
Role of FGFR2c and Its PKCε Downstream Signaling in the Control of EMT and Autophagy in Pancreatic Ductal Adenocarcinoma Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Treatments
2.2. Immunofluorescence
2.3. Western Blot Analysis
2.4. Transmission Electron Microscopy
2.5. Primers
2.6. RNA Extraction and cDNA Synthesis
2.7. PCR Amplification and Real-Time Quantitation
2.8. Statistics
3. Results
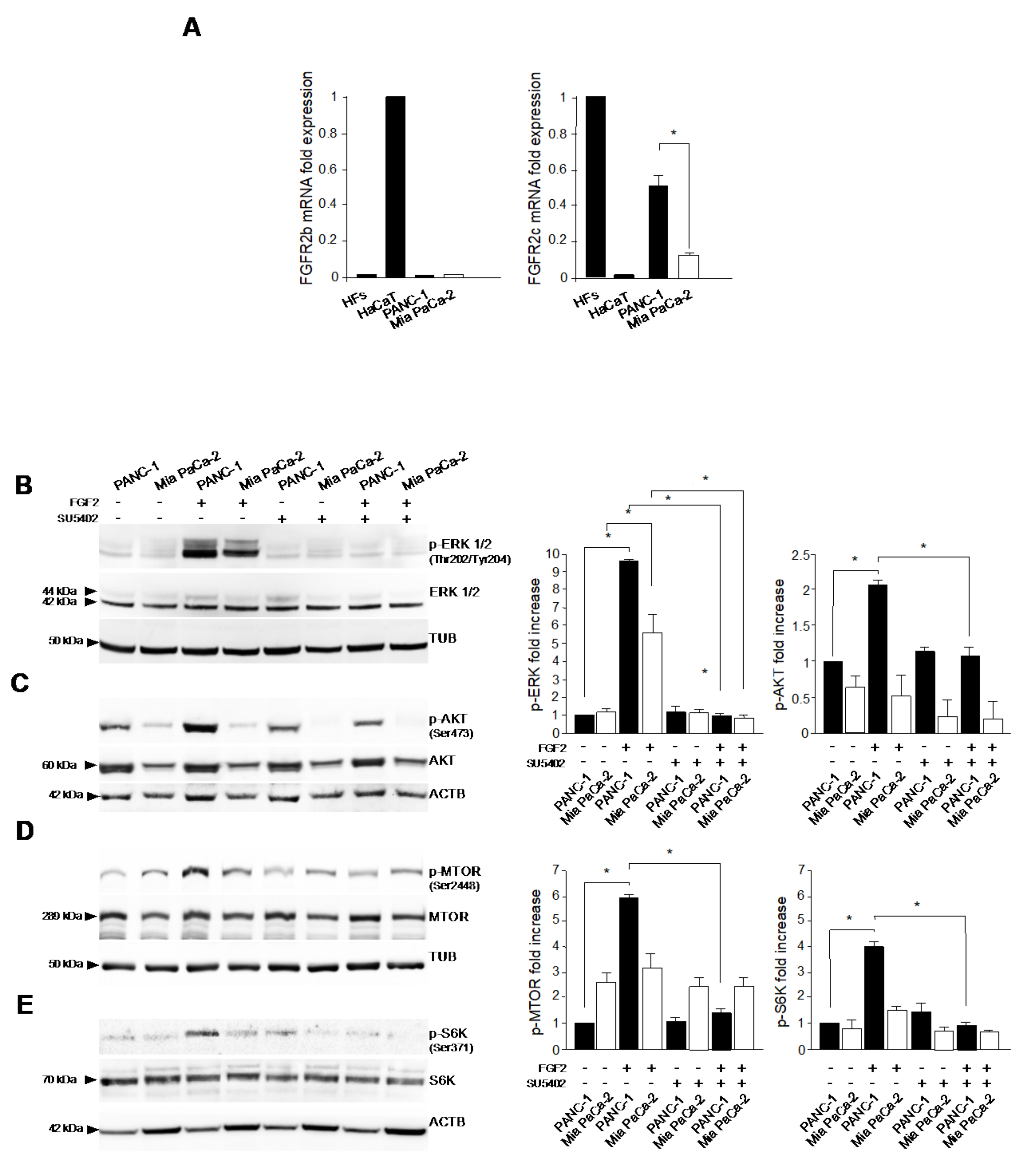
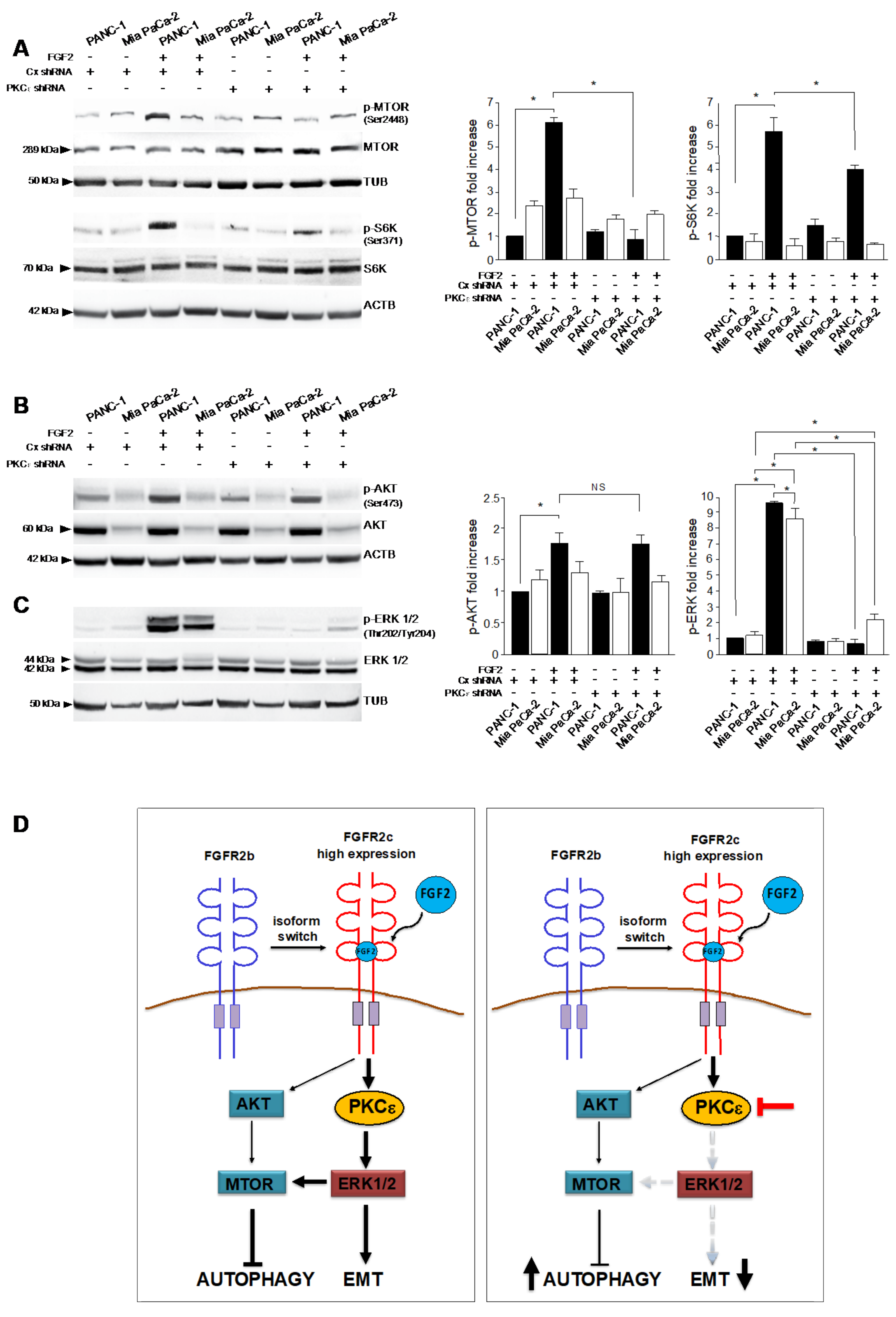
3.1. FGFR2c Aberrant Expression Affects the Intracellular Signaling
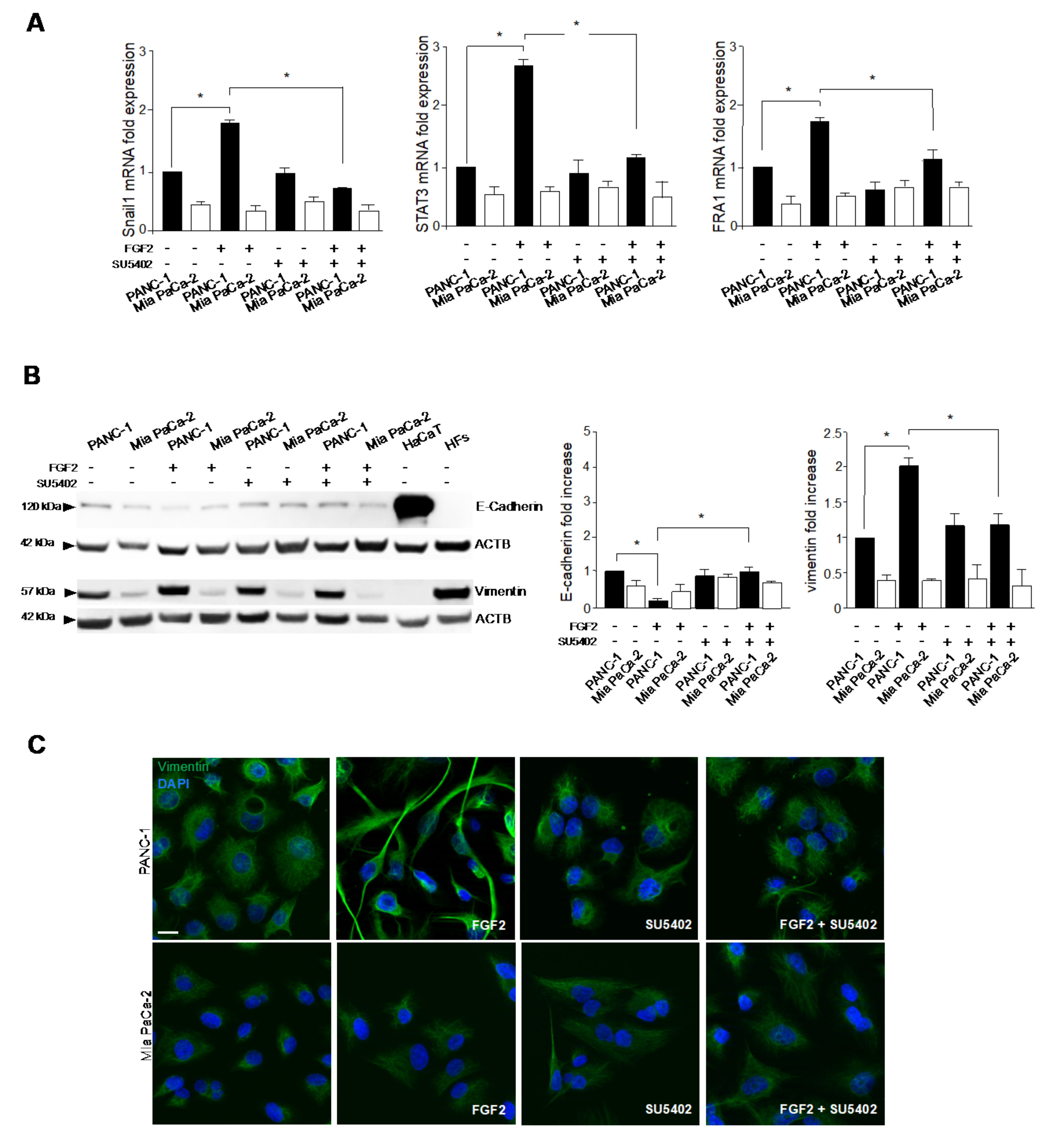
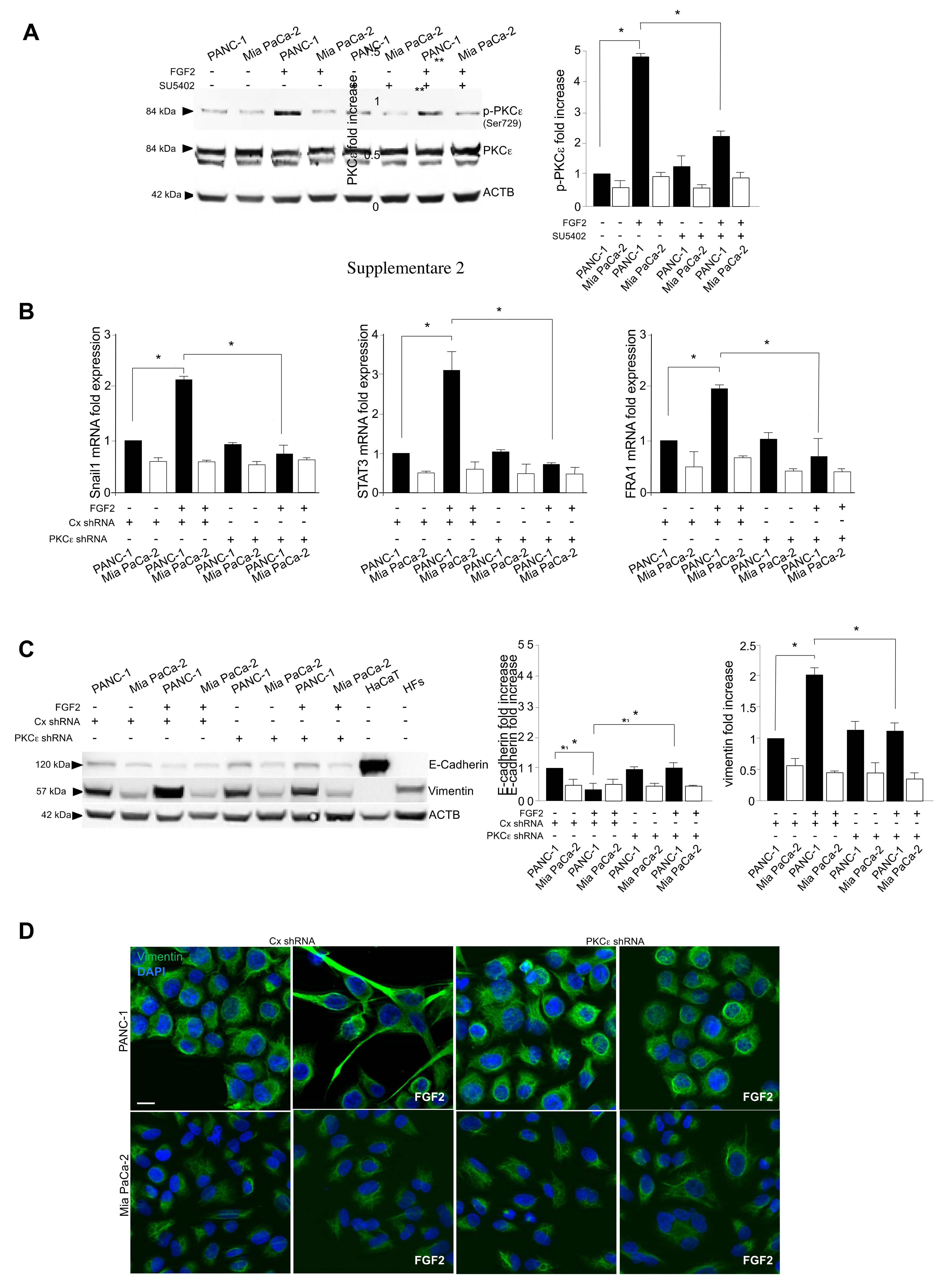
3.2. FGFR2c Expression Enhances the EMT Phenotype in Response to FGF2
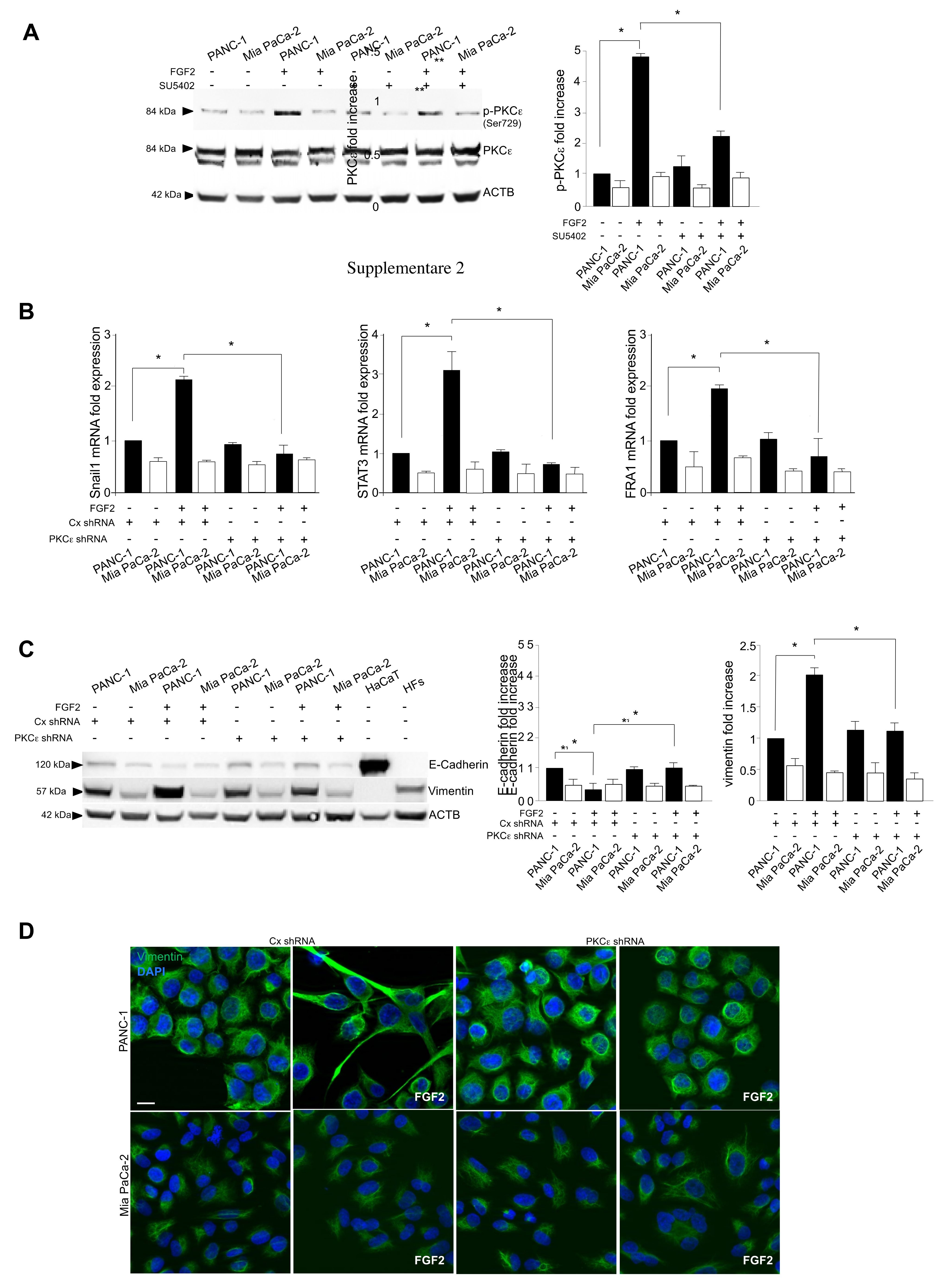
3.3. The Activation of PKCε Is the Key Molecular Event Downstream FGFR2c Underlying EMT Induction
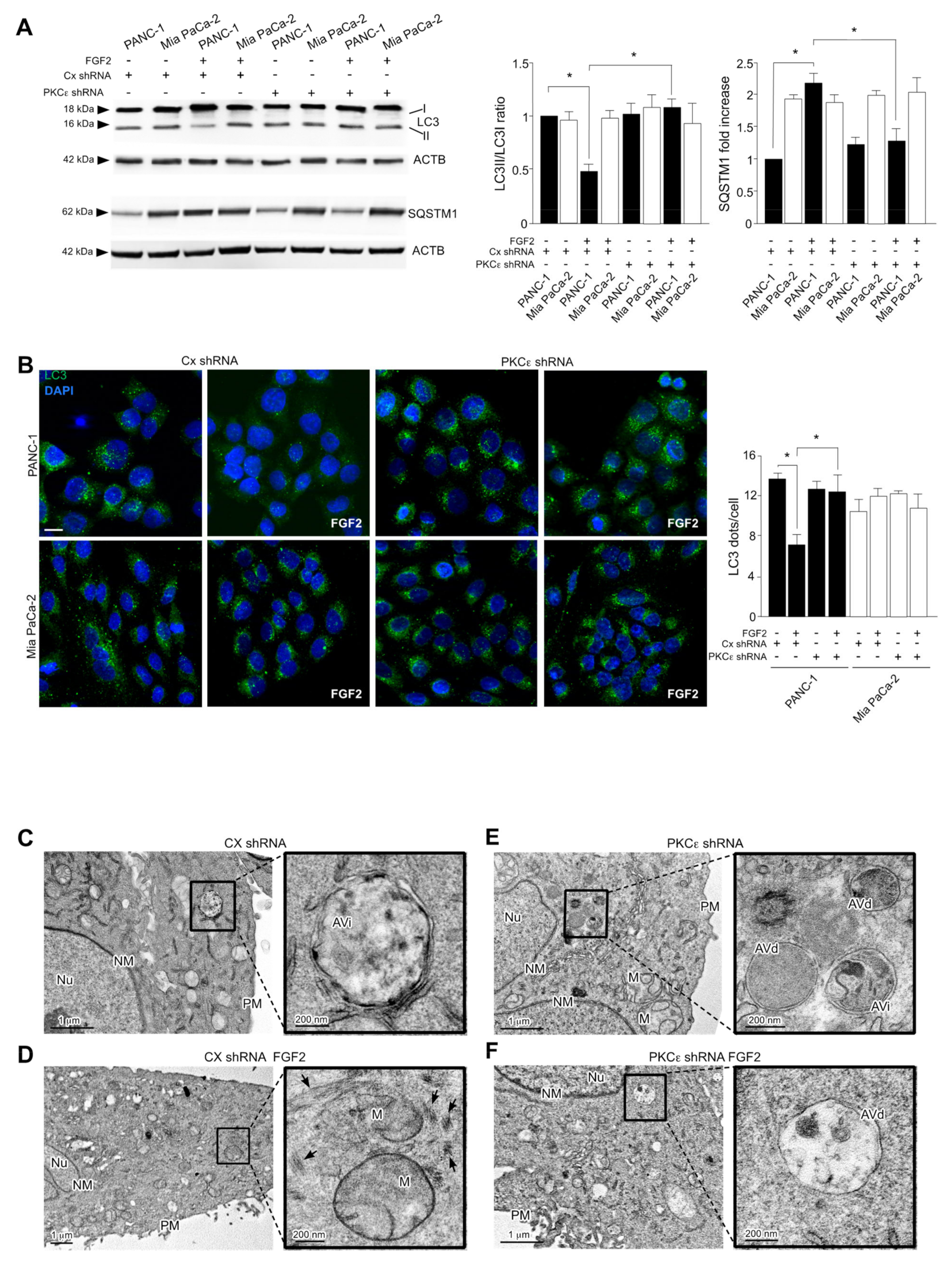
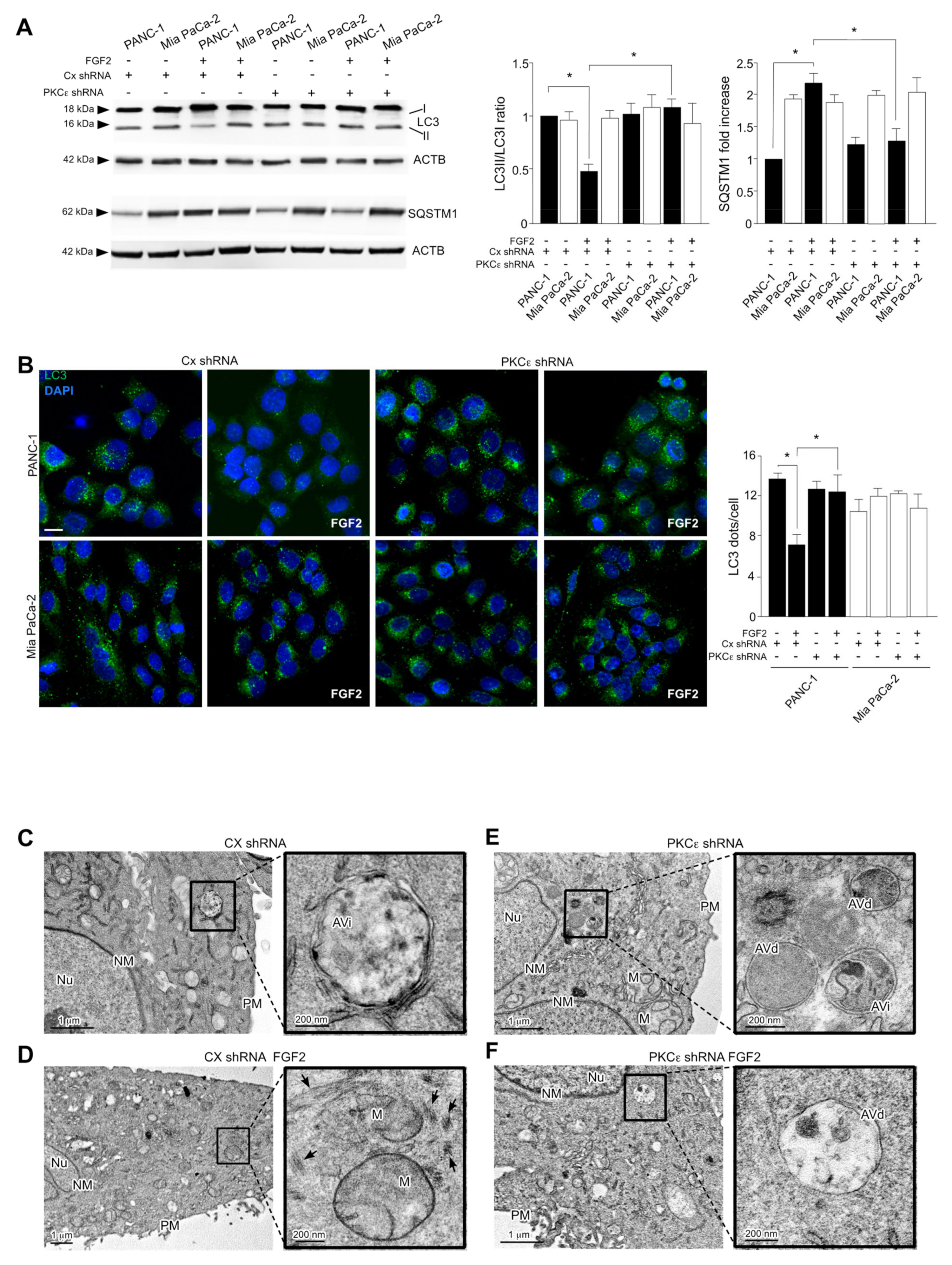
3.4. PKCε Signaling Negatively Impacts on the Autophagic Process
3.5. PKCε Signaling Interferes with Autophagy Converging on ERK1/2 Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singhi, A.D.; Wood, L.D. Early detection of pancreatic cancer using DNA-based molecular approaches. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 457–468. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Novel therapeutic approaches for pancreatic cancer by combined targeting of RAF→MEK→ERK signaling and autophagy survival response. Ann. Transl. Med. 2019, 7, S153. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.M.; Scotti Buzhardt, M.L.; Erdogan, E.; Li, S.; Inman, K.S.; Fields, A.P.; Murray, N.R. A small molecule inhibitor of atypical protein kinase C signaling inhibits pancreatic cancer cell transformed growth and invasion. Oncotarget 2015, 6, 15297–15310. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, S.; Fan, H.; Ji, D.; Chen, C.; Sheng, W. GINS2 promotes EMT in pancreatic cancer via specifically stimulating ERK/MAPK signaling. Cancer Gene Ther. 2021, 28, 839–849. [Google Scholar] [CrossRef]
- Safa, A.R. Epithelial-mesenchymal transition: A hallmark in pancreatic cancer stem cell migration, metastasis formation, and drug resistance. J. Cancer Metastas. Treat. 2020, 6, 36. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; Andre, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakov, N. Protein Kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef]
- Ranieri, D.; Nanni, M.; Persechino, F.; Torrisi, M.R.; Belleudi, F. Role of PKCε in the epithelial-mesenchymal transition induced by FGFR2 isoform switch. Cell Commun. Signal 2020, 18, 76. [Google Scholar] [CrossRef]
- Ranieri, D.; French, D.; Raffa, S.; Guttieri, L.; Torrisi, M.R.; Belleudi, F. Expression of the E5 oncoprotein of HPV16 impacts on the molecular profiles of EMT-related and differentiation genes in ectocervical low-grade lesions. Int. J. Mol. Sci. 2021, 22, 6534. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Matsuda, Y.; Yamamoto, T.; Uchida, E.; Korc, M.; Naito, Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am. J. Pathol. 2012, 180, 1928–1941. [Google Scholar] [CrossRef] [Green Version]
- Ueda, J.; Matsuda, Y.; Yamahatsu, K.; Uchida, E.; Naito, Z.; Korc, M.; Ishiwata, T. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene 2014, 33, 4485–4495. [Google Scholar] [CrossRef] [Green Version]
- Ishiwata, T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Front Biosci. 2018, 23, 626–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.; Mao, M.; Tan, Y.; Mo, X.; Meng, X.; Cao, M.; Zhong, C.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 1876, 188565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Tooze, S. The role of autophagy in pancreatic cancer—Recent advances. Biology 2019, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O′Prey, J.; Morton, J.P.; Nixon, C.C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Basu, A. Regulation of autophagy by protein kinase C-ε in breast cancer cells. Int. J. Mol. Sci. 2020, 21, 4247. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Persechino, F.; Ranieri, D.; Guttieri, L.; Nanni, M.; Torrisi, M.R.; Belleudi, F. Expression profile of fibroblast growth factor receptors, keratinocyte differentiation markers, and epithelial mesenchymal transition-related genes in actinic keratosis: A possible predictive factor for malignant progression? Biology 2021, 10, 331. [Google Scholar] [CrossRef]
- Ranieri, D.; Nanni, M.; Guttieri, L.; Torrisi, M.R.; Belleudi, F. The aberrant expression in epithelial cells of the mesenchymal isoform of FGFR2 controls the negative crosstalk between EMT and autophagy. J. Cell Mol. Med. 2021, 25, 4166–4172. [Google Scholar] [CrossRef]
- Tiong, K.H.; Mah, L.Y.; Leong, C.O. Functional roles of Fibroblast Growth Factor Receptors (FGFRs) signaling in human cancers. Apoptosis 2013, 18, 1447–1468. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.; Kluger, H.; Varsano, T.; Lee, K.; Scheffler, I.; Rimm, D.L.; Ideker, T.; Ronai, Z.A. PKCε promotes oncogenic functions of ATF2 in the nucleus while blocking its apoptotic function at mitochondria. Cell 2012, 148, 543–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karavana, V.N.; Gakiopoulou, H.; Lianos, E.A. Expression of Ser729 phosphorylated PKC epsilon in experimental crescentic glomerulonephritis: An immunohistochemical study. Eur. J. Histochem. 2014, 58, 2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschella, P.C.; Rao, V.U.; McDermott, P.J.; Kuppuswamy, D. Regulation of mTOR and S6K1 activation by the nPKC isoforms, PKCε and PKCδ, in adult cardiac muscle cells. J. Mol. Cell Cardiol. 2007, 43, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Will, M.; Qin, A.C.R.; Toy, W.; Yao, Z.; Rodrik-Outmezguine, V.; Schneider, C.; Huang, X.; Monian, P.; Jiang, X.; de Stanchina, E.; et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS–ERK signaling. Cancer Discov 2014, 4, 334–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [Green Version]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Futakuchi, M.; Heavican, T.; Iqbal, J.; Singh, R.K. Cancer-associated fibroblasts enhance survival and progression of the aggressive pancreatic tumor Via FGF-2 and CXCL8. Cancer Microenviron. 2019, 12, 37–46. [Google Scholar] [CrossRef]
- Nanni, M.; Ranieri, D.; Persechino, F.; Torrisi, M.R.; Belleudi, F. The aberrant expression of the mesenchymal variant of FGFR2 in the epithelial context inhibits autophagy. Cells 2019, 8, 653. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranieri, D.; Guttieri, L.; Raffa, S.; Torrisi, M.R.; Belleudi, F. Role of FGFR2c and Its PKCε Downstream Signaling in the Control of EMT and Autophagy in Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 4993. https://doi.org/10.3390/cancers13194993
Ranieri D, Guttieri L, Raffa S, Torrisi MR, Belleudi F. Role of FGFR2c and Its PKCε Downstream Signaling in the Control of EMT and Autophagy in Pancreatic Ductal Adenocarcinoma Cells. Cancers. 2021; 13(19):4993. https://doi.org/10.3390/cancers13194993
Chicago/Turabian StyleRanieri, Danilo, Luisa Guttieri, Salvatore Raffa, Maria Rosaria Torrisi, and Francesca Belleudi. 2021. "Role of FGFR2c and Its PKCε Downstream Signaling in the Control of EMT and Autophagy in Pancreatic Ductal Adenocarcinoma Cells" Cancers 13, no. 19: 4993. https://doi.org/10.3390/cancers13194993
APA StyleRanieri, D., Guttieri, L., Raffa, S., Torrisi, M. R., & Belleudi, F. (2021). Role of FGFR2c and Its PKCε Downstream Signaling in the Control of EMT and Autophagy in Pancreatic Ductal Adenocarcinoma Cells. Cancers, 13(19), 4993. https://doi.org/10.3390/cancers13194993