and one of the 36 (2.8%) ALL patients. In addition, FLT3/ITD and FLT3/ALM mutations have also been reported in paediatric AML [45,46,47]. While the frequency of the former is approximately 15% lower than in adults [48,49,50], the latter is detected in 7% of cases, similar to that in adults [51]. Direct experiments have shown that the FLT3-ITD mutations alone are insufficient to cause AML but induce a myeloproliferative disease in a murine bone marrow transplant model. However, the combination of FLT3-ITD and promyelocytic leukemia protein (PML)/ retinoic acid receptor alpha (RARalpha) mutations induces an acute promyelocytic leukemia (APL)-like disease in recipient mice over a period of 7–23 weeks with 100% penetrance, favouring the hypothesis that AML is the consequence of cooperation between at least two classes of mutations [52,53]. The overexpression of the wild-type FLT3 collaborates with the NUP98-HOX gene fusions (NUP98-HOXA10 and NUP98-HOXD13) to induce aggressive AMLs in mice [54,55]. Studies have also reported different gene rearrangements in CML and AML patients, examples of which include NUP98/HOXA9 translocations in the combination ETV6/platelet-derived growth factor receptor beta (PDGFR β) or ABL1, respectively [56,57]. Of interest, a high FLT3 expression has also been associated with poor survival in KMT2A- AFF1+ ALL patients, further highlighting the relevance of FLT3 inhibitors for the treatment of ALL [58]. All these observations demonstrate the role of the FLT3 receptor in chronic and acute leukaemia and the identification of the mechanisms underlying its deregulation provides new opportunities in the treatment of these diseases [58,59].
2.2. KIT
KIT is a stem cell growth factor receptor involved in various functions of cell differentiation and survival, as well as melanogenesis, fertility and homeostasis [60]. It has been established that numerous malignancies such as mastocytosis, gastrointestinal tumours, melanomas and human mast cell leukaemia are the result of KIT mutations [61,62,63,64]. Almost 80% of AML cases have been found to have KIT proto-oncogene expression and support cell proliferation [65]. KIT has an additional role of improving the fibronectin attachment of the AML cells, leading to proliferative and antiapoptotic signals [66]. The structural organisation of KIT comprises a single transmembrane helix, a cytoplasmic juxtamembrane domain, a split kinase domain with a kinase insert and five immunoglobulin-like domains. It has been established that a major portion of mutations leading to disease development could be attributed to a few exons, including exon 17 encoding a kinase insert and exon 11 encoding a juxtamembrane domain and the extracellular exon 8 [67].
Amongst the different types of AML, core binding factor-AML has the maximum KIT mutations, with significant variations in reported cases. There has been evidence of KIT mutations having an adverse impact on relapse and survival in adult t(8;21) AML [68]. A study by Pollard et al. [69] reported a 20% prevalence of KIT mutations in their study population and suggested that, in paediatric patients with core binding factor-AML, KIT mutations did not have prognostic significance. This may be due to the fact that most studies of KIT mutations in children are smaller in size and reflect different settings of therapeutic treatments of AML patients. Molecular targeted therapies for leukaemia have led to the development of a vast array of drugs. KIT can be employed as a potential therapeutic target in two ways: first, by increasing the susceptibility of malignant cells to chemotherapeutic agents via synchronising the cells into the cell cycle and, secondly, by annulling the chemoresistance of leukaemia cells to apoptosis with the use of chemicals capable of blocking the KIT receptor activity [70,71]. Dasatinib is an FDA-approved small molecule compound that inhibits KIT [72]. Some other inhibitors with activity against KIT are SU6668 and SU5416, which are small-molecule indolinone-RTK inhibitors [73]. KIT inhibitors are further effective when used in combination with chemotherapy rather than alone, as cancers have multiple pathways of growth and proliferation [74]. With focused and in-depth research, targeting molecular abnormalities such as KIT can lead to less toxic treatments with higher efficiency than the conventional chemotherapy.
2.3. DDRs
DDRs belong to the RTK family and consist of two members DDR1 and DDR2, nonintegrin collagen-type receptors that signal in response to the binding of a collagen protein [75]. Although DDRs are mainly expressed during embryonic development, their role in adults is very limited. DDRs are either overexpressed and/or mutated in haematological cancers, their overexpression is correlated with a bad prognosis and their importance depends on the type and stage of each cancer type [76,77]. A study aimed at characterising gene expression signatures in ALL cells linked with known genotypic abnormalities in adult patients revealed that DDR1 was one among the kinases that was highly expressed in cases without molecular rearrangements, as well as in BCR/ABL1-positive ALL [78]. Barisione et al. [79] showed the correlation of DDR1 mRNA levels with CLL outcomes, suggesting the functional role of DDR1 in the development of this cancer. Moreover, B-ALL study cases in adults without AFF4/AFF1 and TCF3/PBX1 molecular rearrangements showed high transcriptional levels of the DDR1 gene [78]. Tomasson et al. [80] reported DDR1 somatic mutations (DDR1A803V) in patients with de novo AML. These somatic mutation maps to the activation loop on its kinase domain, indicating that this DDR1 mutant may effectively disrupt kinase function. A high-throughput DNA sequencing study was conducted by Loriaux et al. to determine whether aberrantly activated TKs other than FLT3 and KIT receptors contribute to acute myeloid leukaemia (AML) pathogenesis [81]. Their results showed around 30 nonsynonymous sequence variations in 22 different kinases, and their analyses further identified five AML patients with nonsynonymous somatic mutations in the DDR1 juxtamembrane domain.
2.4. Eph Receptor Family
Eph receptors form the largest group of RTKs, and they are activated by ephrin ligands either attached to the cell membrane through glycosylphosphatidylinositol anchors or embedded there with their transmembrane domains. These receptors form an important cell communication system, with significant roles in both normal physiology and pathogenesis. There are fourteen Eph group members encoded in the human genome, which are further divided into A and B subtypes [82]. The EphA subtype consists of nine members (EPHA1–EPHA8 AND EPHA10) and are activated mostly by five ephrin-A ligands (EPHRIN-A1–EPHRIN-A5). The five EphB receptors (EPHB1–EPHB4 and EPHB6) interact predominantly with three ephrin-B ligands (EPHRIN-B1–EPHRIN-B3). The unusual feature that distinguishes them from most other RTKs is that Eph receptors bind to ephrin ligands expressed on the plasma membrane of a neighbouring cell and often form multimeric structures. Both the Eph receptors and ephrins can initiate signal transduction in each receptor- and ligand-presenting cell upon cell–cell contact formation, triggering both forward (through ephrin-ligated Eph receptors) and reverse (through Eph-bound ephrins) responses. The abnormal expression of the proteins of the Eph/ephrin system has been detected in many types of haematopoietic malignancies, and they have been reported to be either up- or downregulated based on the cancer type (Table 2). For example, EPHA3, originally identified in the LK63 pre-B ALL cell line, was later reported to be expressed in different cell lines of T-cell leukaemia and has been shown to induce both adhesive and repulsive responses in different cell types [83,84,85,86]. Altogether, the Eph-associated protein tyrosine phosphatase activity proved to not only control Eph receptor phosphorylation levels but, also, to switch the response of ephrin contact from repulsion to adhesion, thus playing a significant role in the pathology of some types of leukemic cells [86]. Several studies have shown the copy number variation of EPHA3 to be linked with various types of haematological malignancies, including AML, suggesting that these copy number variations could also be used as a diagnostic marker [87,88]. While aberrant EPHB4/EPHRIN-B2 expression has been found in different leukaemia and lymphoma cell lines [89], the overexpression of EPHB6 has been reported in both myeloid and lymphoid leukemic cells [90,91,92]. In a study by Nakanishi et al., the authors performed a screening study in K562 cells to determine whether ALL1 fusion proteins are involved in the regulation of EPH genes. These cells producing recombinant ALL1/AF4 or ALL1/AF9 fusion protein showed the transcriptional upregulation of EPHA7 accompanied by ERK phosphorylation [93,94]. Thus, anti-EPHA7 antibodies may play a prominent role in leukaemia linked with these translocations.
Table 2.
Examples of the regulation of Eph family members in haematopoietic malignancies.
2.5. SRC Kinases
The SRC family comprise genes encoding nine structurally related NRTKs, including SRC, BLK, FGR, FYN, HCY, LCK, LYN, YES and YRK, which play a major role in several physiological processes, like proliferation, angiogenesis, migration, differentiation, invasion and immune function [102]. At least some of them are distinctive proto-oncogenes, triggering unregulated cell proliferation as a result of mutation or overexpression in cancer. SRC activation may be due to interactions with cell membrane receptors [103] or a result of post-translational modifications and mutations [104], and we earlier reported that targeting the synthetic lethal interaction between the SRC kinase and the EPHB6 receptor may benefit cancer treatment, as this molecule is downregulated in multiple malignancies [105]. The role of the SRC kinase in CML was first established by Danhauser-Riedl et al. [106], who showed that LYN and haematopoietic cell kinase (HCK)-SRC-related kinases were activated, along with BCR-ABL1 fusion, in myeloid cells. Dos Santos et al. [107] found an interesting link between the LYN kinase and the mTOR pathway, asserting LYN as a therapeutic target for AML. It has been found that the SRC kinase uses different signalling pathways to induce both CML and B-ALL [108]. These results suggested that Philadelphia-positive acute leukaemia patients can be aided by the simultaneous co-inhibition of SRC and ABL1 kinases. Recent studies have given further insights into the importance of SRC kinases in different malignancies [109,110]. Weir et al. [109] found TL02-59 as one of the promising compounds in AML treatment, thereby establishing FGR as a novel therapeutic target in this malignancy. HCK was found to be involved in erythroid cell differentiation by being a part of the RAS-ERK and PI3K/AKT pathways. The elevated expression of HCK was observed in CD34+ haematopoietic stem and progenitor cell (HSPC) subsets of AML patients, paving the way for investigating HCK inhibitors [111]. Ingley [112] have reported the distribution of LYN throughout the plasma membrane and cytosol of AML cells with the most prominent expression amongst the SRC family members. Thus, the expression levels of HCK, FGR and LYN have a direct relation with the poor prognosis of the AML disease [110]. On the other hand, in B-CLL, LYN has been suggested as the only SRC kinase (compared to others, such as SRC, FYN, c-FGR and LCK) to be responsible for the abnormal cellular tyrosine phosphorylation and is the proximal kinase to initiate a signalling cascade in the BCR pathway [113]. Therefore, strategies to target BCR signalling as an emerging therapeutic approach have been well-discussed in the literature [114,115]. In brief, the BCR stimulation by antigen leads to the phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) by LYN, following which, SYK is recruited and activated through tyrosine phosphorylation. The imbalanced activity within the BCR signalling axis has been responsible for the deregulation of the gene expression in CLL [116]. The LYN overexpression in CLL was linked with a significant downregulation of microRNA-337-5p. This suggests that the aberrant expression of microRNA-337-5p could be involved in the post-transcriptional regulation of LYN mRNA [117]. Taken together, all these studies indicate the potential importance of SRC kinase family members as novel targets for leukaemia treatment.
2.6. SYK Family
SYK is a cytoplasmic TK acting to couple BCR to intracellular signalling pathways in B lymphocytes [118]. SYK is a pro-survival factor, and therefore, its knockdown or inhibition leads to apoptosis in the cell subsets of CLL and ALL of the B cell lineage that express active SYK [119,120,121,122]. The observed effect might be due to the significant downstream reduction of major inflammatory mediators such as tumor necrosis factor alpha (TNFα), interleukins (ILs)-1, 6 and 18 that suggest the role of SYK in mediating the proinflammatory response [116]. Further, the inhibition of SYK mobilises CLL cells from tissue compartments to the blood by antagonising signals that restrain CLL cells within tissues, thus inducing cell death [123]. Understanding the molecular characterisations of these mobilised CLL cells and their sensitivity to chemotherapy play an important role in the identification and use of SYK inhibitors. SYK is expressed in a majority of AML patients and is a critical regulator of FLT3 in AML [124]. Efforts to use SYK inhibitors therapeutically in AML patients have shown promising results, especially in combination with standard chemotherapeutics such as cytosine arabinoside (AraC). Moreover, the inhibition of SYK has been reported to increase the sensitivity of leukaemia stem cells to AraC [125]. SYK is also reported to exhibit its cell survival function by signalling through BCR in haematological malignancies, implicating SYK as a promising target for the development of clinically relevant inhibitors [126,127,128].
2.7. JAK/STAT Signalling
JAKs are cytoplasmic TKs, including four family members: JAK1, JAK2, JAK3 and TYK2, which govern cell survival, proliferation, differentiation, haematopoiesis, immune response and apoptotic death response [129,130]. While the members of JAK1, JAK2 and TYK2 are ubiquitously expressed, JAK3 expression is exclusively restricted to the haematopoietic lineage. The frequency of alterations in JAK members among different haematological malignancies is depicted in Figure 3.
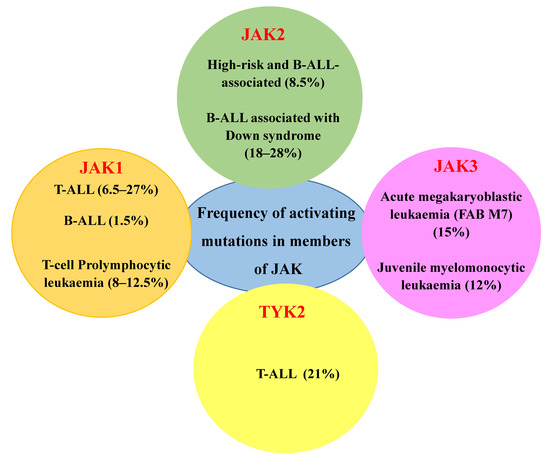
Figure 3.
Frequency of activating mutations in members of JAK among different haematological malignancies [131].
Mutations in the JAK/STAT signalling pathway are hallmark features of several types of leukaemia, such as T-cell prolymphocytic leukaemia, paediatric-ALL and adult T-cell lymphoblastic leukaemia, and the identification of these mutations paves the way for understanding the pathogenesis and in developing targeted therapies for these aggressive malignancies [132,133,134,135]. Chimeric proteins with constitutive kinase activity produced by fusion genes resulting from chromosomal translocations, such as ETV6-JAK2, have also been reported in human leukaemia [136]. The ETV6 gene located on the chromosome 12p13 has been reported to be involved in chromosomal translocations observed in a variety of human leukaemia since 1994 [56,137,138]. Inherent mutations are the most common factors attributed towards the development of malignancies, especially in myeloproliferative neoplasms. It has been reported that the acquisition of the constitutively active Janus kinase 2 (JAK2) V617F mutant occurs in 39–57% of primary myelofibrosis, 41–72% of essential thrombocytosis, 81–99% of Polycythaemia vera and <5% of acute myeloid leukaemia patients [139]. The activation of the transcription factor STAT5 is required for JAK2 V617F-mediated transformation, and therefore, the development of drugs that inhibits STAT5 can be an effective treatment option for myeloproliferative neoplasia [140]. A comprehensive sequence analysis by Vincente et al. identified mutations and copy number variations in IL7R-JAK signalling pathway members among 27.7% of T-ALL samples screened, with JAK3 mutations being the most frequent among others [141,142]. Another study by Degryse and Cools established the involvement of JAK kinases in T-ALL by affirming that the inhibition of both JAK-1 and JAK-3 can be used for T-ALL treatment [143]. This study also provided insights into the use of numerous JAK inhibitors that are already available to be potentially repurposed for T-ALL therapy [143]. The JAK/STAT signalling pathway is activated in response to the AML infiltration, and the inhibition of JAK1/2 resulted in significant anti-leukaemic activity in vitro [144]. In most leukaemia cases, JAK-2 has been the key regulator. However, JAK-3 activating mutations have also been observed in natural killer cell/T-cell lymphoma (35.4%) [145]. Findings have backed the persistent involvement of JAK-STAT pathways in different malignancies, which can open up novel targeted therapeutic approaches aiming to manipulate molecules associated with JAK kinases.
3. TK Inhibitors in Leukaemia and Lymphoma Treatment
Both leukaemia and lymphoma affect circulating leukocytes and bone marrow; however, lymphoma in general has higher survival rates than leukaemia. Thus, AML has only around 10% five-year survival rates in adults above 60 years of age and is accountable for more than half of leukaemia-related deaths in children. With novel chemical compounds emerging in cancer treatment every year, there has been a substantial contribution of small molecule inhibitors towards the development of new targeted therapies. Overall, small molecule inhibitors have been effective in suppressing disease progression in many types of cancers [146]. Protein kinases, as described previously, are key regulators of multiple aspects of cell biology, and their aberrant behaviours often lead to serious pathological conditions. Dysfunction in protein kinase signalling pathways has a major role in the development of different types of cancers, as well as cardiovascular diseases [3,147]. Kinase inhibitors are consequently amongst the most sought-after compounds in the development of targeted therapies with small molecule inhibitors. A brief account of different types of inhibitors based on the target site and mechanism of action is summarised in (Table 3).
Table 3.
Classification of protein kinase inhibitors.
The inhibitors are classified as covalent/irreversible and noncovalent/reversible, based on their binding to kinases. The covalent inhibitors largely bind to the lysine or cysteine residues around the ATP-binding site, and it has been proposed that such molecules are possibly toxic in nature [149]. Based on the ability to bind to the hinge region, noncovalent inhibitors are categorised into type 1, type 2 and type 3 kinase inhibitors. The unique binding sites of type 3 inhibitors enable them to have a higher degree of selectivity when compared to type 1 and type 2 inhibitors [150,151]. A major portion of the inhibitors are those that target the ATP-binding site, whereas the remaining ones target selected allosteric sites. Certain attributes of a kinase inhibitor, such as pharmacokinetic and pharmacodynamic properties and the degree of oncogenic addiction to the specific kinase, determine their therapeutic efficacy [152]. Small molecule inhibitors were first synthesised as early as 1984 by Hidaka et al., who synthesised naphthalene sulphonamide, which acted as the basis for the development of next-line molecules [153]. Kinase inhibitors such as imatinib, previously known as ST1571, an ABL1 TK inhibitor, has been proven to be quite effective as an anti-leukaemic agent [154]. Imatinib became the first economically successful FDA-approved small molecule protein kinase inhibitor for the treatment of CML patients in 2001 [155]. Consequently, there has been a steady increase in the number of FDA-approved kinase inhibitors, with more than 70 drugs making it to the list now. Table 4 depicts different drugs/small molecule inhibitors for protein TKs that have been approved by the FDA for clinical use.
Table 4.
List of a few examples of small molecule protein kinase inhibitors approved for clinical use.
Consistent with the important role of TK signalling in malignancy, amongst their inhibitors approved for clinical protocols, most are being used for treating various types of cancers [149]. For example, the advent of imatinib treatment has been revolutionary in patients with CML, assisting in their management and improving their overall survival. Other TK inhibitors, including dasatinib, nilotinib and bosutinib, are mostly prescribed in cases where the patient does not respond to imatinib. Research findings have strongly supported the fact that inhibitors of protein kinases have proven to be extremely useful in targeted therapy against different types of leukaemia, either alone or in combination with other drugs [167]. To give a few examples, midostaurin was approved in the United States of America for the treatment of adult patients with newly diagnosed, FLT3 mutation-positive AML, aggressive systemic mastocytosis, systemic mastocytosis with associated haematological neoplasm or mast cell leukaemia [164]. In addition, midostaurin was also used in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation [164]. Cytarabine can also be used in combination with sunitinib against AML cell lines with the FL3-ITD mutation and tandutinib for the treatment of AML patients [168]. Trametinib, a MEK inhibitor, has been reported to exhibit strong synergy when combined with midostaurin in cells with mutated and wild-type FLT3 [169]. Moreover, a recent study in the first-line setting for CLL has demonstrated the efficacy of single-agent ibrutinib, which significantly prolonged progression-free survival compared with chlorambucil, with an 85% reduction in the risk of disease progression or death [170]. The further investigation of functions of TKs in cancer cells can support the development of new inhibitory therapeutic compounds. Certain compounds, such as BTK inhibitor ibrutinib, have also been proven to be effective against mantle cell lymphoma patients. Mantle cell lymphoma has a destructive clinical progression with no effective therapy; however, ibrutinib shows complete response rates ranging from 19–23% in this malignancy. Further, second-generation BTK inhibitors have emerged, such as acalabrutinib, tirabrutinib and zanubrutinib, which are superior to ibrutinib in terms of efficacy and toxicity profiles [171]. Although the list of TK inhibitors has significantly increased over the past decades, the main challenge with these drugs is that they mostly have limited specificity and efficiency, which increases the risk of undesirable off-target effects and emerging evading pathways. This might ultimately lead to severe side effects in the patients and to the development of treatment-resistant leukaemic cell populations. Thus, mutations of protein kinases, instability of the inhibitors, residual and acquired drug resistance, general toxicity and off-target effects are some of the major concerns associated with the action of even the last-generation drugs [168]. Another important limitation of kinase inhibitors is the out-of-pocket expenses associated with their usage. For example, continuous therapy with ibrutinib used in the treatment of some lymphomas costs about USD 150,000 per year for each patient, and treatment with imatinib (standard therapy in CML) costs roughly USD 100,000 per patient per year lifelong. It has been observed that post the entry of generic imatinib, there has been a steady decline in its expenditure, but there is still a sustained surge in dasatinib and nilotinib expenditure in patients with CML [172]. The development of new-generation drugs with more specificity and less side effects greatly benefit patients. New-generation drugs are preferred over the older versions due to their ability to reduce serious limitations, such as low potency and specificity in addition to their better ability to cross the blood–brain barrier. An example would be the development of nilotinib, a second-generation drug with enhanced properties in its structure leading to higher binding affinity of the drug to ABL1, when compared to its first-generation counterpart imatinib. Imatinib was the first drug to specifically target the ABL1 kinase, and nilotinib works with lesser harmful side effects [173]. Thus, the development of next-generation kinase inhibitors based on the advances in cancer biology and structural biology research helps in generating more effective compounds with better therapeutic windows.
4. Conclusions
The action of TKs is indispensable in cell regulation, with functions ranging from controlling growth, cell cycle, adhesion and migration to governing apoptotic responses. With evidence of their contributions to human malignancies, these molecules are popular targets in drug discovery and in existing cancer treatment protocols. In a nutshell, we discussed here the role of TKs in the development and progression of different types of leukaemia. The overexpression and mutation of both receptor and NRTKs lead to impaired regulation in pathways, often supporting the initiation and progression of leukaemic diseases. FLT3 and KIT have been implicated in cases of acute and chronic leukaemia, opening up the prospects of treatment with higher efficacy if the underlying mechanisms of deregulation are fully investigated. While DDRs have been shown to be involved in lymphocytic leukaemia, mostly, SRC kinases are potential drivers in both lymphocytic and myeloid leukaemia. Eph receptors and their ligands are known to have important functions in physiology and pathogenesis; their upregulation or downregulation is associated with multiple haematologic malignancies. SYK is another kinase associated with intracellular signalling pathways in B cells contributing to leukaemia and lymphomas. JAKs are key regulators within the immune system and haematopoiesis. Therefore, their inherent mutations can often lead to different types of cancer, including ALL and Non-Hodgkin’s lymphoma. With the advent of targeted therapies and the supporting advanced technology, medicine has witnessed a surge in novel and more effective treatment options for cancer. Small molecule compounds, especially TK inhibitors, have been developed, with more agents being granted FDA approval every year. Further detailed analyses of the action of TKs in leukaemia and screening new compounds targeting these molecules should further facilitate detecting effective and less toxic inhibitors aimed at combating leukaemic diseases either alone or in combination with conventional chemotherapies for haematological malignancies.
Author Contributions
Conception and design of the review paper: A.F., V.G., F.J.V. and K.K.B. and writing of the review paper: K.K.B., A.B. and F.S.V. All authors have read and agreed to the published version of the manuscript.
Funding
There are no funding sources associated with this paper.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Abbreviations
| ABL1 | Abelson tyrosine-protein kinase |
| ACK | Activated Cdc42-associated kinase |
| ALL | Acute lymphoblastic leukaemia |
| ALM | Activation loop mutations |
| AML | Acute myeloid leukaemia |
| APL | Acute promyelocytic leukaemia |
| ATP | Adenosine triphosphate |
| B- ALL | B-cell acute lymphoblastic leukaemia |
| BCR | Breakpoint cluster region |
| BTK | Bruton’s tyrosine kinase |
| CDC | Cell division cycle |
| CDK | Cyclin-dependent kinases |
| c-KIT | CD117, also called KIT or C-kit receptor |
| CLL | Chronic lymphocytic leukaemia |
| c-Met | mesenchymal–epithelial transition factor |
| CMGC | CDK/MAPK/GSK/CDK-like kinase group |
| CML | Chronic myelogenous leukaemia |
| DDR | Discoidin domain receptor |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EPH | erythropoietin-producing human haepatocellular receptors |
| Eph A | Ephrin type A receptors |
| EphB | Ephrin type B receptors |
| ERBB2 | Receptor tyrosine protein kinase erbB-2 |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and drug administration |
| FGFR | Fibroblast growth factor receptor |
| FGR | Gardner-Rasheed feline sarcoma viral (v-fgr) oncogene homologue |
| FYN | FYN oncogene related to SRC, FGR and YES |
| GSK | Glycogen synthase kinase |
| HCK | Haematopoietic cell kinase |
| HDAC | Histone deacetylase |
| HER | Human epidermal growth factor receptor |
| HSPC | Haematopoietic stem and progenitor cell |
| ITAMs | Immunoreceptor tyrosine-based activation motifs |
| ITDs | Internal tandem duplications |
| JAK | Janus kinase |
| JAK-STAT | Janus kinase signal transducer and activator of transcription |
| JMML | Juvenile myelomonocytic leukaemia |
| MAPK/ERK | Mitogen-activated protein kinases/Extracellular signal-regulated kinases |
| mTOR | The mammalian target of rapamycin |
| NRTK | Nonreceptor tyrosine kinase |
| NSCLC | Non-small cell lung carcinoma |
| PARP | Poly (ADP-ribose) polymerase |
| PDGFR β | Platelet-derived growth factor receptor beta |
| PI3K/AKT | Phosphatidylinositol 3-kinases/Protein kinase B |
| PML | Promyelocytic leukaemia |
| PTP | Protein tyrosine phosphatase |
| PTPN1 | Tyrosine protein phosphatase nonreceptor type 1/protein-tyrosine phosphatase 1B (PTP1B) |
| PTPN2 | Tyrosine protein phosphatase nonreceptor type 2 |
| RAF | RAF proto-oncogene serine/threonine protein kinase |
| RARA | Retinoic acid receptor alpha |
| RTK | Receptor tyrosine kinase |
| SRC | Rous sarcoma oncogene cellular homologue/Proto-oncogene tyrosine protein kinase Src |
| STAT5 | Signal transducer and activator of transcription 5 |
| SYK | Spleen-associated tyrosine kinase |
| T-ALL | T-cell acute lymphoblastic leukemia |
| ETV6 | Translocation-ETS-leukemia/ETV6 |
| TKD | Tyrosine kinase domain |
| TKI | Tyrosine kinase inhibitors |
| TKs | Tyrosine Kinases |
| TNFα | Tumor necrosis factor alpha |
| TYK2 | Tyrosine kinase 2 |
| ZAP-70 | Zeta chain of T-cell receptor-associated protein kinase 70 |
References
- Coussens, L.; Parker, P.J.; Rhee, L.; Yang-Feng, T.L.; Chen, E.; Waterfield, M.D.; Francke, U.; Ullrich, A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 1986, 233, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and Cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [PubMed]
- Köstler, W.J. Targeting Receptor Tyrosine Kinases in Cancer. In Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease; Wheeler, D., Yarden, Y., Eds.; Springer: New York, NY, USA, 2015. [Google Scholar]
- Kittler, H.; Tschandl, P. Driver mutations in the mitogen-activated protein kinase pathway: The seeds of good and evil. Br. J. Dermatol. 2018, 178, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Chalandon, Y.; Schwaller, J. Targeting mutated protein tyrosine kinases and their signaling pathways in hematologic malignancies. Haematologica 2005, 90, 949–968. [Google Scholar]
- Bartram, C.R.; de Klein, A.; Hagemeijer, A.; van Agthoven, T.; van Kessel, A.G.; Bootsma, D.; Grosveld, G.; Ferguson-Smith, M.A.; Davies, T.; Stone, M.; et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1983, 306, 277–280. [Google Scholar] [CrossRef]
- Platanias, L.C. Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667–4679. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol. Cancer 2018, 17, 31. [Google Scholar] [CrossRef]
- Scheijen, B.; Griffin, J.D. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene 2002, 21, 3314–3333. [Google Scholar] [CrossRef]
- Grant, S.K. Therapeutic protein kinase inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, J. Precision therapy for acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef]
- Julio, D.; Ferran, N.; Dolors, C.; Elias, C. Chronic lymphocytic leukemia: From molecular pathogenesis to novel therapeutic strategies. Haematologica 2020, 105, 2205–2217. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef]
- Paulson, R.F.; Bernstein, A. Receptor tyrosine kinases and the regulation of hematopoiesis. Semin. Immunol. 1995, 7, 267–277. [Google Scholar] [CrossRef]
- Ku, M.; Wall, M.; MacKinnon, R.N.; Walkley, C.R.; Purton, L.E.; Tam, C.; Izon, D.; Campbell, L.; Cheng, H.C.; Nandurkar, H. Src family kinases and their role in hematological malignancies. Leuk. Lymphoma 2015, 56, 577–586. [Google Scholar] [CrossRef]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef]
- Fernandez, S.; Desplat, V.; Villacreces, A.; Guitart, A.V.; Milpied, N.; Pigneux, A.; Vigon, I.; Pasquet, J.M.; Dumas, P.Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? Int. J. Mol. Sci. 2019, 20, 3429. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Levine, R.L.; Thompson, C.; Basile, G.; Gilliland, D.G.; Freedman, A.S. Systematic genomic screen for tyrosine kinase mutations in CLL. Leukemia 2008, 22, 1966–1969. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Huang, H.; Lei, X.; Tang, G.; Cao, X.; Peng, J. Recent advances in Bcr-Abl tyrosine kinase inhibitors for overriding T315I mutation. Chem. Biol. Drug Des. 2020. [Google Scholar] [CrossRef]
- Yaghmaie, M.; Yeung, C.C. Molecular Mechanisms of Resistance to Tyrosine Kinase Inhibitors. Curr. Hematol. Malig. Rep. 2019, 14, 395–404. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef]
- Hunter, T.; Cooper, J.A. Protein-tyrosine kinases. Annu. Rev. Biochem. 1985, 54, 897–930. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase - Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Veronese, L.; Tournilhac, O.; Verrelle, P.; Davi, F.; Dighiero, G.; Chautard, E.; Veyrat-Masson, R.; Kwiatkowski, F.; Goumy, C.; Gouas, L.; et al. Strong correlation between VEGF and MCL-1 mRNA expression levels in B-cell chronic lymphocytic leukemia. Leuk. Res. 2009, 33, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, R.; Galeano, A.; Becu-Villalobos, D.; Spinelli, O.; Sapia, S.; Bezares, R.F. Autocrine/paracrine involvement of insulin-like growth factor-I and its receptor in chronic lymphocytic leukaemia. Br. J. Haematol. 2005, 130, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Shanafelt, T.D.; Bone, N.D.; Strege, A.K.; Jelinek, D.F.; Kay, N.E. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: Implication for apoptosis resistance. Leukemia 2005, 19, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: Implications for therapy. Blood 2011, 117, 1928–1937. [Google Scholar] [CrossRef]
- Baskar, S.; Kwong, K.Y.; Hofer, T.; Levy, J.M.; Kennedy, M.G.; Lee, E.; Staudt, L.M.; Wilson, W.H.; Wiestner, A.; Rader, C. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 396–404. [Google Scholar] [CrossRef]
- Doepfner, K.T.; Boller, D.; Arcaro, A. Targeting receptor tyrosine kinase signaling in acute myeloid leukemia. Crit. Rev. Oncol. 2007, 63, 215–230. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. Role of FLT3 in leukemia. Curr. Opin. Hematol. 2002, 9, 274–281. [Google Scholar] [CrossRef]
- Kindler, T.; Lipka, D.B.; Fischer, T. FLT3 as a therapeutic target in AML: Still challenging after all these years. Blood 2010, 116, 5089–5102. [Google Scholar] [CrossRef]
- Carow, C.E.; Levenstein, M.; Kaufmann, S.H.; Chen, J.; Amin, S.; Rockwell, P.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996, 87, 1089–1096. [Google Scholar] [CrossRef]
- Chu, S.H.; Small, D. Mechanisms of resistance to FLT3 inhibitors. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2009, 12, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Sexauer, A.N.; Tasian, S.K. Targeting FLT3 Signaling in Childhood Acute Myeloid Leukemia. Front. Pediatrics 2017, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Alonzo, T.A.; Stirewalt, D.L.; Zwaan, M.; Zimmerman, M.; Reinhardt, D.; Kaspers, G.J.; Heerema, N.A.; Gerbing, R.; Lange, B.J.; et al. Clinical implications of FLT3 mutations in pediatric AML. Blood 2006, 108, 3654–3661. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.C.; Shih, L.Y.; Hung, I.J.; Yang, C.P.; Chen, S.H.; Jaing, T.H.; Liu, H.C.; Wang, L.Y.; Chang, W.H. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia 2003, 17, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Woods, W.G.; Stirewalt, D.L.; Sweetser, D.A.; Buckley, J.D.; Tjoa, T.K.; Bernstein, I.D.; Radich, J.P. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 2001, 97, 89–94. [Google Scholar] [CrossRef]
- Kondo, M.; Horibe, K.; Takahashi, Y.; Matsumoto, K.; Fukuda, M.; Inaba, J.; Kato, K.; Kojima, S.; Matsuyama, T. Prognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemia. Med. Pediatric Oncol. 1999, 33, 525–529. [Google Scholar] [CrossRef]
- Iwai, T.; Yokota, S.; Nakao, M.; Okamoto, T.; Taniwaki, M.; Onodera, N.; Watanabe, A.; Kikuta, A.; Tanaka, A.; Asami, K.; et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. Leukemia 1999, 13, 38–43. [Google Scholar] [CrossRef]
- Meshinchi, S.; Stirewalt, D.L.; Alonzo, T.A.; Zhang, Q.; Sweetser, D.A.; Woods, W.G.; Bernstein, I.D.; Arceci, R.J.; Radich, J.P. Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood 2003, 102, 1474–1479. [Google Scholar] [CrossRef][Green Version]
- Kelly, L.M.; Liu, Q.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Gilliland, D.G. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 2002, 99, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Amaral, S.M.; Curley, D.P.; Ley, T.J.; Gilliland, D.G. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc. Natl. Acad. Sci. USA 2002, 99, 8283–8288. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, L.; Argiropoulos, B.; Pineault, N.; Abramovich, C.; Sly, L.M.; Krystal, G.; Wan, A.; Humphries, R.K. The Flt3 receptor tyrosine kinase collaborates with NUP98-HOX fusions in acute myeloid leukemia. Blood 2006, 108, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, S.; Li, L.; Slape, C.; Nguyen, B.; Novak, R.; Duffield, A.; Huso, D.; Desiderio, S.; Borowitz, M.J.; Aplan, P.; et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 2012, 119, 2883–2894. [Google Scholar] [CrossRef]
- Golub, T.R.; Barker, G.F.; Lovett, M.; Gilliland, D.G. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 1994, 77, 307–316. [Google Scholar] [CrossRef]
- Ahuja, H.G.; Popplewell, L.; Tcheurekdjian, L.; Slovak, M.L. NUP98 gene rearrangements and the clonal evolution of chronic myelogenous leukemia. Genes Chromosomes Cancer 2001, 30, 410–415. [Google Scholar] [CrossRef]
- Chillon, M.C.; Gomez-Casares, M.T.; Lopez-Jorge, C.E.; Rodriguez-Medina, C.; Molines, A.; Sarasquete, M.E.; Alcoceba, M.; Miguel, J.D.; Bueno, C.; Montes, R.; et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia 2012, 26, 2360–2366. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Wang, F.; Wang, M.; Liu, H.; Chen, X.; Cao, P.; Ma, X.; Teng, W.; Zhang, X.; et al. The mutational spectrum of FLT3 gene in acute lymphoblastic leukemia is different from acute myeloid leukemia. Cancer Gene Ther. 2020, 27, 81–88. [Google Scholar] [CrossRef]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef]
- Chatterjee, A.; Ghosh, J.; Kapur, R. Mastocytosis: A mutated KIT receptor induced myeloproliferative disorder. Oncotarget 2015, 6, 18250–18264. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Corless, C.L.; Fletcher, J.A.; Heinrich, M.C. Biology of gastrointestinal stromal tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 3813–3825. [Google Scholar] [CrossRef]
- Ikeda, H.; Kanakura, Y.; Tamaki, T.; Kuriu, A.; Kitayama, H.; Ishikawa, J.; Kanayama, Y.; Yonezawa, T.; Tarui, S.; Griffin, J.D. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood 1991, 78, 2962–2968. [Google Scholar] [CrossRef]
- Bendall, L.J.; Makrynikola, V.; Hutchinson, A.; Bianchi, A.C.; Bradstock, K.F.; Gottlieb, D.J. Stem cell factor enhances the adhesion of AML cells to fibronectin and augments fibronectin-mediated anti-apoptotic and proliferative signals. Leukemia 1998, 12, 1375–1382. [Google Scholar] [CrossRef]
- Malaise, M.; Steinbach, D.; Corbacioglu, S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2009, 4, 77–82. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Zhu, H.H.; Jiang, Q.; Jiang, H.; Zhang, L.P.; Xu, L.P.; Wang, Y.; Liu, Y.R.; Lai, Y.Y.; Shi, H.X.; et al. Prevalence and prognostic significance of c-KIT mutations in core binding factor acute myeloid leukemia: A comprehensive large-scale study from a single Chinese center. Leuk. Res. 2014, 38, 1435–1440. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.B.; Ho, P.A.; Zeng, R.; Ravindranath, Y.; Dahl, G.; Lacayo, N.J.; Becton, D.; Chang, M.; et al. Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 2010, 115, 2372–2379. [Google Scholar] [CrossRef]
- Pietsch, T.; Kyas, U.; Steffens, U.; Yakisan, E.; Hadam, M.R.; Ludwig, W.D.; Zsebo, K.; Welte, K. Effects of human stem cell factor (c-kit ligand) on proliferation of myeloid leukemia cells: Heterogeneity in response and synergy with other hematopoietic growth factors. Blood 1992, 80, 1199–1206. [Google Scholar] [CrossRef]
- Hassan, H.T.; Zander, A. Stem cell factor as a survival and growth factor in human normal and malignant hematopoiesis. Acta Haematol. 1996, 95, 257–262. [Google Scholar] [CrossRef]
- Dos Santos, C.; McDonald, T.; Ho, Y.W.; Liu, H.; Lin, A.; Forman, S.J.; Kuo, Y.H.; Bhatia, R. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood 2013, 122, 1900–1913. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Blanke, C.D.; Druker, B.J.; Corless, C.L. Inhibition of KIT tyrosine kinase activity: A novel molecular approach to the treatment of KIT-positive malignancies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 1692–1703. [Google Scholar] [CrossRef]
- Advani, A.S. Targeting the c-kit receptor in the treatment of acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2006, 1, 101–107. [Google Scholar] [CrossRef]
- Dorison, A.; Dussaule, J.C.; Chatziantoniou, C. The Role of Discoidin Domain Receptor 1 in Inflammation, Fibrosis and Renal Disease. Nephron 2017, 137, 212–220. [Google Scholar] [CrossRef]
- Valiathan, R.R.; Marco, M.; Leitinger, B.; Kleer, C.G.; Fridman, R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [Google Scholar] [CrossRef]
- Borza, C.M.; Pozzi, A. Discoidin domain receptors in disease. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 34, 185–192. [Google Scholar] [CrossRef]
- Chiaretti, S.; Li, X.; Gentleman, R.; Vitale, A.; Wang, K.S.; Mandelli, F.; Foa, R.; Ritz, J. Gene expression profiles of B-lineage adult acute lymphocytic leukemia reveal genetic patterns that identify lineage derivation and distinct mechanisms of transformation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 7209–7219. [Google Scholar] [CrossRef]
- Barisione, G.; Fabbi, M.; Cutrona, G.; De Cecco, L.; Zupo, S.; Leitinger, B.; Gentile, M.; Manzoni, M.; Neri, A.; Morabito, F.; et al. Heterogeneous expression of the collagen receptor DDR1 in chronic lymphocytic leukaemia and correlation with progression. Blood Cancer J. 2017, 6, e513. [Google Scholar] [CrossRef][Green Version]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef]
- Loriaux, M.M.; Levine, R.L.; Tyner, J.W.; Frohling, S.; Scholl, C.; Stoffregen, E.P.; Wernig, G.; Erickson, H.; Eide, C.A.; Berger, R.; et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood 2008, 111, 4788–4796. [Google Scholar] [CrossRef]
- Barquilla, A.; Pasquale, E.B. Eph receptors and ephrins: Therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef]
- Boyd, A.W.; Ward, L.D.; Wicks, I.P.; Simpson, R.J.; Salvaris, E.; Wilks, A.; Welch, K.; Loudovaris, M.; Rockman, S.; Busmanis, I. Isolation and characterization of a novel receptor-type protein tyrosine kinase (hek) from a human pre-B cell line. J. Biol. Chem. 1992, 267, 3262–3267. [Google Scholar] [CrossRef]
- Wicks, I.P.; Wilkinson, D.; Salvaris, E.; Boyd, A.W. Molecular cloning of HEK, the gene encoding a receptor tyrosine kinase expressed by human lymphoid tumor cell lines. Proc. Natl. Acad. Sci. USA 1992, 89, 1611–1615. [Google Scholar] [CrossRef]
- Lawrenson, I.D.; Wimmer-Kleikamp, S.H.; Lock, P.; Schoenwaelder, S.M.; Down, M.; Boyd, A.W.; Alewood, P.F.; Lackmann, M. Ephrin-A5 induces rounding, blebbing and de-adhesion of EphA3-expressing 293T and melanoma cells by CrkII and Rho-mediated signalling. J. Cell Sci. 2002, 115, 1059–1072. [Google Scholar]
- Wimmer-Kleikamp, S.H.; Nievergall, E.; Gegenbauer, K.; Adikari, S.; Mansour, M.; Yeadon, T.; Boyd, A.W.; Patani, N.R.; Lackmann, M. Elevated protein tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood 2008, 112, 721–732. [Google Scholar] [CrossRef]
- Walter, M.J.; Payton, J.E.; Ries, R.E.; Shannon, W.D.; Deshmukh, H.; Zhao, Y.; Baty, J.; Heath, S.; Westervelt, P.; Watson, M.A.; et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc. Natl. Acad. Sci. USA 2009, 106, 12950–12955. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Liu, L.; Zhao, X.; Wu, Q.; Yu, B.; Shao, Y.; Yang, H.; Fu, X.; Wan, J.; Zhang, W. Copy number variations of EphA3 are associated with multiple types of hematologic malignancies. Clin. Lymphoma Myeloma Leuk. 2011, 11, 50–53. [Google Scholar] [CrossRef]
- Steube, K.G.; Meyer, C.; Habig, S.; Uphoff, C.C.; Drexler, H.G. Expression of receptor tyrosine kinase HTK (hepatoma transmembrane kinase) and HTK ligand by human leukemia-lymphoma cell lines. Leuk. Lymphoma 1999, 33, 371–376. [Google Scholar] [CrossRef]
- Shimoyama, M.; Matsuoka, H.; Tamekane, A.; Ito, M.; Iwata, N.; Inoue, R.; Chihara, K.; Furuya, A.; Hanai, N.; Matsui, T. T-cell-specific expression of kinase-defective Eph-family receptor protein, EphB6 in normal as well as transformed hematopoietic cells. Growth Factors 2000, 18, 63–78. [Google Scholar] [CrossRef]
- Muller-Tidow, C.; Schwable, J.; Steffen, B.; Tidow, N.; Brandt, B.; Becker, K.; Schulze-Bahr, E.; Halfter, H.; Vogt, U.; Metzger, R.; et al. High-throughput analysis of genome-wide receptor tyrosine kinase expression in human cancers identifies potential novel drug targets. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.L.; Trinidad, E.M.; de Garcillan, B.; Ballesteros, M.; Castellanos, M.; Cotillo, I.; Munoz, J.J.; Zapata, A.G. Expression profile of Eph receptors and ephrin ligands in healthy human B lymphocytes and chronic lymphocytic leukemia B-cells. Leuk. Res. 2009, 33, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Nakamura, T.; Canaani, E.; Croce, C.M. ALL1 fusion proteins induce deregulation of EphA7 and ERK phosphorylation in human acute leukemias. Proc. Natl. Acad. Sci. USA 2007, 104, 14442–14447. [Google Scholar] [CrossRef] [PubMed]
- Charmsaz, S.; Scott, A.M.; Boyd, A.W. Targeted therapies in hematological malignancies using therapeutic monoclonal antibodies against Eph family receptors. Exp. Hematol. 2017, 54, 31–39. [Google Scholar] [CrossRef]
- Merchant, A.A.; Jorapur, A.; McManus, A.; Liu, R.; Krasnoperov, V.; Chaudhry, P.; Singh, M.; Harton, L.; Agajanian, M.; Kim, M.; et al. EPHB4 is a therapeutic target in AML and promotes leukemia cell survival via AKT. Blood Adv. 2017, 1, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Bai, H.; Fang, Z.H.; Lopez, G.; Yang, H.; Tong, W.; Wang, Z.Z.; Garcia-Manero, G. Aberrant DNA methylation and epigenetic inactivation of Eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood 2010, 115, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Kampen, K.R.; Scherpen, F.J.; Garcia-Manero, G.; Yang, H.; Kaspers, G.J.; Cloos, J.; Zwaan, C.M.; van den Heuvel-Eibrink, M.M.; Kornblau, S.M.; De Bont, E.S. EphB1 Suppression in Acute Myelogenous Leukemia: Regulating the DNA Damage Control System. Mol. Cancer Res. 2015, 13, 982–992. [Google Scholar] [CrossRef] [PubMed]
- El Zawily, A.; McEwen, E.; Toosi, B.; Vizeacoumar, F.S.; Freywald, T.; Vizeacoumar, F.J.; Freywald, A. The EphB6 receptor is overexpressed in pediatric T cell acute lymphoblastic leukemia and increases its sensitivity to doxorubicin treatment. Sci. Rep. 2017, 7, 14767. [Google Scholar] [CrossRef]
- Wrobel, T.; Pogrzeba, J.; Stefanko, E.; Wojtowicz, M.; Jazwiec, B.; Dzietczenia, J.; Mazur, G.; Kuliczkowski, K. Expression of Eph A4, Eph B2 and Eph B4 receptors in AML. Pathol. Oncol. Res. Por 2014, 20, 901–907. [Google Scholar] [CrossRef]
- Tyner, J.W.; Deininger, M.W.; Loriaux, M.M.; Chang, B.H.; Gotlib, J.R.; Willis, S.G.; Erickson, H.; Kovacsovics, T.; O’Hare, T.; Heinrich, M.C.; et al. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc. Natl. Acad. Sci. USA 2009, 106, 8695–8700. [Google Scholar] [CrossRef]
- Oricchio, E.; Nanjangud, G.; Wolfe, A.L.; Schatz, J.H.; Mavrakis, K.J.; Jiang, M.; Liu, X.; Bruno, J.; Heguy, A.; Olshen, A.B.; et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell 2011, 147, 554–564. [Google Scholar] [CrossRef]
- Lieu, C.; Kopetz, S. The SRC family of protein tyrosine kinases: A new and promising target for colorectal cancer therapy. Clin. Colorectal Cancer 2010, 9, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef]
- Paul, J.M.; Toosi, B.; Vizeacoumar, F.S.; Bhanumathy, K.K.; Li, Y.; Gerger, C.; El Zawily, A.; Freywald, T.; Anderson, D.H.; Mousseau, D.; et al. Targeting synthetic lethality between the SRC kinase and the EPHB6 receptor may benefit cancer treatment. Oncotarget 2016, 7, 50027–50042. [Google Scholar] [CrossRef] [PubMed]
- Danhauser-Riedl, S.; Warmuth, M.; Druker, B.J.; Emmerich, B.; Hallek, M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996, 56, 3589–3596. [Google Scholar]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Recher, C. A critical role for Lyn in acute myeloid leukemia. Blood 2008, 111, 2269–2279. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, Y.; Pelletier, S.; Buchdunger, E.; Warmuth, M.; Fabbro, D.; Hallek, M.; Van Etten, R.A.; Li, S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat. Genet. 2004, 36, 453–461. [Google Scholar] [CrossRef]
- Weir, M.C.; Shu, S.T.; Patel, R.K.; Hellwig, S.; Chen, L.; Tan, L.; Gray, N.S.; Smithgall, T.E. Selective Inhibition of the Myeloid Src-Family Kinase Fgr Potently Suppresses AML Cell Growth in Vitro and in Vivo. ACS Chem. Biol. 2018, 13, 1551–1559. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid Src-family kinases is associated with poor prognosis in AML and influences Flt3-ITD kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef]
- Roversi, F.M.; Pericole, F.V.; Machado-Neto, J.A.; da Duarte, A.S.S.; Longhini, A.L.; Corrocher, F.A.; Palodetto, B.; Ferro, K.P.; Rosa, R.G.; Baratti, M.O.; et al. Hematopoietic cell kinase (HCK) is a potential therapeutic target for dysplastic and leukemic cells due to integration of erythropoietin/PI3K pathway and regulation of erythropoiesis: HCK in erythropoietin/PI3K pathway. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 450–461. [Google Scholar] [CrossRef]
- Ingley, E. Functions of the Lyn tyrosine kinase in health and disease. Cell Commun. Signal. 2012, 10, 21. [Google Scholar] [CrossRef]
- Contri, A.; Brunati, A.M.; Trentin, L.; Cabrelle, A.; Miorin, M.; Cesaro, L.; Pinna, L.A.; Zambello, R.; Semenzato, G.; Donella-Deana, A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J. Clin. Investig. 2005, 115, 369–378. [Google Scholar] [CrossRef]
- Takata, M.; Sabe, H.; Hata, A.; Inazu, T.; Homma, Y.; Nukada, T.; Yamamura, H.; Kurosaki, T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 1994, 13, 1341–1349. [Google Scholar] [CrossRef]
- Stevenson, F.K.; Krysov, S.; Davies, A.J.; Steele, A.J.; Packham, G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2011, 118, 4313–4320. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Gandhi, V. Protein kinases: Emerging therapeutic targets in chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2012, 21, 409–423. [Google Scholar] [CrossRef]
- Hussein, K.; von Neuhoff, N.; Busche, G.; Buhr, T.; Kreipe, H.; Bock, O. Opposite expression pattern of Src kinase Lyn in acute and chronic haematological malignancies. Ann. Hematol. 2009, 88, 1059–1067. [Google Scholar] [CrossRef]
- Geahlen, R.L. Getting Syk: Spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 2014, 35, 414–422. [Google Scholar] [CrossRef]
- Rinaldi, A.; Kwee, I.; Taborelli, M.; Largo, C.; Uccella, S.; Martin, V.; Poretti, G.; Gaidano, G.; Calabrese, G.; Martinelli, G.; et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br. J. Haematol. 2006, 132, 303–316. [Google Scholar] [CrossRef]
- Baudot, A.D.; Jeandel, P.Y.; Mouska, X.; Maurer, U.; Tartare-Deckert, S.; Raynaud, S.D.; Cassuto, J.P.; Ticchioni, M.; Deckert, M. The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCdelta and proteasome-dependent regulation of Mcl-1 expression. Oncogene 2009, 28, 3261–3273. [Google Scholar] [CrossRef]
- Buchner, M.; Fuchs, S.; Prinz, G.; Pfeifer, D.; Bartholome, K.; Burger, M.; Chevalier, N.; Vallat, L.; Timmer, J.; Gribben, J.G.; et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009, 69, 5424–5432. [Google Scholar] [CrossRef]
- Young, R.M.; Hardy, I.R.; Clarke, R.L.; Lundy, N.; Pine, P.; Turner, B.C.; Potter, T.A.; Refaeli, Y. Mouse models of non-Hodgkin lymphoma reveal Syk as an important therapeutic target. Blood 2009, 113, 2508–2516. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Sharman, J.; Sweetenham, J.; Johnston, P.B.; Vose, J.M.; Lacasce, A.; Schaefer-Cutillo, J.; De Vos, S.; Sinha, R.; Leonard, J.P.; et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010, 115, 2578–2585. [Google Scholar] [CrossRef]
- Puissant, A.; Fenouille, N.; Alexe, G.; Pikman, Y.; Bassil, C.F.; Mehta, S.; Du, J.; Kazi, J.U.; Luciano, F.; Ronnstrand, L.; et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell 2014, 25, 226–242. [Google Scholar] [CrossRef]
- Polak, A.; Bialopiotrowicz, E.; Krzymieniewska, B.; Wozniak, J.; Stojak, M.; Cybulska, M.; Kaniuga, E.; Mikula, M.; Jablonska, E.; Gorniak, P.; et al. SYK inhibition targets acute myeloid leukemia stem cells by blocking their oxidative metabolism. Cell Death Dis. 2020, 11, 956. [Google Scholar] [CrossRef]
- Rickert, R.C. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol. 2013, 13, 578–591. [Google Scholar] [CrossRef]
- Hahn, C.K.; Berchuck, J.E.; Ross, K.N.; Kakoza, R.M.; Clauser, K.; Schinzel, A.C.; Ross, L.; Galinsky, I.; Davis, T.N.; Silver, S.J.; et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell 2009, 16, 281–294. [Google Scholar] [CrossRef]
- Feldman, A.L.; Sun, D.X.; Law, M.E.; Novak, A.J.; Attygalle, A.D.; Thorland, E.C.; Fink, S.R.; Vrana, J.A.; Caron, B.L.; Morice, W.G.; et al. Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas. Leukemia 2008, 22, 1139–1143. [Google Scholar] [CrossRef]
- Rane, S.G.; Reddy, E.P. Janus kinases: Components of multiple signaling pathways. Oncogene 2000, 19, 5662–5679. [Google Scholar] [CrossRef]
- Xiang, Z.; Zhao, Y.; Mitaksov, V.; Fremont, D.H.; Kasai, Y.; Molitoris, A.; Ries, R.E.; Miner, T.L.; McLellan, M.D.; DiPersio, J.F.; et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood 2008, 111, 4809–4812. [Google Scholar] [CrossRef]
- Springuel, L.; Renauld, J.C.; Knoops, L. JAK kinase targeting in hematologic malignancies: A sinuous pathway from identification of genetic alterations towards clinical indications. Haematologica 2015, 100, 1240–1253. [Google Scholar] [CrossRef]
- Nicolae, A.; Xi, L.; Pham, T.H.; Pham, T.A.; Navarro, W.; Meeker, H.G.; Pittaluga, S.; Jaffe, E.S.; Raffeld, M. Mutations in the JAK/STAT and RAS signaling pathways are common in intestinal T-cell lymphomas. Leukemia 2016, 30, 2245–2247. [Google Scholar] [CrossRef]
- Wahnschaffe, L.; Braun, T.; Timonen, S.; Giri, A.K.; Schrader, A.; Wagle, P.; Almusa, H.; Johansson, P.; Bellanger, D.; Lopez, C.; et al. JAK/STAT-Activating Genomic Alterations Are a Hallmark of T-PLL. Cancers 2019, 11, 1833. [Google Scholar] [CrossRef]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffe, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef]
- Bernard, O.A.; Romana, S.P.; Poirel, H.; Berger, R. Molecular cytogenetics of t(12;21) (p13;q22). Leuk. Lymphoma 1996, 23, 459–465. [Google Scholar] [CrossRef]
- Golub, T.R.; McLean, T.; Stegmaier, K.; Carroll, M.; Tomasson, M.; Gilliland, D.G. The TEL gene and human leukemia. Biochim. Biophys. Acta 1996, 1288, M7-10. [Google Scholar] [CrossRef]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Tago, K.; Abe, M.; Sonoda, Y.; Kasahara, T. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J. Biol. Chem. 2010, 285, 5296–5307. [Google Scholar] [CrossRef]
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; La Starza, R.; et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 1301–1310. [Google Scholar] [CrossRef]
- Flex, E.; Petrangeli, V.; Stella, L.; Chiaretti, S.; Hornakova, T.; Knoops, L.; Ariola, C.; Fodale, V.; Clappier, E.; Paoloni, F.; et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 2008, 205, 751–758. [Google Scholar] [CrossRef]
- Degryse, S.; Cools, J. JAK kinase inhibitors for the treatment of acute lymphoblastic leukemia. J. Hematol. Oncol. 2015, 8, 91. [Google Scholar] [CrossRef]
- Habbel, J.; Arnold, L.; Chen, Y.; Mollmann, M.; Bruderek, K.; Brandau, S.; Duhrsen, U.; Hanoun, M. Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro. Blood Adv. 2020, 4, 3000–3010. [Google Scholar] [CrossRef]
- Koo, G.C.; Tan, S.Y.; Tang, T.; Poon, S.L.; Allen, G.E.; Tan, L.; Chong, S.C.; Ong, W.S.; Tay, K.; Tao, M.; et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012, 2, 591–597. [Google Scholar] [CrossRef]
- Al-Hussaini, M.; DiPersio, J.F. Small molecule inhibitors in acute myeloid leukemia: From the bench to the clinic. Expert Rev. Hematol. 2014, 7, 439–464. [Google Scholar] [CrossRef]
- Force, T.; Kuida, K.; Namchuk, M.; Parang, K.; Kyriakis, J.M. Inhibitors of protein kinase signaling pathways: Emerging therapies for cardiovascular disease. Circulation 2004, 109, 1196–1205. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef]
- Converso, A.; Hartingh, T.; Garbaccio, R.M.; Tasber, E.; Rickert, K.; Fraley, M.E.; Yan, Y.; Kreatsoulas, C.; Stirdivant, S.; Drakas, B.; et al. Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 1240–1244. [Google Scholar] [CrossRef]
- Vanderpool, D.; Johnson, T.O.; Ping, C.; Bergqvist, S.; Alton, G.; Phonephaly, S.; Rui, E.; Luo, C.; Deng, Y.L.; Grant, S.; et al. Characterization of the CHK1 allosteric inhibitor binding site. Biochemistry 2009, 48, 9823–9830. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Hidaka, H.; Inagaki, M.; Kawamoto, S.; Sasaki, Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry 1984, 23, 5036–5041. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.; Johnson, J.R.; Duan, J.; Gobburu, J.; Rahman, A.; Benson, K.; Leighton, J.; Kim, S.K.; Wood, R.; et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 935–942. [Google Scholar]
- Dagher, R.; Cohen, M.; Williams, G.; Rothmann, M.; Gobburu, J.; Robbie, G.; Rahman, A.; Chen, G.; Staten, A.; Griebel, D.; et al. Approval summary: Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3034–3038. [Google Scholar]
- Kim, T.D.; Dorken, B.; le Coutre, P. Nilotinib for the treatment of chronic myeloid leukemia. Expert Rev. Hematol. 2008, 1, 29–39. [Google Scholar] [CrossRef]
- Amsberg, G.K.; Koschmieder, S. Profile of bosutinib and its clinical potential in the treatment of chronic myeloid leukemia. Oncotargets Ther. 2013, 6, 99–106. [Google Scholar] [CrossRef]
- Shamroe, C.L.; Comeau, J.M. Ponatinib: A new tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Ann. Pharmacother. 2013, 47, 1540–1546. [Google Scholar] [CrossRef]
- Zabriskie, M.S.; Vellore, N.A.; Gantz, K.C.; Deininger, M.W.; O’Hare, T. Radotinib is an effective inhibitor of native and kinase domain-mutant BCR-ABL1. Leukemia 2015, 29, 1939–1942. [Google Scholar] [CrossRef]
- Cameron, F.; Sanford, M. Ibrutinib: First global approval. Drugs 2014, 74, 263–271. [Google Scholar] [CrossRef]
- Shah, A.; Mangaonkar, A. Idelalisib: A Novel PI3Kdelta Inhibitor for Chronic Lymphocytic Leukemia. Ann. Pharmacother. 2015, 49, 1162–1170. [Google Scholar] [CrossRef]
- Markham, A.; Dhillon, S. Acalabrutinib: First Global Approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef]
- Kim, E.S. Midostaurin: First Global Approval. Drugs 2017, 77, 1251–1259. [Google Scholar] [CrossRef]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First Approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Ling, Y.; Xie, Q.; Zhang, Z.; Zhang, H. Protein kinase inhibitors for acute leukemia. Biomark. Res. 2018, 6, 8. [Google Scholar] [CrossRef]
- Morales, M.L.; Arenas, A.; Ortiz-Ruiz, A.; Leivas, A.; Rapado, I.; Rodriguez-Garcia, A.; Castro, N.; Zagorac, I.; Quintela-Fandino, M.; Gomez-Lopez, G.; et al. MEK inhibition enhances the response to tyrosine kinase inhibitors in acute myeloid leukemia. Sci. Rep. 2019, 9, 18630. [Google Scholar] [CrossRef]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef]
- Owen, C.; Berinstein, N.L.; Christofides, A.; Sehn, L.H. Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr. Oncol. 2019, 26, e233–e240. [Google Scholar] [CrossRef]
- Kenzik, K.M.; Bhatia, R.; Bhatia, S. Expenditures for First- and Second-Generation Tyrosine Kinase Inhibitors before and After Transition of Imatinib to Generic Status. JAMA Oncol. 2020, 6, 542–546. [Google Scholar] [CrossRef]
- Zhang, B.S.; Chen, Y.P.; Lv, J.L.; Yang, Y. Comparison of the Efficacy of Nilotinib and Imatinib in the Treatment of Chronic Myeloid Leukemia. J. Coll. Physicians Surg. Pak. 2019, 29, 631–634. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).