
RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient

 , , and
, , and
Simple Summary
Abstract
1. Introduction
2. Results
2.1. A Secondary RAC1(P34R) Mutation Is Acquired in PTC following Dabrafenib Treatment
2.2. Tumor Histopathology Indicated an Anaplastic Phenotype at Progression
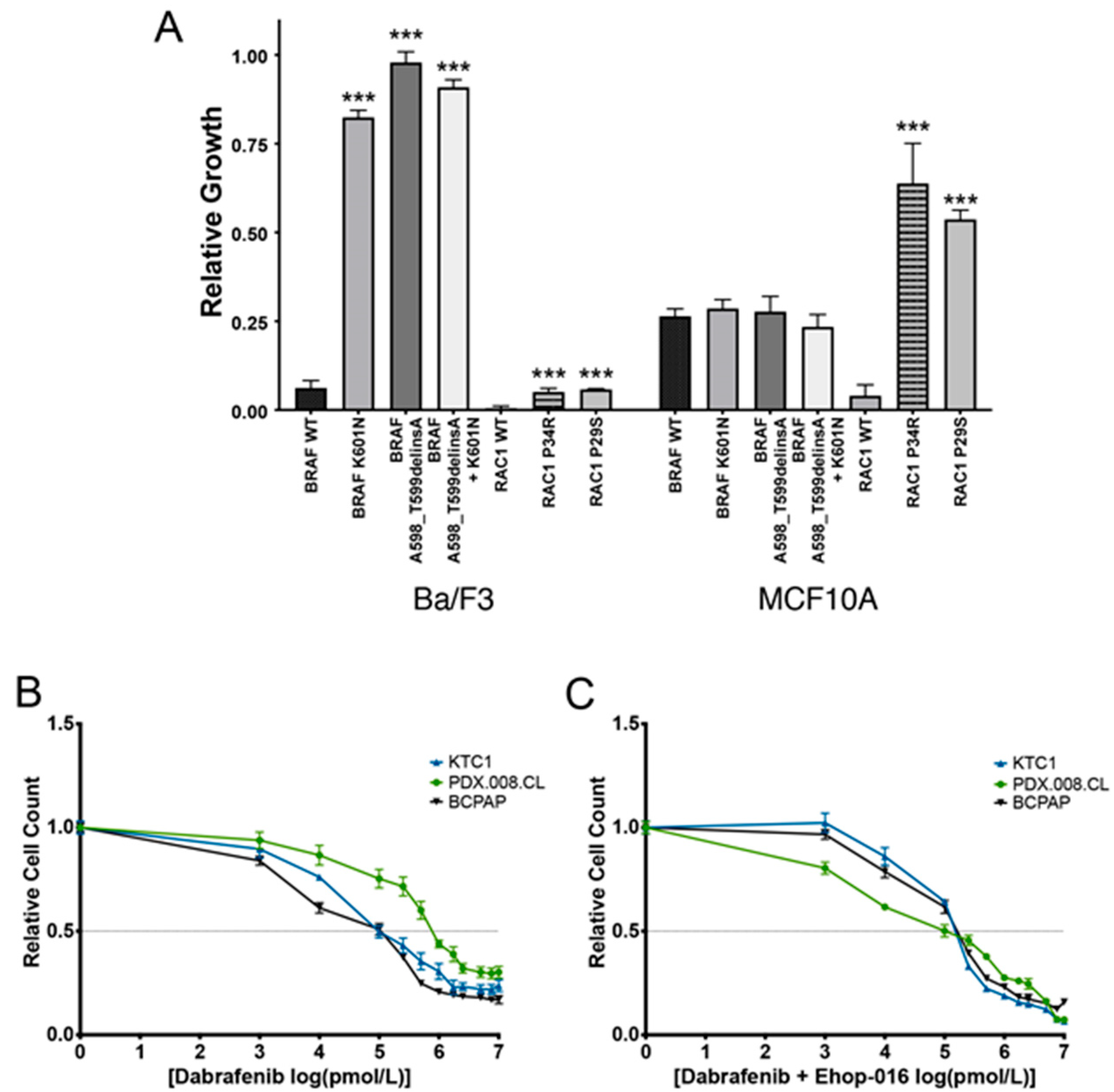
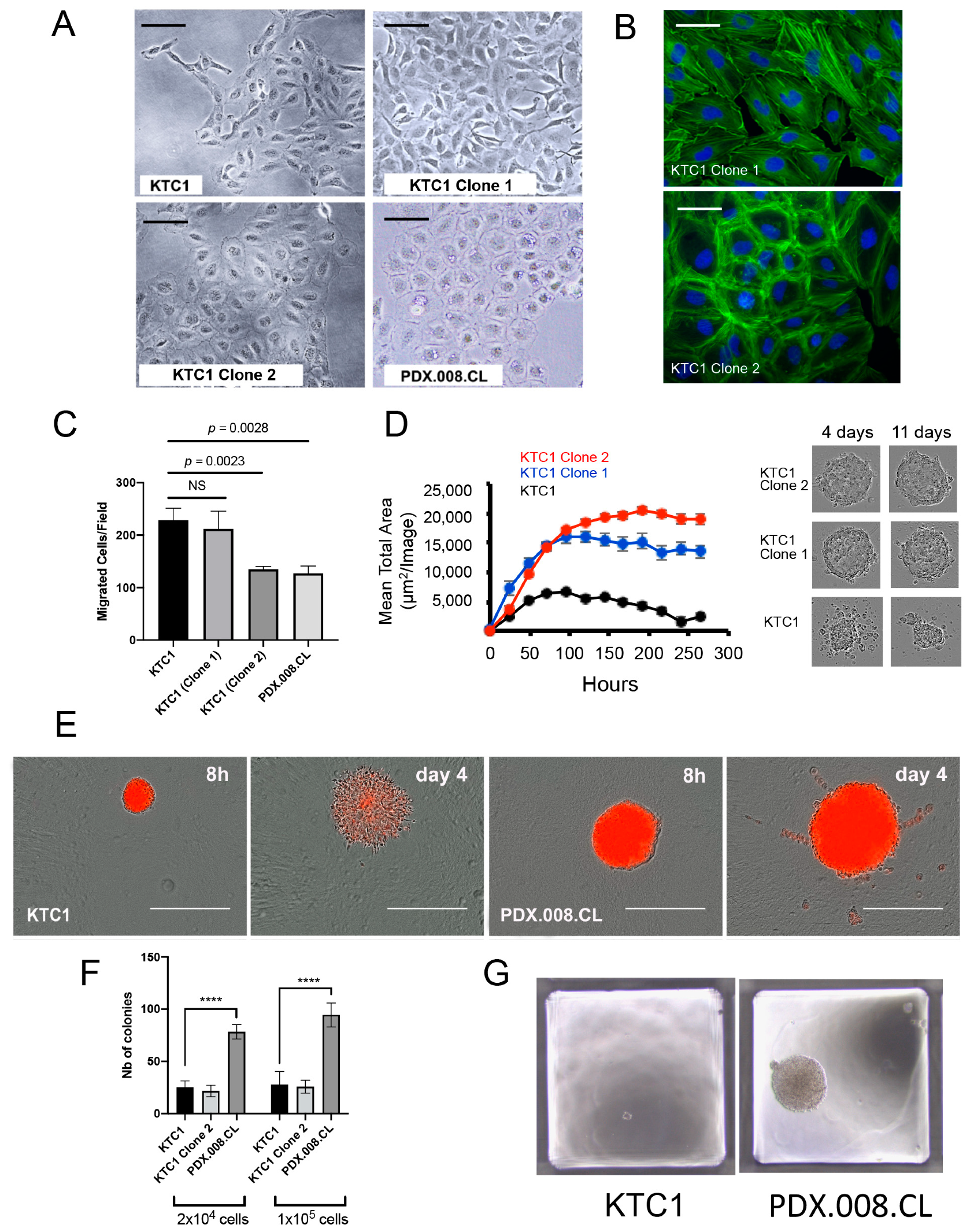
2.3. Expression of Mutated BRAF and RAC1 Proteins Increases Cell Growth
2.4. Concurrent BRAF and RAC1 Alterations Drive BRAF Inhibitor Resistance
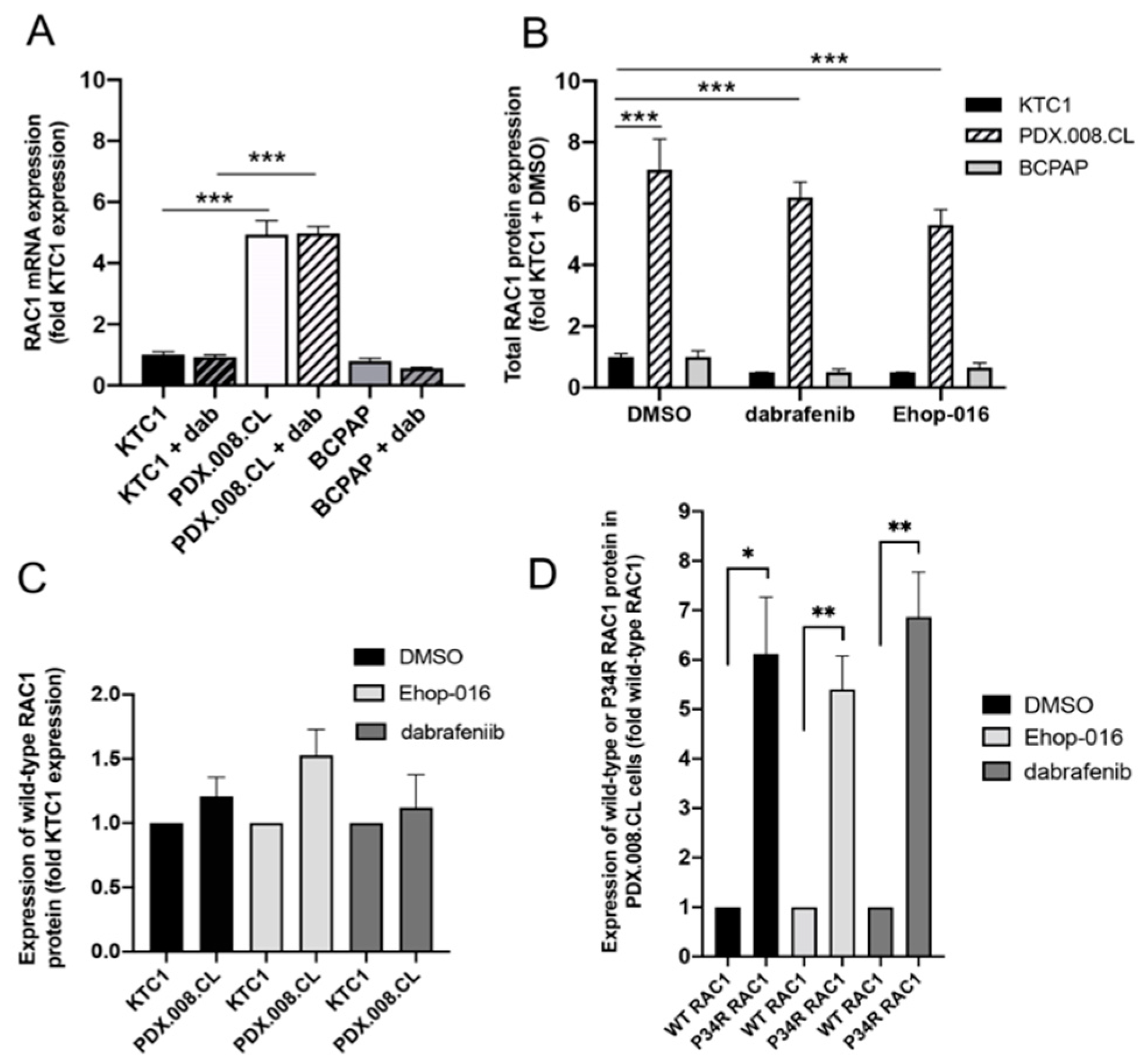
2.5. RAC1 mRNA and RAC1(P34R) Protein Are Over-Expressed in PDX.008.CL Cells in Comparison to Other BRAF-Mutated PTC Cells
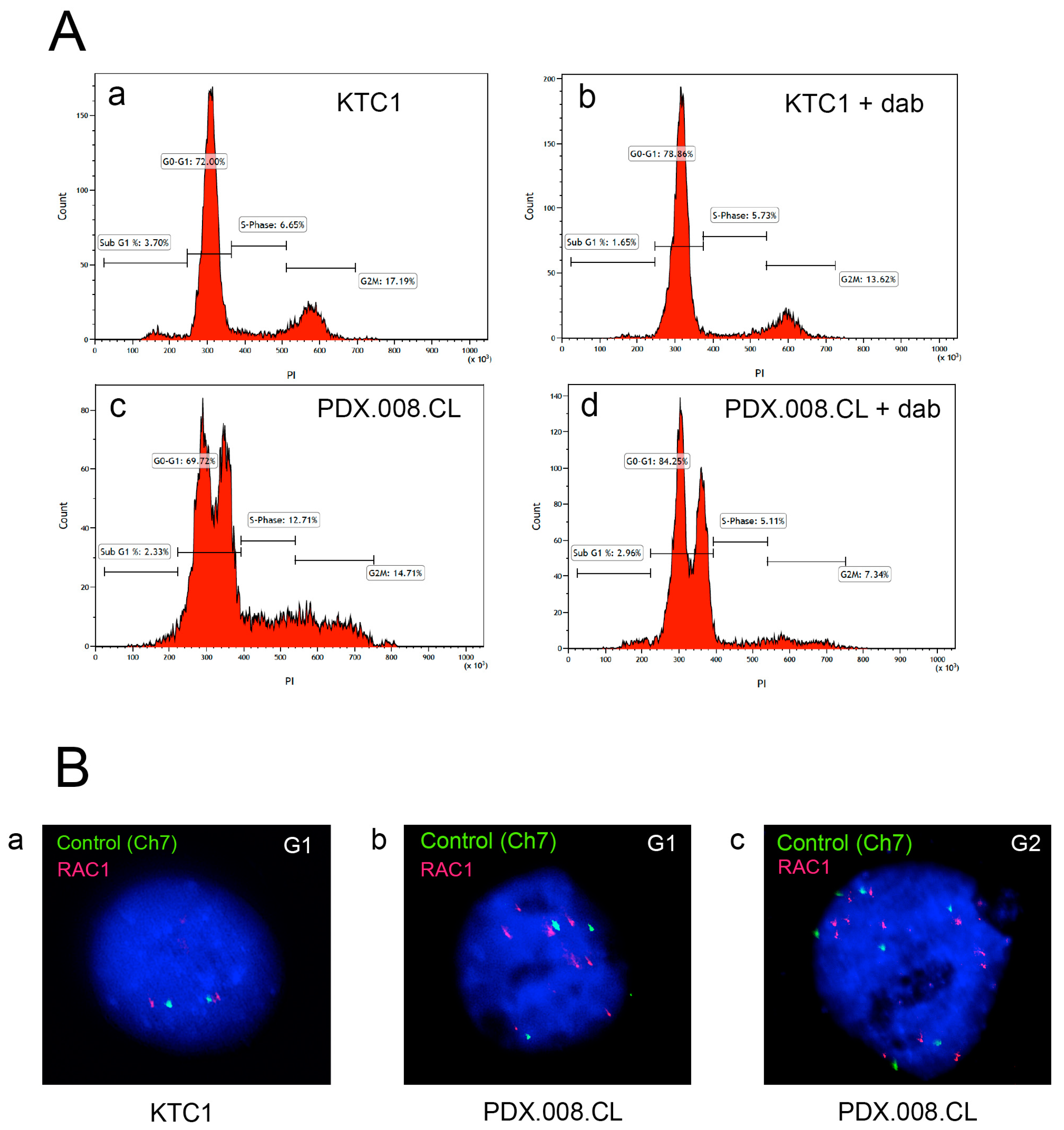
2.6. The PDX.008.CL Cells Are Aneuploid
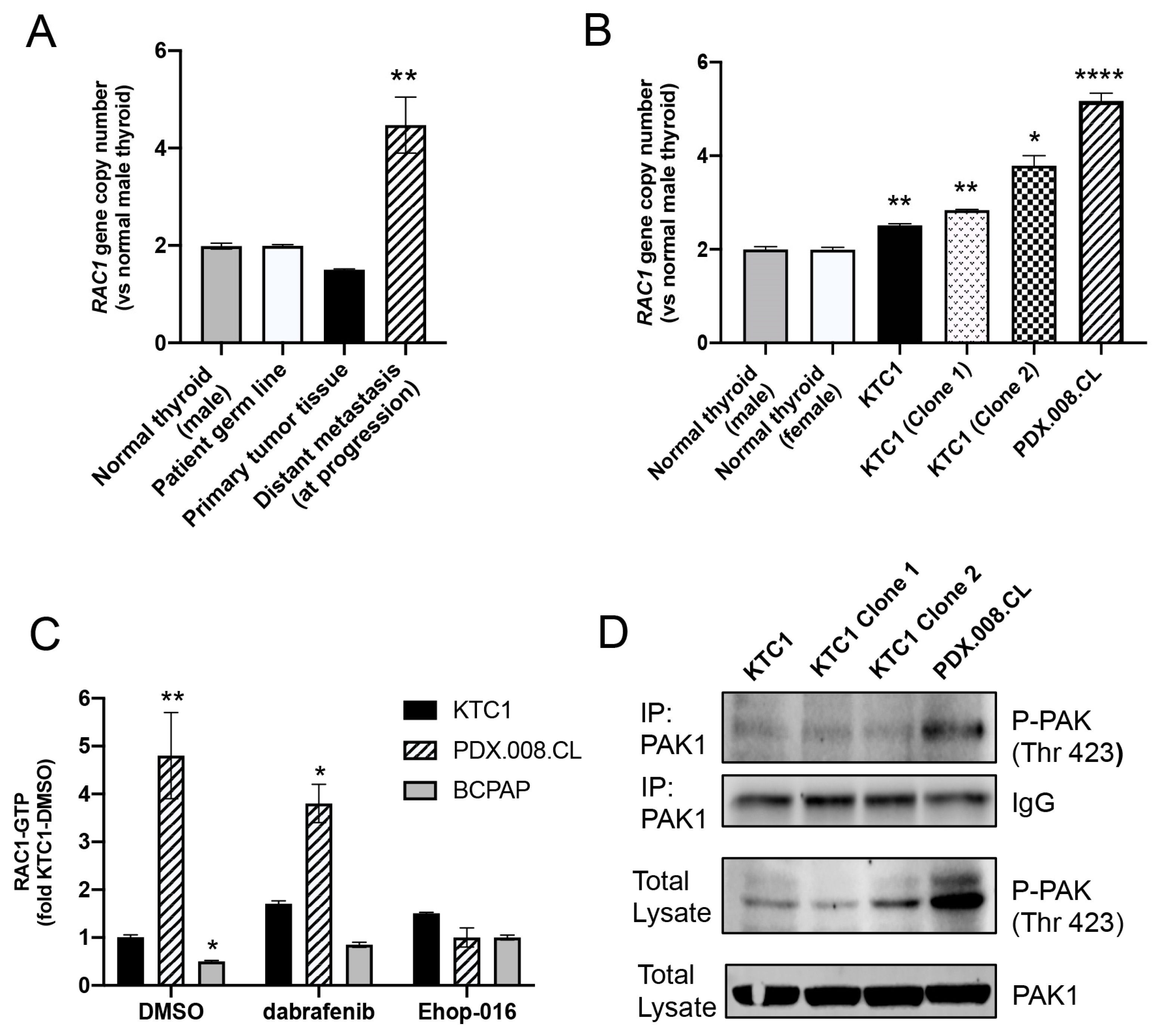
2.7. RAC1 Gene Copy Number Is Increased in the Patient’s Resistant Tumor Tissue and PDX.008.CL Cells
2.8. PDX.008.CL Cells Show High RAC1 and PAK1 Activation
2.9. Increased RAC1 Copy Numbers Induce Morphological and Growth Changes
2.10. Proto-Oncogenes Located on Chromosome 7 Are Amplified and Their Expression Is Upregulated in the PDX.008.CL Cell Line
3. Discussion
4. Materials and Methods
4.1. DNA Sequencing, Mapping, and Variant Calling
4.2. Somatic Variant Analysis
4.3. Cell Line Establishment
4.4. PDX.008.CL Cell Line RAC1(P34R) Sequencing
4.5. Other Cell Lines
4.6. Stable Transduction Experiments
4.7. Pharmacological Inhibitors
4.8. Conventional Western Blotting
4.9. Quantitative Western Blotting
4.10. RAC1 Activity Assay
4.11. PAK1 Activity Assay
4.12. RT-qPCR Analysis
4.13. Transient Viability Assays
4.14. Actin Staining
4.15. Cell Growth Assays
4.16. Migration and Invasion Assays
4.17. Spheroid Formation, 3-D Growth, and Invasion
4.18. Soft Agar Colony Formation Assay
4.19. Flow Cytometry
4.20. Gene Copy Number Assay
4.21. Fluorescence In Situ Hybridization (FISH)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Shah, J.P. Thyroid carcinoma: Epidemiology, histology, and diagnosis. Clin. Adv. Hematol. Oncol. 2015, 13 (Suppl. 4), 3–6. [Google Scholar]
- Carhill, A.A.; Litofsky, D.R.; Ross, D.S.; Jonklaas, J.; Cooper, D.S.; Brierley, J.D.; Ladenson, P.W.; Ain, K.B.; Fein, H.G.; Haugen, B.R.; et al. Long-Term Outcomes Following Therapy in Differentiated Thyroid Carcinoma: NTCTCS Registry Analysis 1987–2012. J. Clin. Endocrinol. Metab. 2015, 100, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Fallahi, P.; Ferrari, S.M.; Carpi, A.; Berti, P.; Materazzi, G.; Minuto, M.; Guastalli, M.; Miccoli, P. Dedifferentiated thyroid cancer: A therapeutic challenge. Biomed. Pharm. 2008, 62, 559–563. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Patel, A.; Danysh, B.P.; Dadu, R.; Kopetz, S.; Falchook, G. BRAF inhibitors: Experience in thyroid cancer and general review of toxicity. Horm. Cancer 2015, 6, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Zager, J.S. Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin. Drug Metab. Toxicol. 2013, 9, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Bjursten, S.; Vannas, C.; Filges, S.; Puls, F.; Pandita, A.; Fagman, H.; Stahlberg, A.; Levin, M. Response to BRAF/MEK Inhibition in A598_T599insV BRAF Mutated Melanoma. Case Rep. Oncol. 2019, 12, 872–879. [Google Scholar] [CrossRef]
- Trudel, S.; Odolczyk, N.; Dremaux, J.; Toffin, J.; Regnier, A.; Sevestre, H.; Zielenkiewicz, P.; Arnault, J.P.; Gubler, B. The clinical response to vemurafenib in a patient with a rare BRAFV600DK601del mutation-positive melanoma. BMC Cancer 2014, 14, 727. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Wei, L.; Wirth, L.J.; Daniels, G.A.; De Souza, J.A.; Timmers, C.D.; Sexton, J.L.; Beshara, M.; Nichols, D.; Snyder, N.; et al. Results of randomized phase II trial of dabrafenib versus dabrafenib plus trametinib in BRAF-mutated paillary thyroid carcinoma. J. Clin. Oncol. 2017, 35, 6022. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Ryder, M.; Jimenez, C. Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef] [PubMed]
- Danysh, B.P.; Rieger, E.Y.; Sinha, D.K.; Evers, C.V.; Cote, G.J.; Cabanillas, M.E.; Hofmann, M.C. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget 2016, 7, 30907–30923. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.H.; Konda, B.; Sipos, J.; Liu, T.; Webb, A.; Ringel, M.D.; Timmers, C.D.; Shah, M.H. KRAS G12V Mutation in Acquired Resistance to Combined BRAF and MEK Inhibition in Papillary Thyroid Cancer. J. Natl. Compr. Canc. Netw. 2019, 17, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; Dadu, R.; Iyer, P.; Wanland, K.B.; Busaidy, N.L.; Ying, A.; Gule-Monroe, M.; Wang, J.R.; Zafereo, M.; Hofmann, M.C. Acquired Secondary RAS Mutation in BRAF(V600E)-Mutated Thyroid Cancer Patients Treated with BRAF Inhibitors. Thyroid 2020, 30, 1288–1296. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal 2010, 8, 23. [Google Scholar] [CrossRef]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef]
- Bauer, N.N.; Chen, Y.W.; Samant, R.S.; Shevde, L.A.; Fodstad, O. Rac1 activity regulates proliferation of aggressive metastatic melanoma. Exp. Cell Res. 2007, 313, 3832–3839. [Google Scholar] [CrossRef]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Malicherova, B.; Burjanivova, T.; Minarikova, E.; Kasubova, I.; Pecova, T.; Bobrovska, M.; Homola, I.; Lasabova, Z.; Plank, L. Detection of driver mutations in FFPE samples from patients with verified malignant melanoma. Neoplasma 2019, 66, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1(P29S) Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9. [Google Scholar] [CrossRef]
- Ng, P.K.; Li, J.; Jeong, K.J.; Shao, S.; Chen, H.; Tsang, Y.H.; Sengupta, S.; Wang, Z.; Bhavana, V.H.; Tran, R.; et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 2018, 33, 450–462.e10. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G.; Morris, S.; Dong, H.J.; Chernoff, J.; Bokoch, G.M. Regulation of human leukocyte p21-activated kinases through G protein--coupled receptors. Science 1995, 269, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Adam, L.; Wang, R.A.; Mandal, M.; Nguyen, D.; Sahin, A.; Chernoff, J.; Hung, M.C.; Kumar, R. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J. Biol. Chem. 2000, 275, 36238–36244. [Google Scholar] [CrossRef]
- McCarty, S.K.; Saji, M.; Zhang, X.; Jarjoura, D.; Fusco, A.; Vasko, V.V.; Ringel, M.D. Group I p21-activated kinases regulate thyroid cancer cell migration and are overexpressed and activated in thyroid cancer invasion. Endocr. Relat. Cancer 2010, 17, 989–999. [Google Scholar] [CrossRef]
- McCarty, S.K.; Saji, M.; Zhang, X.; Knippler, C.M.; Kirschner, L.S.; Fernandez, S.; Ringel, M.D. BRAF activates and physically interacts with PAK to regulate cell motility. Endocr. Relat. Cancer 2014, 21, 865–877. [Google Scholar] [CrossRef][Green Version]
- Knippler, C.M.; Saji, M.; Rajan, N.; Porter, K.; La Perle, K.M.D.; Ringel, M.D. MAPK- and AKT-activated thyroid cancers are sensitive to group I PAK inhibition. Endocr. Relat. Cancer 2019, 26, 699–712. [Google Scholar] [CrossRef]
- Lei, M.; Robinson, M.A.; Harrison, S.C. The active conformation of the PAK1 kinase domain. Structure 2005, 13, 769–778. [Google Scholar] [CrossRef]
- Zenke, F.T.; King, C.C.; Bohl, B.P.; Bokoch, G.M. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J. Biol. Chem. 1999, 274, 32565–32573. [Google Scholar] [CrossRef] [PubMed]
- Sells, M.A.; Knaus, U.G.; Bagrodia, S.; Ambrose, D.M.; Bokoch, G.M.; Chernoff, J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 1997, 7, 202–210. [Google Scholar] [CrossRef]
- Mogilner, A.; Keren, K. The shape of motile cells. Curr. Biol. 2009, 19, R762–R771. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Wang, P.; Yang, J.; Ma, J.; Liu, C.; Zhou, M. Prognostic and Clinicopathological Value of Rac1 in Cancer Survival: Evidence from a Meta-Analysis. J. Cancer 2018, 9, 2571–2579. [Google Scholar] [CrossRef]
- Puli, O.R.; Danysh, B.P.; McBeath, E.; Sinha, D.K.; Hoang, N.M.; Powell, R.T.; Danysh, H.E.; Cabanillas, M.E.; Cote, G.J.; Hofmann, M.C. The Transcription Factor ETV5 Mediates BRAFV600E-Induced Proliferation and TWIST1 Expression in Papillary Thyroid Cancer Cells. Neoplasia 2018, 20, 1121–1134. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Mehta, A.; Boufraqech, M.; Davis, S.; Wang, J.; Tian, Z.; Yu, Z.; Boxer, M.B.; Kiefer, J.A.; et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 2015, 6, 9073–9085. [Google Scholar] [CrossRef]
- Garcia, C.; Buffet, C.; El Khattabi, L.; Rizk-Rabin, M.; Perlemoine, K.; Ragazzon, B.; Bertherat, J.; Cormier, F.; Groussin, L. MET overexpression and activation favors invasiveness in a model of anaplastic thyroid cancer. Oncotarget 2019, 10, 2320–2334. [Google Scholar] [CrossRef]
- Stagni, C.; Zamuner, C.; Elefanti, L.; Zanin, T.; Bianco, P.D.; Sommariva, A.; Fabozzi, A.; Pigozzo, J.; Mocellin, S.; Montesco, M.C.; et al. BRAF Gene Copy Number and Mutant Allele Frequency Correlate with Time to Progression in Metastatic Melanoma Patients Treated with MAPK Inhibitors. Mol. Cancer Ther. 2018, 17, 1332–1340. [Google Scholar] [CrossRef]
- Menzer, C.; Menzies, A.M.; Carlino, M.S.; Reijers, I.; Groen, E.J.; Eigentler, T.; de Groot, J.W.B.; van der Veldt, A.A.M.; Johnson, D.B.; Meiss, F.; et al. Targeted Therapy in Advanced Melanoma With Rare BRAF Mutations. J. Clin. Oncol. 2019, 37, 3142–3151. [Google Scholar] [CrossRef]
- Hallmeyer, S.; Gonzalez, R.; Lawson, D.H.; Cranmer, L.D.; Linette, G.P.; Puzanov, I.; Taback, B.; Cowey, C.L.; Ribas, A.; Daniels, G.A.; et al. Vemurafenib treatment for patients with locally advanced, unresectable stage IIIC or metastatic melanoma and activating exon 15 BRAF mutations other than V600E. Melanoma Res. 2017, 27, 585–590. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.B. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef]
- Byeon, H.K.; Na, H.J.; Yang, Y.J.; Ko, S.; Yoon, S.O.; Ku, M.; Yang, J.; Kim, J.W.; Ban, M.J.; Kim, J.H.; et al. Acquired resistance to BRAF inhibition induces epithelial-to-mesenchymal transition in BRAF (V600E) mutant thyroid cancer by c-Met-mediated AKT activation. Oncotarget 2017, 8, 596–609. [Google Scholar] [CrossRef][Green Version]
- Duquette, M.; Sadow, P.M.; Husain, A.; Sims, J.N.; Antonello, Z.A.; Fischer, A.H.; Song, C.; Castellanos-Rizaldos, E.; Makrigiorgos, G.M.; Kurebayashi, J.; et al. Metastasis-associated MCL1 and P16 copy number alterations dictate resistance to vemurafenib in a BRAFV600E patient-derived papillary thyroid carcinoma preclinical model. Oncotarget 2015, 6, 42445–42467. [Google Scholar] [CrossRef]
- Qiu, R.G.; Chen, J.; Kirn, D.; McCormick, F.; Symons, M. An essential role for Rac in Ras transformation. Nature 1995, 374, 457–459. [Google Scholar] [CrossRef]
- Kissil, J.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Bender Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.; et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007, 67, 8089–8094. [Google Scholar] [CrossRef] [PubMed]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pedersen, E.; Basse, A.; Lefever, T.; Peyrollier, K.; Kapoor, S.; Mei, Q.; Karlsson, R.; Chrostek-Grashoff, A.; Brakebusch, C. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene 2010, 29, 3362–3373. [Google Scholar] [CrossRef] [PubMed]
- Araiza-Olivera, D.; Feng, Y.; Semenova, G.; Prudnikova, T.Y.; Rhodes, J.; Chernoff, J. Suppression of RAC1-driven malignant melanoma by group A PAK inhibitors. Oncogene 2018, 37, 944–952. [Google Scholar] [CrossRef]
- Wang, J.; Rao, Q.; Wang, M.; Wei, H.; Xing, H.; Liu, H.; Wang, Y.; Tang, K.; Peng, L.; Tian, Z.; et al. Overexpression of Rac1 in leukemia patients and its role in leukemia cell migration and growth. Biochem. Biophys. Res. Commun. 2009, 386, 769–774. [Google Scholar] [CrossRef]
- Zhou, Y.; Liao, Q.; Han, Y.; Chen, J.; Liu, Z.; Ling, H.; Zhang, J.; Yang, W.; Oyang, L.; Xia, L.; et al. Rac1 overexpression is correlated with epithelial mesenchymal transition and predicts poor prognosis in non-small cell lung cancer. J. Cancer 2016, 7, 2100–2109. [Google Scholar] [CrossRef]
- Ji, J.; Feng, X.; Shi, M.; Cai, Q.; Yu, Y.; Zhu, Z.; Zhang, J. Rac1 is correlated with aggressiveness and a potential therapeutic target for gastric cancer. Int. J. Oncol. 2015, 46, 1343–1353. [Google Scholar] [CrossRef]
- Kamai, T.; Yamanishi, T.; Shirataki, H.; Takagi, K.; Asami, H.; Ito, Y.; Yoshida, K. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 2004, 10, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, L.; Hamaker, M.; Small, D.; Duffield, A.S. FLT3-ITD cooperates with Rac1 to modulate the sensitivity of leukemic cells to chemotherapeutic agents via regulation of DNA repair pathways. Haematologica 2019, 104, 2418–2428. [Google Scholar] [CrossRef]
- Skvortsov, S.; Dudas, J.; Eichberger, P.; Witsch-Baumgartner, M.; Loeffler-Ragg, J.; Pritz, C.; Schartinger, V.H.; Maier, H.; Hall, J.; Debbage, P.; et al. Rac1 as a potential therapeutic target for chemo-radioresistant head and neck squamous cell carcinomas (HNSCC). Br. J. Cancer 2014, 110, 2677–2687. [Google Scholar] [CrossRef]
- Jordan, P.; Brazao, R.; Boavida, M.G.; Gespach, C.; Chastre, E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999, 18, 6835–6839. [Google Scholar] [CrossRef] [PubMed]
- Goka, E.T.; Chaturvedi, P.; Lopez, D.T.M.; Garza, A.; Lippman, M.E. RAC1b Overexpression Confers Resistance to Chemotherapy Treatment in Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef]
- Faria, M.; Matos, P.; Pereira, T.; Cabrera, R.; Cardoso, B.A.; Bugalho, M.J.; Silva, A.L. RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS ONE 2017, 12, e0172689. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Carmo, F.; Bugalho, M.J. RAC1b overexpression in papillary thyroid carcinoma: A role to unravel. Eur. J. Endocrinol. 2013, 168, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Lintz, M.; Munoz, A.; Reinhart-King, C.A. The Mechanics of Single Cell and Collective Migration of Tumor Cells. J. Biomech. Eng. 2017, 139, 021005. [Google Scholar] [CrossRef] [PubMed]
- Molina, O.; Abad, M.A.; Sole, F.; Menendez, P. Aneuploidy in Cancer: Lessons from Acute Lymphoblastic Leukemia. Trends Cancer 2021, 7, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; Wooten, E.C.; Bernardi, R.J.; Martin, T.D.; Chen, T.; et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 173, 499–514.e23. [Google Scholar] [CrossRef]
- Ferrer-Roca, O.; Perez-Gomez, J.A.; Cigudosa, J.C.; Gomez, E.; Estevez, M. Genetic heterogeneity of benign thyroid lesions. Static and flow cytometry, karyotyping and in situ hybridization analysis. Anal. Cell Pathol. 1998, 16, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Belge, G.; Roque, L.; Soares, J.; Bruckmann, S.; Thode, B.; Fonseca, E.; Clode, A.; Bartnitzke, S.; Castedo, S.; Bullerdiek, J. Cytogenetic investigations of 340 thyroid hyperplasias and adenomas revealing correlations between cytogenetic findings and histology. Cancer Genet. Cytogenet. 1998, 101, 42–48. [Google Scholar] [CrossRef]
- Roque, L.; Serpa, A.; Clode, A.; Castedo, S.; Soares, J. Significance of trisomy 7 and 12 in thyroid lesions with follicular differentiation: A cytogenetic and in situ hybridization study. Lab. Investig. 1999, 79, 369–378. [Google Scholar]
- Barril, N.; Carvalho-Sales, A.B.; Tajara, E.H. Detection of numerical chromosome anomalies in interphase cells of benign and malignant thyroid lesions using fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2000, 117, 50–56. [Google Scholar] [CrossRef]
- Teyssier, J.R.; Liautaud-Roger, F.; Ferre, D.; Patey, M.; Dufer, J. Chromosomal changes in thyroid tumors. Relation with DNA content, karyotypic features, and clinical data. Cancer Genet. Cytogenet. 1990, 50, 249–263. [Google Scholar] [CrossRef]
- Herrmann, M.E.; Lalley, P.A. Significance of trisomy 7 in thyroid tumors. Cancer Genet. Cytogenet. 1992, 62, 144–149. [Google Scholar] [CrossRef]
- Taruscio, D.; Carcangiu, M.L.; Ried, T.; Ward, D.C. Numerical chromosomal aberrations in thyroid tumors detected by double fluorescence in situ hybridization. Genes Chromosomes Cancer 1994, 9, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Roque, L.; Clode, A.L.; Gomes, P.; Rosa-Santos, J.; Soares, J.; Castedo, S. Cytogenetic findings in 31 papillary thyroid carcinomas. Genes Chromosomes Cancer 1995, 13, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Dettori, T.; Frau, D.V.; Lai, M.L.; Mariotti, S.; Uccheddu, A.; Daniele, G.M.; Tallini, G.; Faa, G.; Vanni, R. Aneuploidy in oncocytic lesions of the thyroid gland: Diffuse accumulation of mitochondria within the cell is associated with trisomy 7 and progressive numerical chromosomal alterations. Genes Chromosomes Cancer 2003, 38, 22–31. [Google Scholar] [CrossRef]
- Braun, R.; Ronquist, S.; Wangsa, D.; Chen, H.; Anthuber, L.; Gemoll, T.; Wangsa, D.; Koparde, V.; Hunn, C.; Habermann, J.K.; et al. Single Chromosome Aneuploidy Induces Genome-Wide Perturbation of Nuclear Organization and Gene Expression. Neoplasia 2019, 21, 401–412. [Google Scholar] [CrossRef]
- Kim, H.; Nguyen, N.P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef]
- Palechor-Ceron, N.; Suprynowicz, F.A.; Upadhyay, G.; Dakic, A.; Minas, T.; Simic, V.; Johnson, M.; Albanese, C.; Schlegel, R.; Liu, X. Radiation induces diffusible feeder cell factor(s) that cooperate with ROCK inhibitor to conditionally reprogram and immortalize epithelial cells. Am. J. Pathol. 2013, 183, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, R.E.; Klopper, J.P.; Korch, C.; Pugazhenthi, U.; Benezra, M.; Knauf, J.A.; Fagin, J.A.; Marlow, L.A.; Copland, J.A.; Smallridge, R.C.; et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 2008, 93, 4331–4341. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N.; et al. Comprehensive Genetic Characterization of Human Thyroid Cancer Cell Lines: A Validated Panel for Preclinical Studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, J.; Tanaka, K.; Otsuki, T.; Moriya, T.; Kunisue, H.; Uno, M.; Sonoo, H. All-trans-retinoic acid modulates expression levels of thyroglobulin and cytokines in a new human poorly differentiated papillary thyroid carcinoma cell line, KTC-1. J. Clin. Endocrinol. Metab. 2000, 85, 2889–2896. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HGNC | Chr | Start (GRCh37/hg19) | Ref (cDNA) | Alt | Type | Allele Frequency | HGNC_AAS | Oncogenic Prediction Score (CScape) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre_1 ST (FFPE) T200.1 | Pre_2 LN (FFPE) T200.1 | Post (M) FNA (WES) | ||||||||
| BRAF | 7 | 140453191 | TAC | - | DEL | 17.7 | 33.7 | 18.2 | BRAF_T599del | n/a |
| BRAF | 7 | 140453132 | A | T | SNV | 0 | 35.6 | 24.7 | BRAF_K601N | 0.66 |
| RAC1 | 7 | 6426908 | C | G | SNV | 0 | 0 | 25.6 | RAC1_P34R | 0.85 |
| PIK3CA | 3 | 18942499 | G | T | SNV | 0 | 6.6 | 0 | PIK3CA_C769Fl | 0.92 |
| POS1 | 6 | 117687245 | C | A | SNV | 0 | 5.0 | 0 | ROS1_L936M | 0.71 |
| BRCA1 | 17 | 41244936 | C | T | SNV | 0 | 8.1 | 0 | BRCA1_P871L | 0.43 |
| XPO1 | 2 | 61713042 | G | T | SNV | 0 | 5.1 | 0 | XPO1_R790I | 0.95 |
| NBN | 8 | 90990479 | G | C | SNV | 7.0 | 9.7 | 0 | NBN_E185Q | 0.79 |
| NBN | 8 | 90958530 | A | G | SNV | 9.3 | 0 | 0 | NBN_splice_region_variant | n/a |
| BRCA1 | 17 | 41223094 | A | G | SNV | 7.5 | 0 | 0 | BRCA1_S1634G | 0.74 |
| BRCA1 | 17 | 41244000 | A | G | SNV | 7.9 | 0 | 0 | BRCA1_K1183R | 0.37 |
| FLT3 | 13 | 28624294 | C | T | SNV | 6.3 | 0 | 0 | FLT3_T227M | 0.53 |
| TSHR | 1 | 81528564 | G | T | SNV | 8.3 | 0 | 0 | TSHR_splice_region_variant | n/a |
| SETD2 | 3 | 47125385 | C | T | SNV | 12.8 | 0 | 0 | SETD2_P1962L | 0.46 |
| ATR | 3 | 142217564 | C | A | SNV | 4.9 | 0 | 0 | ATR_A1811A | 0.30 |
| ATR | 3 | 142274740 | A | - | DEL | 8.6 | 0 | 0 | ATR_I774fs | n/a |
| MED12 | X | 70361760 | A | - | DEL | 4.6 | 0 | 0 | MED12_Q2149fs | n/a |
| GABRA6 | 5 | 161114522 | A | G | SNV | 7.1 | 0 | 0 | intron | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagheri-Yarmand, R.; Busaidy, N.L.; McBeath, E.; Danysh, B.P.; Evans, K.W.; Moss, T.J.; Akcakanat, A.; Ng, P.K.S.; Knippler, C.M.; Golden, J.A.; et al. RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient. Cancers 2021, 13, 4950. https://doi.org/10.3390/cancers13194950
Bagheri-Yarmand R, Busaidy NL, McBeath E, Danysh BP, Evans KW, Moss TJ, Akcakanat A, Ng PKS, Knippler CM, Golden JA, et al. RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient. Cancers. 2021; 13(19):4950. https://doi.org/10.3390/cancers13194950
Chicago/Turabian StyleBagheri-Yarmand, Rozita, Naifa L. Busaidy, Elena McBeath, Brian P. Danysh, Kurt W. Evans, Tyler J. Moss, Argun Akcakanat, Patrick K. S. Ng, Christina M. Knippler, Jalyn A. Golden, and et al. 2021. "RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient" Cancers 13, no. 19: 4950. https://doi.org/10.3390/cancers13194950
APA StyleBagheri-Yarmand, R., Busaidy, N. L., McBeath, E., Danysh, B. P., Evans, K. W., Moss, T. J., Akcakanat, A., Ng, P. K. S., Knippler, C. M., Golden, J. A., Williams, M. D., Multani, A. S., Cabanillas, M. E., Shaw, K. R., Meric-Bernstam, F., Shah, M. H., Ringel, M. D., & Hofmann, M. C. (2021). RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient. Cancers, 13(19), 4950. https://doi.org/10.3390/cancers13194950