
Type 1 Nuclear Receptor Activity in Breast Cancer: Translating Preclinical Insights to the Clinic


Simple Summary
Abstract
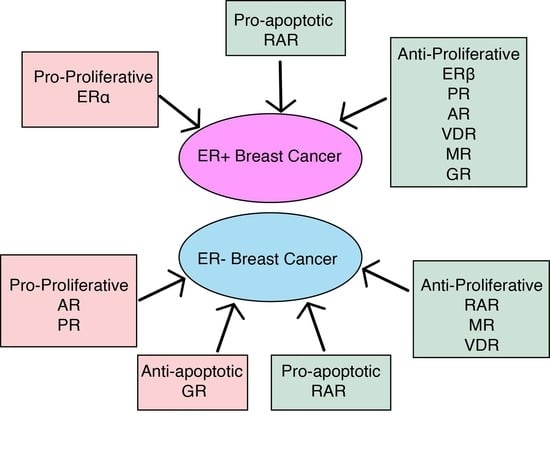
1. Nuclear Receptors in Breast Cancer
2. Nuclear Receptor Autoregulation and Crosstalk
3. Oestrogen Receptor Signalling
4. Progesterone Receptor Signalling
5. Androgen Receptor Signalling
6. Glucocorticoid Receptor Signalling
7. Other Nuclear Receptors
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beatson, G.T. On the treatment of inoperable cases of carcinoma of the mamma. Lancet 1896, 2, 1–14. [Google Scholar]
- Jensen, E.V.; Jacobson, H.I.; Walf, A.A.; Frye, C.A. Estrogen action: A historic perspective on the implications of considering al-ternative approaches. Physiol. Behav. 2010, 99, 151–162. [Google Scholar] [CrossRef]
- Bagamasbad, P.; Denver, R.J. Mechanisms and significance of nuclear receptor auto- and cross-regulation. Gen. Comp. Endocrinol. 2011, 170, 3–17. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, B.W. 90 YEARS of PROGESTERONE: Reminiscing on the origins of the field of progesterone and estrogen receptor action. J. Mol. Endocrinol. 2020, 65, C1–C4. [Google Scholar] [CrossRef]
- Conzen, S.D. Minireview: Nuclear Receptors and Breast Cancer. Mol. Endocrinol. 2008, 22, 2215–2228. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estébanez-Perpiña, E.; Brunsveld, L. Nuclear receptor crosstalk—Defining the mecha-nisms for therapeutic innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, B.W. Coregulators: From whence came these ‘master genes’. Mol. Endocrinol. 2007, 21, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.J. Family Matters: Collaboration and Conflict Among the Steroid Receptors Raises a Need for Group Therapy. Endocrinology 2016, 157, 4553–4560. [Google Scholar] [CrossRef]
- A Lamb, C.; I Vanzulli, S.; Lanari, C. Hormone receptors in breast cancer: More than estrogen receptors. Medicina 2019, 79, 540–545. [Google Scholar] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Siersbæk, R.D.; Kumar, S.; Carroll, J. Signaling pathways and steroid receptors modulating estrogen receptor α function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Doan, T.B.; Graham, J.D.; Clarke, C. Emerging functional roles of nuclear receptors in breast cancer. J. Mol. Endocrinol. 2017, 58, R169–R190. [Google Scholar] [CrossRef]
- Schmidt, D.; Wilson, M.D.; Spyrou, C.; Brown, G.D.; Odom, D.T. Europe PMC Funders Group ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods 2014, 48, 240–248. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.; Tarulli, G.; Serandour, A.A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nat. Cell Biol. 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Hickey, T.E.; Selth, L.A.; Chia, K.M.; Laven-Law, G.; Milioli, H.H.; Roden, D.; Jindal, S.; Hui, M.; Finlay-Schultz, J.; Ebrahimie, E.; et al. The androgen receptor is a tumor suppressor in estrogen receptor–positive breast cancer. Nat. Med. 2021, 27, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Taylor, C.; Brown, G.D.; Papachristou, E.; Carroll, J.; D’Santos, C.S. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat. Protoc. 2016, 11, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Papachristou, E.K.; Kishore, K.; Holding, A.N.; Harvey, K.; Roumeliotis, T.I.; Chilamakuri, C.S.R.; Omarjee, S.; Chia, K.M.; Swarbrick, A.; Lim, E.; et al. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Gburcik, V.; Picard, D. The cell-specific activity of the estrogen receptor a may be fine-tuned by phosphorylation-induced structural gymnastics. Nucl. Recept. Signal. 2006, 4, e005. [Google Scholar] [CrossRef]
- Ström, A.; Hartman, J.; Foster, J.S.; Kietz, S.; Wimalasena, J.; Gustafsson, J. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc. Natl. Acad. Sci. USA 2004, 101, 1566–1571. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the fork-head protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef]
- Group, C.; Cancer, B. Type and timing of menopausal hormone therapy and breast cancer risk: Individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019, 394, 1159–1168. [Google Scholar]
- Althuis, M.D.; Fergenbaum, J.H.; Garcia-Closas, M.; A Brinton, L.; Madigan, M.P.; E Sherman, M. Etiology of hormone receptor-defined breast cancer: A systematic review of the literature. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1558–1568. [Google Scholar]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Turner, N.C.; Jungsil PALOMA3 Study Group; Andre, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.; Stemmer, S.; Burris, H.; Yap, Y.-S.; Sonke, G.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.; Winer, E.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Cheng, J.; Bloomquist, E.; Sanchez, J.; Wedam, S.B.; Singh, H.; Amiri-Kordestani, L.; Ibrahim, A.; Sridhara, R.; Goldberg, K.B.; et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: A US Food and Drug Administration pooled analysis. Lancet Oncol. 2020, 21, 250–260. [Google Scholar] [CrossRef]
- Schettini, F.; Giudici, F.; Giuliano, M.; Cristofanilli, M.; Arpino, G.; del Mastro, L.; Puglisi, F.; de Placido, S.; Paris, I.; de Placido, P.; et al. Overall Survival of CDK4/6-Inhibitor-Based Treatments in Clinically Relevant Subgroups of Metastatic Breast Cancer: Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2020, 112, 1089–1097. [Google Scholar] [CrossRef]
- Schettini, F.; Giuliano, M.; Giudici, F.; Conte, B.; De Placido, P.; Venturini, S.; Rognoni, C.; Di Leo, A.; Locci, M.; Jerusalem, G.; et al. Endocrine-Based Treatments in Clinically-Relevant Subgroups of Hormone Receptor-Positive/HER2-Negative Metastatic Breast Cancer: Systematic Review and Meta-Analysis. Cancers 2021, 13, 1458. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): One cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Turner, S.; Chia, S.; Kanakamedala, H.; Hsu, W.; Park, J.; Chandiwana, D.; Ridolfi, A.; Yu, C.; Zarate, J.P.; Rugo, H.S. Effectiveness of Alpelisib + Fulvestrant Compared with Real-World Standard Treatment Among Patients with HR+, HER2-, PIK3CA-Mutated Breast Cancer. Oncologist 2021, 26, e1133–e1142. [Google Scholar] [CrossRef]
- Carroll, J.; A Meyer, C.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The Dynamic Structure of the Estrogen Receptor. J. Amino Acids 2011, 2011, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.; Brown, G.D.; Gojis, O.; Ellis, I.; Green, A.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Bin Mohamed, Y.; Orlov, Y.; Velkov, S.; Thoreau, H.; Mei, P.H.; et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef]
- Fuqua, S.A.W.; Gu, G.; Rechoum, Y. Estrogen receptor (ER) α mutations in breast cancer: Hidden in plain sight. Breast Cancer Res. Treat. 2014, 144, 11–19. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; DE Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.; Zotenko, E.; Locke, W.; Korbie, D.; Millar, E.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 2015, 6, 7758. [Google Scholar] [CrossRef]
- Atlas, T.C.G. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G Mutation in Estrogen Receptor-α: A Novel Mechanism for Acquired Endocrine Resistance in Breast Cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2016, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients re-ceiving fulvestrant. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhou, W.; Hafner, M.; Blake, R.A.; Chalouni, C.; Chen, I.; De Bruyn, T.; Giltnane, J.M.; Hartman, S.; Heidersbach, A.; et al. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178, 949–963.e18. [Google Scholar] [CrossRef]
- Puyang, X.; Furman, C.; Zheng, G.Z.; Wu, Z.J.; Banka, D.; B, K.A.; Agoulnik, S.; Bolduc, D.M.; Buonamici, S.; Caleb, B.; et al. Discovery of Selective Estrogen Receptor Covalent Antagonists for the Treatment of ERαWT and ERαMUT Breast Cancer. Cancer Discov. 2018, 8, 1176–1193. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Wang, J.S.; Pluard, T.; Johnston, S.; Morikawa, A.A.; Dees, C.E.; Jones, R.H.; Haley, B.; Armstrong, A.; Cohen, A.L.; et al. Abstract PD8-06: Phase I/II trial of H3B-6545, a novel selective estrogen receptor covalent antagonist (SER-CA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. Cancer Res. 2021, 81, PD8-06 LP. [Google Scholar]
- Stone, A.; Valdes-Mora, F.; Gee, J.M.W.; Farrow, L.; McClelland, R.A.; Fiegl, H.; Dutkowski, C.; McCloy, R.; Sutherland, R.L.; Musgrove, E.A.; et al. Tamoxifen-Induced Epigenetic Silencing of Oestrogen-Regulated Genes in Anti-Hormone Resistant Breast Cancer. PLoS ONE 2012, 7, e40466. [Google Scholar] [CrossRef]
- Fan, M.; Yan, P.S.; Hartman-Frey, C.; Chen, L.; Paik, H.; Oyer, S.L.; Salisbury, J.D.; Cheng, A.; Li, L.; Abbosh, P.H.; et al. Diverse Gene Expression and DNA Methylation Profiles Correlate with Differential Adaptation of Breast Cancer Cells to the Antiestrogens Tamoxifen and Fulvestrant. Cancer Res. 2006, 66, 11954–11966. [Google Scholar] [CrossRef]
- Phuong, N.T.T.; Kim, S.K.; Lim, S.C.; Kim, H.S.; Kim, T.H.; Lee, K.Y.; Ahn, S.-G.; Yoon, J.-H.; Kang, K.W. Role of PTEN promoter methylation in tamoxifen-resistant breast cancer cells. Breast Cancer Res. Treat. 2010, 130, 73–83. [Google Scholar] [CrossRef]
- Maier, S.; Nimmrich, I.; Koenig, T.; Eppenberger-Castori, S.; Bohlmann, I.; Paradiso, A.; Spyratos, F.; Thomssen, C.; Mueller, V.; Nährig, J.; et al. DNA-methylation of the homeodomain transcription factor PITX2 reliably predicts risk of distant disease recur-rence in tamoxifen-treated, node-negative breast cancer patients--Technical and clinical validation in a multi-centre setting in collaboration wi. Eur. J. Cancer 2007, 43, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Pathiraja, T.N.; Nayak, S.R.; Xi, Y.; Jiang, S.; Garee, J.P.; Edwards, D.P.; Lee, A.V.; Chen, J.; Shea, M.J.; Santen, R.J.; et al. Epigenetic Reprogramming of HOXC10 in Endocrine-Resistant Breast Cancer. Sci. Transl. Med. 2014, 6, 229ra41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Achinger-Kawecka, J.; Stirzaker, C.; Chia, K.; Portman, N.; Campbell, E.; Du, Q.; Laven-Law, G.; Nair, S.S.; Yong, A.; Wilkinson, A.; et al. Epigenetic therapy suppresses endocrine-resistant breast tumour growth by re-wiring ER-mediated 3D chromatin interactions. bioRxiv 2021. [Google Scholar] [CrossRef]
- Svotelis, A.; Gévry, N.; Grondin, G.; Gaudreau, L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle 2010, 9, 364–370. [Google Scholar] [CrossRef]
- Nayak, S.R.; Harrington, E.; Boone, D.; Hartmaier, R.; Chen, J.; Pathiraja, T.N.; Cooper, K.L.; Fine, J.L.; Sanfilippo, J.; Davidson, N.E.; et al. A Role for Histone H2B Variants in Endocrine-Resistant Breast Cancer. Horm. Cancer 2015, 6, 214–224. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treat-ment with a nonsteroidal aromat. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.-J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor–Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 81. [Google Scholar] [CrossRef]
- Pasqualini, J.R. Breast cancer and steroid metabolizing enzymes: The role of progestogens. Maturitas 2009, 65, S17–S21. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C.; Hess, K.; Jeitziner, R. Progesterone and Overlooked Endocrine Pathways in Breast Cancer Pathogenesis. Endocrinology 2015, 156, 3442–3450. [Google Scholar] [CrossRef]
- Carroll, J.S.; Hickey, T.E.; Tarulli, G.A.; Williams, M.; Tilley, W.D. Deciphering the divergent roles of progestogens in breast can-cer. Nat. Rev. Cancer 2016, 17, 54–64. [Google Scholar] [CrossRef]
- Stanczyk, F.Z.; Hapgood, J.; Winer, S.; Mishell, D.R. Progestogens Used in Postmenopausal Hormone Therapy: Differences in Their Pharmacological Properties, Intracellular Actions, and Clinical Effects. Endocr. Rev. 2012, 34, 171–208. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.; Paish, E.C.; Powe, D.G.; Gee, J.; Nicholson, R.I.; Lee, A.H.; Robertson, J.F.; Ellis, I. Biologic and Clinical Characteristics of Breast Cancer With Single Hormone Receptor–Positive Phenotype. J. Clin. Oncol. 2007, 25, 4772–4778. [Google Scholar] [CrossRef]
- Graham, J.D.; Yeates, C.; Balleine, R.L.; Harvey, S.S.; Milliken, J.S.; Bilous, A.M.; Clarke, C. Progesterone receptor A and B protein expression in human breast cancer. J. Steroid Biochem. Mol. Biol. 1996, 56, 93–98. [Google Scholar] [CrossRef]
- Horwitz, K.; McGuire, W. Estrogen control of progesterone receptor in human breast cancer. Correlation with nuclear processing of estrogen receptor. J. Biol. Chem. 1978, 253, 2223–2228. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Manson, J.E.; Anderson, G.L.; Cauley, J.A.; Aragaki, A.K.; Stefanick, M.L.; Lane, D.S.; Johnson, K.C.; Wactawski-Wende, J.; Chen, C.; et al. Estrogen Plus Progestin and Breast Cancer Incidence and Mortality in the Women’s Health Initiative Ob-servational Study. JNCI J. Natl. Cancer Inst. 2013, 105, 526–535. [Google Scholar] [CrossRef]
- Hankinson, S.E.S.E. Towards an integrated model for breast cancer etiology: The lifelong interplay of genes, lifestyle, and hor-mones. Breast Cancer Res. 2004, 6, 213–218. [Google Scholar] [CrossRef]
- Lydon, J.P.; Ge, G.; Kittrell, F.S.; Medina, D.; O’Malley, B.W. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Res. 1999, 59, 4276–4284. [Google Scholar]
- Collett, K.; Hartveit, F.; Skjaerven, R.; O Maehle, B. Prognostic role of oestrogen and progesterone receptors in patients with breast cancer: Relation to age and lymph node status. J. Clin. Pathol. 1996, 49, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Bardou, V.-J.; Arpino, G.; Elledge, R.M.; Osborne, C.K.; Clark, G.M. Progesterone Receptor Status Significantly Improves Outcome Prediction Over Estrogen Receptor Status Alone for Adjuvant Endocrine Therapy in Two Large Breast Cancer Databases. J. Clin. Oncol. 2003, 21, 1973–1979. [Google Scholar] [CrossRef] [PubMed]
- Viale, G.G. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a ran-domized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: BIG 1-98. J. Clin. Oncol. 2007, 25, 3846–3852. [Google Scholar] [CrossRef]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; Van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.U.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of Breast Cancer by Immunohistochemistry to Investigate a Relationship between Subtype and Short and Long Term Survival: A Collaborative Analysis of Data for 10,159 Cases from 12 Studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef]
- Van Belle, V.V. Qualitative assessment of the progesterone receptor and HER2 improves the Nottingham Prognostic Index up to 5 years after breast cancer diagnosis. J. Clin. Oncol. 2010, 28, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- A Purdie, C.; Quinlan, P.; Jordan, L.B.; Ashfield, A.; Ogston, S.; A Dewar, J.; Thompson, A.M. Progesterone receptor expression is an independent prognostic variable in early breast cancer: A population-based study. Br. J. Cancer 2013, 110, 565–572. [Google Scholar] [CrossRef]
- Campbell, E.J.; Tesson, M.; Doogan, F.; Mohammed, Z.M.; Mallon, E.; Edwards, J. The combined endocrine receptor in breast cancer, a novel approach to traditional hormone receptor inter-pretation and a better discriminator of outcome than ER and PR alone. Br. J. Cancer 2016, 115, 967–973. [Google Scholar] [CrossRef]
- Voduc, K.D.; Cheang, M.C.U.; Tyldesley, S.; Gelmon, K.; Nielsen, T.O.; Kennecke, H. Breast Cancer Subtypes and the Risk of Local and Regional Relapse. J. Clin. Oncol. 2010, 28, 1684–1691. [Google Scholar] [CrossRef]
- Singhal, H.; Greene, M.E.; Tarulli, G.; Zarnke, A.L.; Bourgo, R.J.; Laine, M.; Chang, Y.-F.; Ma, S.; Dembo, A.G.; Raj, G.V.; et al. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci. Adv. 2016, 2, e1501924. [Google Scholar] [CrossRef] [PubMed]
- Vignon, F.; Bardon, S.; Chalbos, D.; Rochefort, H. Antiestrogenic Effect of R5020, a Synthetic Progestin in Human Breast Cancer Cells in Culture. J. Clin. Endocrinol. Metab. 1983, 56, 1124–1130. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Swarbrick, A.; Lee, C.S.L.; Cornish, A.L.; Sutherland, R.L. Mechanisms of Cyclin-Dependent Kinase Inactivation by Progestins. Mol. Cell. Biol. 1998, 18, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Hardy, D.B.; Mendelson, C.R. Progesterone Receptor Inhibits Proliferation of Human Breast Cancer Cells via Induction of MAPK Phosphatase 1 (MKP-1/DUSP1). J. Biol. Chem. 2011, 286, 43091–43102. [Google Scholar] [CrossRef] [PubMed]
- Kabos, P.; Finlay-Schultz, J.; Li, C.; Kline, E.; Finlayson, C.; Wisell, J.; Manuel, C.A.; Edgerton, S.M.; Harrell, J.C.; Elias, A.; et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res. Treat. 2012, 135, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Badwe, R.R. Single-injection depot progesterone before surgery and survival in women with operable breast cancer: A random-ized controlled trial. J. Clin. Oncol. 2011, 29, 2845–2851. [Google Scholar] [CrossRef]
- Abrams, J.; Aisner, J.; Cirrincione, C.; Berry, D.A.; Muss, H.B.; Cooper, M.R.; Henderson, I.C.; Panasci, L.; Kirshner, J.; Ellerton, J.; et al. Dose-Response Trial of Megestrol Acetate in Advanced Breast Cancer: Cancer and Leukemia Group B Phase III Study. J. Clin. Oncol. 1999, 17, 64. [Google Scholar] [CrossRef]
- Alexieva-Figusch, J.; Van Gilse, H.A.; Hop, W.C.J.; Phoa, C.H.; Der Wijst, J.B.-V.; Treurniet, R.E. Progestin therapy in advanced breast cancer: Megestrol acetate—an evaluation of 160 treated cases. Cancer 1980, 46, 2369–2372. [Google Scholar] [CrossRef]
- Muss, H.B.; Paschold, E.H.; Black, W.R.; Cooper, M.R.; Capizzi, R.L.; Christian, R.; Cruz, J.M.; Jackson, D.V.; Stuart, J.J.; Richards, F. Megestrol acetate v tamoxifen in advanced breast cancer: A phase III trial of the Piedmont Oncology Association (POA). Semin. Oncol. 1985, 12, 55–61. [Google Scholar]
- Pannuti, F.; Martoni, A.; Di Marco, A.; Piana, E.; Saccani, F.; Becchi, G.; Mattioli, G.; Barbanti, F.; Marra, G.; Persiani, W.; et al. Prospective, randomized clinical trial of two different high dosages of medroxyprogesterone acetate (MAP) in the treatment of metastatic breast cancer. Eur. J. Cancer (1965) 1979, 15, 593–601. [Google Scholar] [CrossRef]
- Robertson, J.; Williams, M.; Todd, J.; Nicholson, R.; Morgan, D.; Blamey, R. Factors predicting the response of patients with advanced breast cancer to endocrine (Megace) therapy. Eur. J. Cancer Clin. Oncol. 1989, 25, 469–475. [Google Scholar] [CrossRef]
- Bines, J.; Dienstmann, R.; Obadia, R.M.; Branco, L.G.P.; Quintella, D.C.; Castro, T.M.; Camacho, P.G.; Soares, F.A.; Costa, M.E.F. Activity of megestrol acetate in postmenopausal women with advanced breast cancer after nonsteroidal aromatase inhibitor failure: A phase II trial. Ann. Oncol. 2014, 25, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Birrell, S.; Bentel, J.; Hickey, T.; Ricciardelli, C.; Weger, M.; Horsfall, D.; Tilley, W. Androgens induce divergent proliferative responses in human breast cancer cell lines. J. Steroid Biochem. Mol. Biol. 1995, 52, 459–467. [Google Scholar] [CrossRef]
- Espie, M. Megestrol Acetate in Advanced Breast Carcinoma. Oncology 1994, 51, 8–12. [Google Scholar] [CrossRef]
- Ingle, J.N.; Ahmann, D.L.; Green, S.J.; Bisel, H.F.; Frytak, S.; Edmonson, J.H.; Kvols, L.K.; Nichols, W.C.; Creagan, E.T.; Hahn, R.G.; et al. Randomized Clinical Trial of Diethylstilbestrol versus Tamoxifen in Postmenopausal Women with Advanced Breast Cancer. N. Engl. J. Med. 1981, 304, 16–21. [Google Scholar] [CrossRef]
- Izuo, M.; Iino, Y.; Endo, K. Oral high-dose medroxyprogesterone acetate (MAP) in treatment of advanced breast cancer. Breast Cancer Res. Treat. 1981, 1, 125–130. [Google Scholar] [CrossRef]
- Jonat, W.; Howell, A.; Blomqvist, C.; Eiermann, W.; Winblad, G.; Tyrrell, C.; Mauriac, L.; Roche, H.; Lundgren, S.; Hellmund, R.; et al. A randomised trial comparing two doses of the new selective aromatase inhibitor anastrozole (Arimidex) with megestrol acetate in postmenopausal patients with advanced breast cancer. Eur. J. Cancer 1996, 32, 404–412. [Google Scholar] [CrossRef]
- Mattsson, W. Current status of high dose progestin treatment in advanced breast cancer. Breast Cancer Res. Treat. 1983, 3, 231–235. [Google Scholar] [CrossRef]
- Morgan, L.R. Megestrol acetate v tamoxifen in advanced breast cancer in postmenopausal patients. Semin. Oncol. 1985, 12, 43–47. [Google Scholar] [PubMed]
- Clarke, C.L.; Graham, J.D. Non-Overlapping Progesterone Receptor Cistromes Contribute to Cell-Specific Transcriptional Outcomes. PLoS ONE 2012, 7, e35859. [Google Scholar] [CrossRef]
- Yin, P.; Roqueiro, D.; Huang, L.; Owen, J.K.; Xie, A.; Navarro, A.; Monsivais, D.; Coon V, J.S.; Kim, J.J.; Dai, Y.; et al. Genome-Wide Progesterone Receptor Binding: Cell Type-Specific and Shared Mechanisms in T47D Breast Cancer Cells and Primary Leiomyoma Cells. PLoS ONE 2012, 7, e29021. [Google Scholar] [CrossRef]
- Baird, R.; Carroll, J. Understanding Oestrogen Receptor Function in Breast Cancer and its Interaction with the Progesterone Receptor. New Preclinical Findings and their Clinical Implications. Clin. Oncol. 2015, 28, 1–3. [Google Scholar] [CrossRef][Green Version]
- Bardon, S.; Vigkpn, F.; Chalbos, D.; Rochefort, H. RU486, A Progestin and Glucocorticoid Antagonist, Inhibits the Growth of Breast Cancer Cells via the Progesterone Receptor*. J. Clin. Endocrinol. Metab. 1985, 60, 692–697. [Google Scholar] [CrossRef]
- Horwitz, K.B. The antiprogestin RU38 486: Receptor-mediated progestin versus antiprogestin actions screened in estrogen-insensitive T47Dco human breast cancer cells. Endocrinology 1985, 116, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Lee, C.S.; Cornish, A.L.; Swarbrick, A.; Sutherland, R.L. Antiprogestin inhibition of cell cycle progression in T-47D breast cancer cells is accompanied by induction of the cyclin-dependent kinase inhibitor p21. Mol. Endocrinol. 1997, 11, 54–66. [Google Scholar] [CrossRef][Green Version]
- Klijn, J.G.; Setyono-Han, B.; A Foekens, J. Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer. Steroids 2000, 65, 825–830. [Google Scholar] [CrossRef]
- Bakker, G.H.; Setyono-Han, B.; Henkelman, M.S.; De Jong, F.H.; Lamberts, S.W.; Van Der Schoot, P.; Klijn, J.G. Comparison of the actions of the antiprogestin mifepristone (RU486), the progestin megestrol acetate, the LHRH analog buserelin, and ovariectomy in treatment of rat mammary tumors. Cancer Treat. Rep. 1987, 71, 1021–1027. [Google Scholar] [PubMed]
- Michna, H.; Schneider, M.R.; Nishino, Y.; El Etreby, M.F. Antitumor activity of the antiprogestins ZK 98.299 and RU 38.486 in hormone dependent rat and mouse mammary tumors: Mechanistic studies. Breast Cancer Res. Treat. 1989, 14, 275–288. [Google Scholar] [CrossRef]
- Bakker, G.; Setyono-Han, B.; Portengen, H.; De Jong, F.; Foekens, J.; Klijn, J. Treatment of breast cancer with different antiprogestins: Preclinical and clinical studies. J. Steroid Biochem. Mol. Biol. 1990, 37, 789–794. [Google Scholar] [CrossRef]
- Iwasaki, K.; Underwood, B.; Herman, M.; Dinda, S.; Kodali, S.; Kloosterboer, H.; Hurd, C.; Moudgil, V. Effects of antiprogestins on the rate of proliferation of breast cancer cells. Mol. Cell. Biochem. 1999, 198, 141–149. [Google Scholar] [CrossRef]
- Perrault, D.; Eisenhauer, A.E.; Pritchard, I.K.; Panasci, L.; Norris, B.; Vandenberg, T.; Fisher, B. Phase II study of the progesterone antagonist mifepristone in patients with untreated metastatic breast carcino-ma: A National Cancer Institute of Canada Clinical Trials Group study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1996, 14, 2709–2712. [Google Scholar] [CrossRef] [PubMed]
- Jonat, W.; Bachelot, T.; Ruhstaller, T.; Kuss, I.; Reimann, U.; Robertson, J.F.R. Randomized phase II study of lonaprisan as second-line therapy for progesterone receptor-positive breast cancer. Ann. Oncol. 2013, 24, 2543–2548. [Google Scholar] [CrossRef]
- Robertson, J.; Willsher, P.; Winterbottom, L.; Blamey, R.; Thorpe, S. Onapristone, a progesterone receptor antagonist, as first-line therapy in primary breast cancer. Eur. J. Cancer 1999, 35, 214–218. [Google Scholar] [CrossRef]
- Lim, E.; Ni, M.; Cao, S.; Hazra, A.; Tamimi, R.M.; Brown, M. Importance of Breast Cancer Subtype in the Development of Androgen-Receptor-Directed Therapy. Curr. Breast Cancer Rep. 2014, 6, 71–78. [Google Scholar] [CrossRef]
- Hickey, T.E.; Robinson, J.L.L.; Carroll, J.S.; Tilley, W.D. Minireview: The Androgen Receptor in Breast Tissues. Mol. Endocrinol. 2012, 26, 1252–1267. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.; Abduljabbar, R.; Ashankyty, I.; Elmouna, A.; Jerjees, D.; Ali, S.; Buluwela, L.; Diez-Rodriguez, M.; Caldas, C.; Green, A.; et al. Prognostic significance of androgen receptor expression in invasive breast cancer: Transcriptomic and protein expression analysis. Breast Cancer Res. Treat. 2016, 159, 215–227. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Bianco-Miotto, T.; Jindal, S.; Butler, L.M.; Leung, S.; McNeil, C.M.; O’Toole, S.A.; Ebrahimie, E.; Millar, E.K.A.; Sakko, A.J.; et al. The Magnitude of Androgen Receptor Positivity in Breast Cancer Is Critical for Reliable Prediction of Dis-ease Outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2328–2341. [Google Scholar] [CrossRef]
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.; Harris, J.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L.; et al. Androgen Receptor Inhibits Estrogen Receptor-α Activity and Is Prognostic in Breast Cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef]
- Truong, T.H.; A Lange, C. Deciphering Steroid Receptor Crosstalk in Hormone-Driven Cancers. Endocrinology 2018, 159, 3897–3907. [Google Scholar] [CrossRef] [PubMed]
- Panet-Raymond, V.; Gottlieb, B.; Beitel, L.K.; Pinsky, L.; A Trifiro, M. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol. Cell. Endocrinol. 2000, 167, 139–150. [Google Scholar] [CrossRef]
- A Greeve, M.; Allan, R.K.; Harvey, J.M.; Bentel, J.M. Inhibition of MCF-7 breast cancer cell proliferation by 5alpha-dihydrotestosterone; a role for p21(Cip1/Waf1). J. Mol. Endocrinol. 2004, 32, 793–810. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, N.C.; Gordon, M.A.; Babbs, B.L.; Spoelstra, N.S.; Butterfield, K.T.C.; Torkko, K.C.; Phan, V.T.; Barton, V.N.; Rogers, T.J.; Sartorius, C.A.; et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol. Cancer Res. 2016, 14, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Castellano, I.; Allia, E.; Accortanzo, V.; Vandone, A.M.; Chiusa, L.; Arisio, R.; Durando, A.; Donadio, M.; Bussolati, G.; Coates, A.S.; et al. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res. Treat. 2010, 124, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Tormey, U.C.; Lippman, M.E.; Edwards, B.K.; Cassidy, J.G. Evaluation of Tamoxifen Doses with and Without Fluoxymesterone in Advanced Breast Cancer. Ann. Intern. Med. 1983, 98, 139. [Google Scholar] [CrossRef]
- Ingle, J.N.; Long, H.J.; Twito, D.I.; Schaid, D.J.; Cullinan, S.A.; Gerstner, J.G.; Krook, J.E.; Mailliard, J.A.; Tschetter, L.K.; Windschitl, H.E.; et al. Combination hormonal therapy with tamoxifen plus fluoxymesterone versus tamoxifen alone in postmenopau-sal women with metastatic breast cancer. An updated analysis. Cancer 1991, 67, 886–891. [Google Scholar] [CrossRef]
- Macedo, L.F.; Guo, Z.; Tilghman, S.L.; Sabnis, G.; Qiu, Y.; Brodie, A. Role of Androgens on MCF-7 Breast Cancer Cell Growth and on the Inhibitory Effect of Letrozole. Cancer Res. 2006, 66, 7775–7782. [Google Scholar] [CrossRef]
- Chia, K.M.; Milioli, H.; Portman, N.; Laven-Law, G.; Coulson, R.; Yong, A.; Segara, D.; Parker, A.; E Caldon, C.; Deng, N.; et al. Non-canonical AR activity facilitates endocrine resistance in breast cancer. Endocrine-Related Cancer 2019, 26, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; He, S.; Wang, D.; Patel, H.K.; Miller, C.P.; Brown, J.L.; Hattersley, G.; Saeh, J.C. Selective Androgen Receptor Modulator RAD140 Inhibits the Growth of Androgen/Estrogen Receptor–Positive Breast Cancer Models with a Distinct Mechanism of Action. Clin. Cancer Res. 2017, 23, 7608–7620. [Google Scholar] [CrossRef]
- Lim, E.; Tarulli, G.; Portman, N.; Hickey, T.; Tilley, W.D.; Palmieri, C. Pushing estrogen receptor around in breast cancer. Endocrine-Related Cancer 2016, 23, T227–T241. [Google Scholar] [CrossRef] [PubMed]
- Overmoyer, B.; Sanz-Altamira, P.; Taylor, R.P.; Hancock, M.L.; Dalton, J.T.; Johnston, M.A.; Steiner, M.S. Enobosarm: A targeted therapy for metastatic, androgen receptor positive, breast cancer. J. Clin. Oncol. 2014, 32, 568. [Google Scholar] [CrossRef]
- Palmieri, C.; Linden, H.M.; Birrell, S.; Lim, E.; Schwartzberg, L.S.; Rugo, H.S.; Cobb, P.W.; Jain, K.; Vogel, C.L.; O’Shaughnessy, J.; et al. Efficacy of enobosarm, a selective androgen receptor (AR) targeting agent, correlates with the degree of AR positivity in advanced AR+/estrogen receptor (ER)+ breast cancer in an international phase 2 clinical study. J. Clin. Oncol. 2021, 39, 1020. [Google Scholar] [CrossRef]
- Vetter, M.; Rothgiesser, K.; Li, Q.; Hawle, H.; Schönfeld, W.; Ribi, K.; Riniker, S.; Von Moos, R.; Müller, A.; Thürlimann, B. SAKK 21/12: A stratified, multicenter phase II trial of transdermal CR1447 in endocrine responsive-HER2 negative and triple negative-androgen receptor positive metastatic or locally advanced breast cancer. Ann. Oncol. 2019, 30, iii52. [Google Scholar] [CrossRef]
- Yin, A.Y.; Htun, M.; Swerdloff, R.S.; Diaz-Arjonilla, M.; Dudley, R.E.; Faulkner, S.; Bross, R.; Leung, A.; Baravarian, S.; Hull, L.; et al. Reexamination of Pharmacokinetics of Oral Testosterone Undecanoate in Hypogonadal Men With a New Self-Emulsifying Formulation. J. Androl. 2011, 33, 190–201. [Google Scholar] [CrossRef]
- Swerdloff, R.S.; Wang, C.; White, W.B.; Kaminetsky, J.; Gittelman, M.C.; Longstreth, J.A.; Dudley, R.E.; Danoff, T.M. A New Oral Testosterone Undecanoate Formulation Restores Testosterone to Normal Concentrations in Hy-pogonadal Men. J. Clin. Endocrinol. Metab. 2020, 105, 2515–2531. [Google Scholar] [CrossRef]
- Vera-Badillo, F.E.; Templeton, A.J.; De Gouveia, P.; Diaz-Padilla, I.; Bedard, P.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen Receptor Expression and Outcomes in Early Breast Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2013, 106, djt319. [Google Scholar] [CrossRef]
- Proverbs-Singh, T.; Feldman, J.L.; Morris, M.J.; Autio, K.A.; Traina, T.A. Targeting the androgen receptor in prostate and breast cancer: Several new agents in development. Endocrine-Related Cancer 2015, 22, R87–R106. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of target-ed therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Brumec, M.; Sobočan, M.; Takač, I.; Arko, D. Clinical Implications of Androgen-Positive Triple-Negative Breast Cancer. Cancers 2021, 13, 1642. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.M.; et al. Enzalutamide for the Treatment of Androgen Receptor–Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Abramson, V.G.; Colleoni, M.; Traina, T.A.; A Holmes, F.; Garcia-Estevez, L.; Hart, L.; Awada, A.; Zamagni, C.; Morris, P.G.; et al. A Randomized Placebo Controlled Phase II Trial Evaluating Exemestane with or without Enzalutamide in Patients with Hormone Receptor–Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 6149–6157. [Google Scholar] [CrossRef]
- Christenson, J.L.; O’Neill, K.I.; Williams, M.M.; Spoelstra, N.S.; Jones, K.L.; Trahan, G.D.; Reese, J.; Van Patten, E.T.; Elias, A.; Eisner, J.R.; et al. Activity of Combined Androgen Receptor Antagonism and Cell Cycle Inhibition in Androgen Receptor Positive Triple Negative Breast Cancer. Mol. Cancer Ther. 2021, 20, 1062–1071. [Google Scholar] [CrossRef]
- Gucalp, A.; Boyle, L.A.; Alano, T.; Arumov, A.; Gounder, M.M.; Patil, S.; Feigin, K.; Edelweiss, M.; D’Andrea, G.; Bromberg, J.; et al. Phase II trial of bicalutamide in combination with palbociclib for the treatment of androgen receptor (+) meta-static breast cancer. J. Clin. Oncol. 2020, 38, 1017. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A. Targeting the androgen receptor in triple-negative breast cancer. Curr. Probl. Cancer 2016, 40, 141–150. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Chandler, B.; Olsen, E.; Wilder-Romans, K.; Moubadder, L.; Liu, M.; Pesch, A.; Zhang, A.; Ritter, C.; Ward, S.T.; et al. Seviteronel, a Novel CYP17 Lyase Inhibitor and Androgen Receptor Antagonist, Radiosensitizes AR-Positive Triple Negative Breast Cancer Cells. Front. Endocrinol. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Abduljabbar, R.; Negm, O.H.; Lai, C.-F.; Jerjees, D.A.; Al-Kaabi, M.; Hamed, M.R.; Tighe, P.; Buluwela, L.; Mukherjee, A.; Green, A.; et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res. Treat. 2015, 150, 335–346. [Google Scholar] [CrossRef]
- Noureddine, L.; Trédan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4446. [Google Scholar] [CrossRef]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the Glucocorticoid Receptor Is Associated with Poor Prognosis in Estrogen Receptor-Negative Breast Cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef]
- West, D.C.; Pan, D.; Tonsing-Carter, E.; Hernandez, K.M.; Pierce, C.F.; Styke, S.C.; Bowie, K.; Garcia, T.I.; Kocherginsky, M.; Conzen, S.D. GR and ER Coactivation Alters the Expression of Differentiation Genes and Associates with Improved ER+ Breast Cancer Outcome. Mol. Cancer Res. 2016, 14, 707–719. [Google Scholar] [CrossRef]
- Miranda, T.B.; Voss, T.C.; Sung, M.; Baek, S.; John, S.; Hawkins, M.; Grøntved, L.; Schiltz, R.L.; Hager, G.L. Reprogramming the chromatin landscape: Interplay of the estrogen and glucocorticoid receptors at the ge-nomic level. Cancer Res. 2013, 73, 5130–5139. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.C.; Schiltz, R.L.; Sung, M.-H.; Yen, P.; Stamatoyannopoulos, J.A.; Biddie, S.; Johnson, T.A.; Miranda, T.B.; John, S.; Hager, G.L. Dynamic Exchange at Regulatory Elements during Chromatin Remodeling Underlies Assisted Loading Mechanism. Cell 2011, 146, 544–554. [Google Scholar] [CrossRef]
- Keith, B.D. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 2008, 8, 1–19. [Google Scholar] [CrossRef]
- Lietzen, L.W.; Ahern, T.; Christiansen, P.; Jensen, A.B.; Sørensen, H.T.; Lash, T.L.; Cronin-Fenton, D.P. Glucocorticoid prescriptions and breast cancer recurrence: A Danish nationwide prospective cohort study. Ann. Oncol. 2014, 25, 2419–2425. [Google Scholar] [CrossRef]
- Skor, M.N.; Wonder, E.L.; Kocherginsky, M.; Goyal, A.; Hall, B.A.; Cai, Y.; Conzen, S.D. Glucocorticoid Receptor Antagonism as a Novel Therapy for Triple-Negative Breast Cancer. Clin. Cancer Res. 2013, 19, 6163–6172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lan, X.; Wu, D.; Sunkel, B.; Ye, Z.; Huang, J.; Liu, Z.; Clinton, S.K.; Jin, V.X.; Wang, Q. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat. Commun. 2015, 6, 8323. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Zannini, A.; Ingallina, E.; Bertolio, R.; Marotta, C.; Neri, C.; Cappuzzello, E.; Forcato, M.; Rosato, A.; et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat. Commun. 2017, 8, 14073. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Muenst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef]
- Nanda, R.; Stringer-Reasor, E.M.; Saha, P.; Kocherginsky, M.; Gibson, J.; Libao, B.; Hoffman, P.C.; Obeid, E.; Merkel, D.E.; Khramtsova, G.; et al. A randomized phase I trial of nanoparticle albumin-bound paclitaxel with or without mifepristone for advanced breast cancer. SpringerPlus 2016, 5, 947. [Google Scholar] [CrossRef]
- Sasano, H.; Frost, A.R.; Saitoh, R.; Matsunaga, G.; Nagura, H.; Krozowski, Z.S.; Silverberg, S.G. Localization of mineralocorticoid receptor and 11 beta-hydroxysteroid dehydrogenase type II in human breast and its disorders. Anticancer. Res. 1997, 17, 2001–2007. [Google Scholar]
- Leo, J.C.L.; Guo, C.; Woon, C.T.; Aw, S.E.; Lin, V.C.L. Glucocorticoid and Mineralocorticoid Cross-Talk with Progesterone Receptor to Induce Focal Adhesion and Growth Inhibition in Breast Cancer Cells. Endocrinology 2004, 145, 1314–1321. [Google Scholar] [CrossRef]
- Doan, T.B.; Cheung, V.; Clyne, C.D.; Hilton, H.N.; Eriksson, N.; Young, M.J.; Funder, J.W.; Muscat, G.E.O.; Fuller, P.J.; Clarke, C.L.; et al. A tumour suppressive relationship between mineralocorticoid and retinoic acid receptors activates a transcrip-tional program consistent with a reverse Warburg effect in breast cancer. Breast Cancer Res. 2020, 22, 122. [Google Scholar] [CrossRef]
- Johansson, H.J.; Sanchez, B.C.; Mundt, F.; Forshed, J.; Kovács, A.; Panizza, E.; Hultin-Rosenberg, L.; Lundgren, B.; Martens, U.; Máthé, G.; et al. Retinoic acid receptor alpha is associated with tamoxifen resistance in breast cancer. Nat. Commun. 2013, 4, 2175. [Google Scholar] [CrossRef]
- Garattini, E.; Bolis, M.; Garattini, S.K.; Fratelli, M.; Centritto, F.; Paroni, G.; Gianni’, M.; Zanetti, A.; Pagani, A.; Fisher, J.N.; et al. Retinoids and breast cancer: From basic studies to the clinic and back again. Cancer Treat. Rev. 2014, 40, 739–749. [Google Scholar] [CrossRef]
- Fettig, L.M.; McGinn, O.; Finlay-Schultz, J.; LaBarbera, D.V.; Nordeen, S.K.; A Sartorius, C. Cross talk between progesterone receptors and retinoic acid receptors in regulation of cytokeratin 5-positive breast cancer cells. Oncogene 2017, 36, 6074–6084. [Google Scholar] [CrossRef][Green Version]
- Xu, H.; Liu, Z.; Shi, H.; Wang, C. Prognostic role of vitamin D receptor in breast cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Beydoun, M.A.; Beydoun, H.A.; Chen, X.; Zonderman, A.B.; Wood, R.J. Vitamin D and breast cancer: A systematic review and meta-analysis of observational studies. Clin. Nutr. ESPEN 2018, 30, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Estébanez, N.; Gómez-Acebo, I.; Palazuelos, C.; Llorca, J.; Dierssen-Sotos, T. Vitamin D exposure and Risk of Breast Cancer: A me-ta-analysis. Sci. Rep. 2018, 8, 9039. [Google Scholar] [CrossRef] [PubMed]
- Dizdar, O.; Harputluoglu, H.; Altundag, K. Vitamin D Intake and Breast Cancer Risk in Postmenopausal Women. Arch. Intern. Med. 2007, 167, 2532. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.; Elser, C.; Ennis, M.; Goodwin, P.J. Blood levels of vitamin D and early stage breast cancer prognosis: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 141, 331–339. [Google Scholar] [CrossRef] [PubMed]
- So, J.Y.; Lee, H.J.; Smolarek, A.K.; Paul, S.; Wang, C.-X.; Maehr, H.; Uskokovic, M.; Zheng, X.; Conney, A.H.; Cai, L.; et al. A Novel Gemini Vitamin D Analog Represses the Expression of a Stem Cell Marker CD44 in Breast Cancer. Mol. Pharmacol. 2010, 79, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Cohen, D.J.; Schwartz, N.; Muktipaty, C.; Koblinski, J.E.; Boyan, B.D.; Schwartz, Z. 24R,25-Dihydroxyvitamin D3 regulates breast cancer cells in vitro and in vivo. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2019, 1863, 1498–1512. [Google Scholar] [CrossRef]
- Narvaez, C.J.; Zinser, G.; Welsh, J. Functions of 1alpha,25-dihydroxyvitamin D(3) in mammary gland: From normal development to breast cancer. Steroids 2001, 66, 301–308. [Google Scholar] [CrossRef]
- Welsh, J. Vitamin D and breast cancer: Past and present. J. Steroid Biochem. Mol. Biol. 2018, 177, 15–20. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Receptor | Receptor Type | Encoding Gene | Ligand |
|---|---|---|---|
| Oestrogen Receptor | Steroid Hormone | ESR1 | Oestradiol, oestrogens |
| ESR2 | Oestradiol, 5α-androstane-3β,17β-diol | ||
| Progesterone Receptor | Steroid Hormone | PGR | Progesterone, progestogens |
| Androgen Receptor | Steroid Hormone | AR | Androgens, 5α-dihydrotestosterone (DHT) |
| Glucocorticoid Receptor | Steroid Hormone | NR3C1 | Glucocorticoids, cortisols |
| Mineralocorticoid Receptor | Steroid Hormone | NR3C2 | Aldosterone |
| Retinoic Acid Receptor | RXR Heterodimer | RARA | All-trans retinoic acid, 9-cis retinoic acid |
| RARB | |||
| RARG | |||
| Vitamin D Receptor | RXR Heterodimer | VDR | Calcitriol, 1,25-dihydroxy vitamin D3 |
| NR Target | Treatment | Class | Combination Treatment | Phase | Stage | BC Subtype | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|---|---|
| ER | Giredestrant | Oral SERD | N/A | II | Neoadjuvant | ER+, HER2- | NCT04436744 |
| N/A | II | Advanced | NCT04576455 | ||||
| Palbociclib | III | NCT04546009 | |||||
| LY3484356 | Abemaciclib, Trastuzumab, Alpelisib, Everolimus | I | Advanced | ER+, HER2- | NCT04188548 | ||
| Rintodestrant | Palbociclib | I | Advanced | ER+, HER2- | NCT03455270 | ||
| ZB716 | Palbociclib | I, II | Advanced | ER+, HER2- | NCT04669587 | ||
| Camizestrant | Palbociclib | III | Advanced | ER+, HER2- | NCT04711252 | ||
| H3B-6545 | SERCA | N/A | I | Advanced | ER+, HER2- | NCT04568902 | |
| Palbociclib | I | NCT04288089 | |||||
| N/A | I, II | NCT03250676 | |||||
| PR | Megestrol acetate | PR Agonist | Letrozole | II | Early, Window | ER+, HER2- | NCT03306472 |
| Prometrium | Letrozole | II | Early, Window | ER+, PR+, HER2- | NCT03906669 | ||
| AR | Enobosarm | SARM | N/A | III | Advanced | ER+, AR+, HER2- | NCT04869943 |
| Severitonel-D | AR Antagonist | Docetaxel | I, II | Advanced | AR+, TNBC | NCT04947189 | |
| Enzalutamide | N/A | II | Advanced | AR+, TNBC | NCT01889238 | ||
| Darolutamide | Capecitabine | II | Advanced | AR+, TNBC | NCT03383679 | ||
| GR | Mifepristone | GR Antagonist | Nab-Paclitaxel | II | Advanced | GR+, TNBC | NCT02788981 |
| Pembrolizumab | II | NCT03225547 | |||||
| N/A | II | Prevention | BRCA1/2mut TNBC | NCT01898312 | |||
| VDR | Vitamin D3 | VDR Agonist | N/A | II | Advanced | TNBC | NCT04677816 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Freelander, A.; Lim, E. Type 1 Nuclear Receptor Activity in Breast Cancer: Translating Preclinical Insights to the Clinic. Cancers 2021, 13, 4972. https://doi.org/10.3390/cancers13194972
Kumar S, Freelander A, Lim E. Type 1 Nuclear Receptor Activity in Breast Cancer: Translating Preclinical Insights to the Clinic. Cancers. 2021; 13(19):4972. https://doi.org/10.3390/cancers13194972
Chicago/Turabian StyleKumar, Sanjeev, Allegra Freelander, and Elgene Lim. 2021. "Type 1 Nuclear Receptor Activity in Breast Cancer: Translating Preclinical Insights to the Clinic" Cancers 13, no. 19: 4972. https://doi.org/10.3390/cancers13194972
APA StyleKumar, S., Freelander, A., & Lim, E. (2021). Type 1 Nuclear Receptor Activity in Breast Cancer: Translating Preclinical Insights to the Clinic. Cancers, 13(19), 4972. https://doi.org/10.3390/cancers13194972