
Mechanisms of Cancer Cell Death: Therapeutic Implications for Pancreatic Ductal Adenocarcinoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathway Inhibition
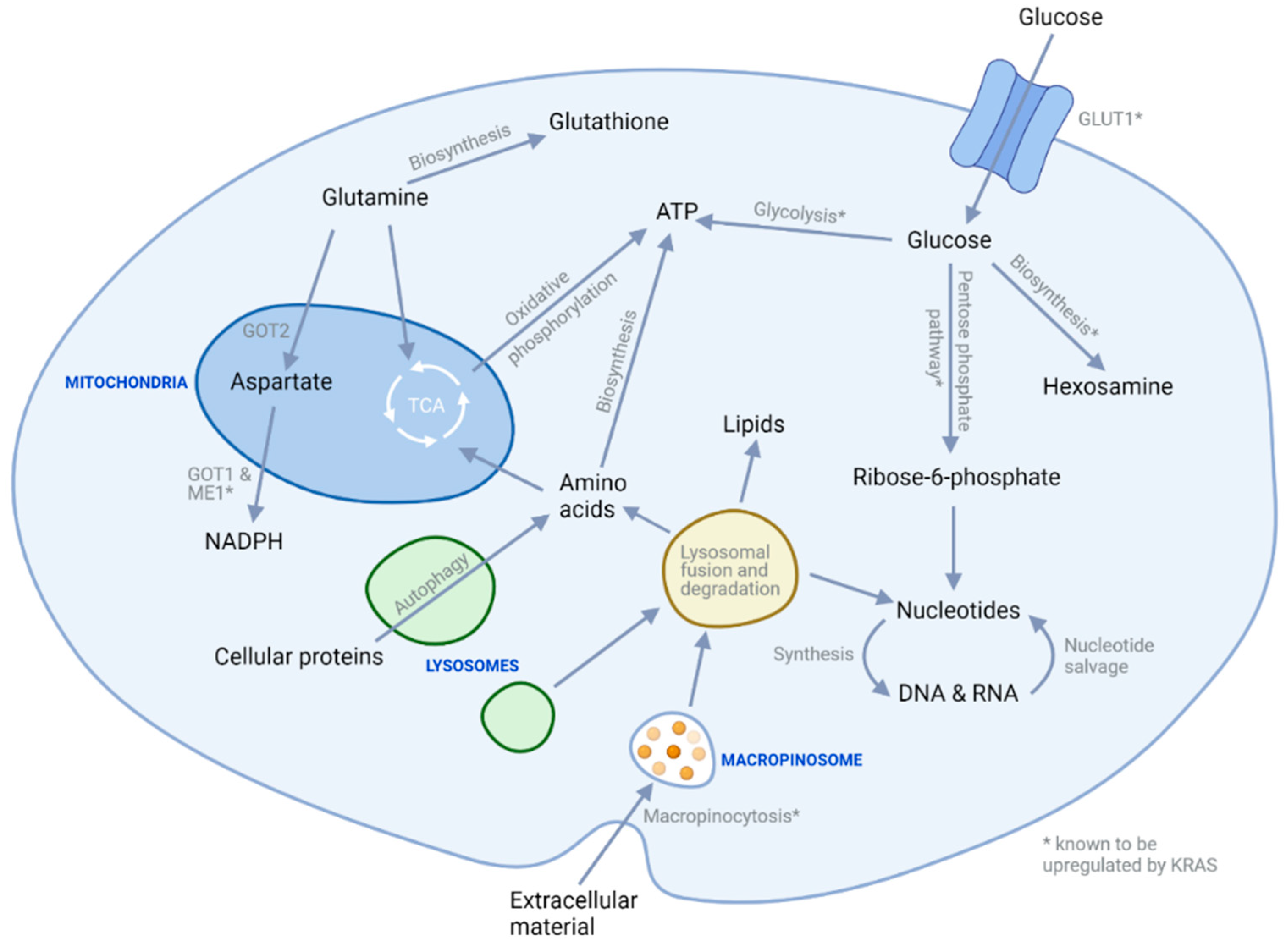
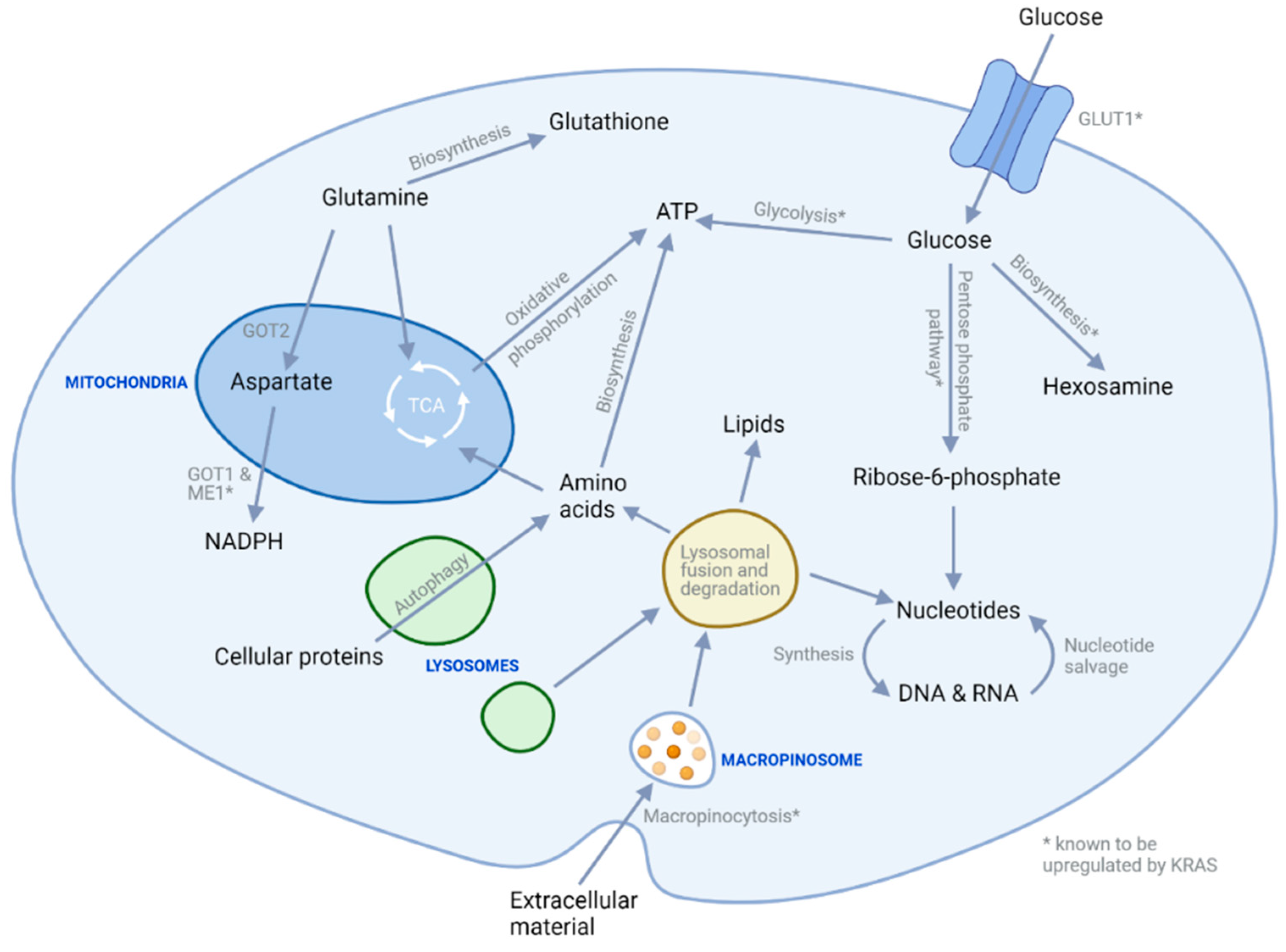
3. Metabolic Targeting
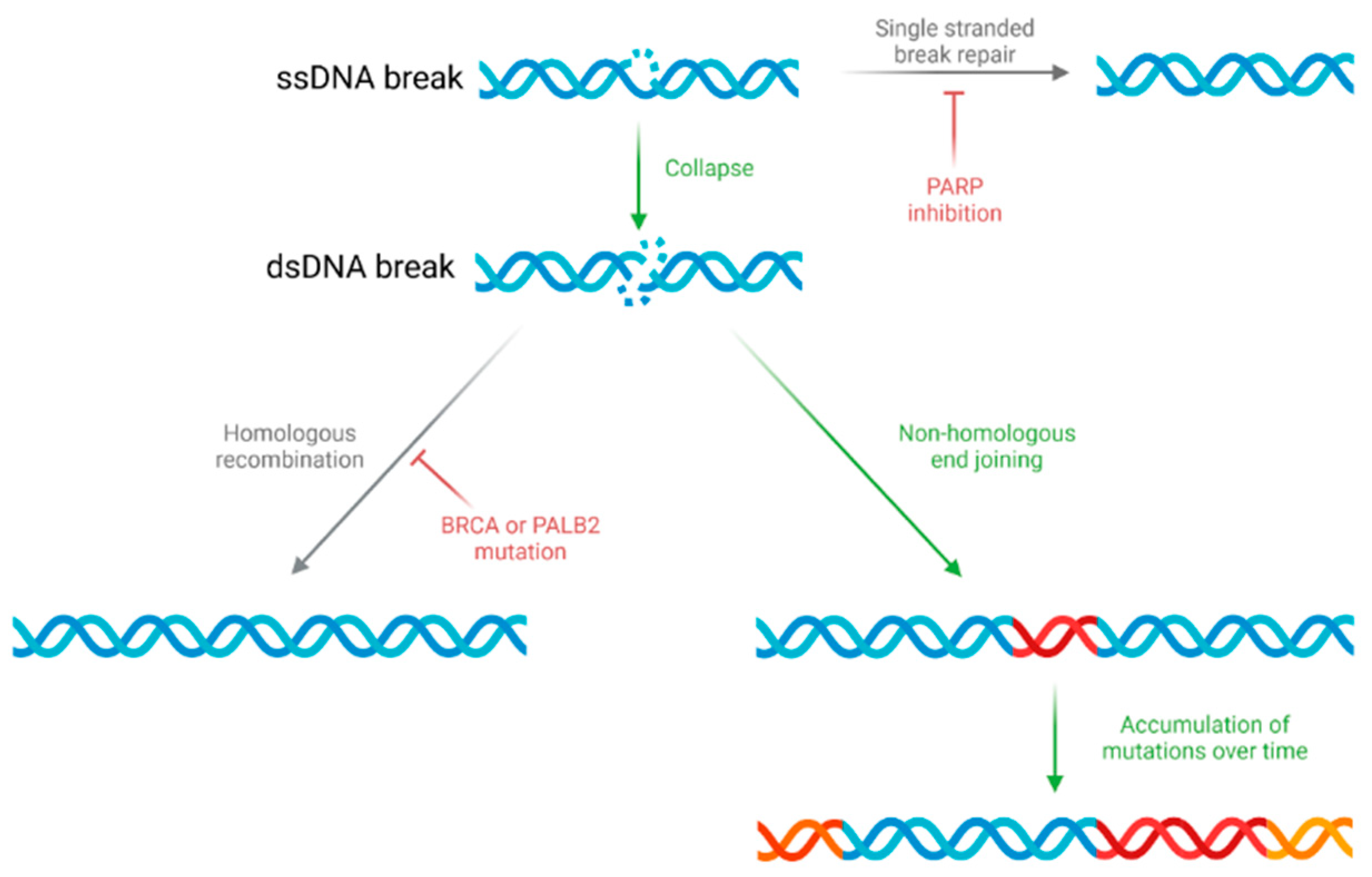
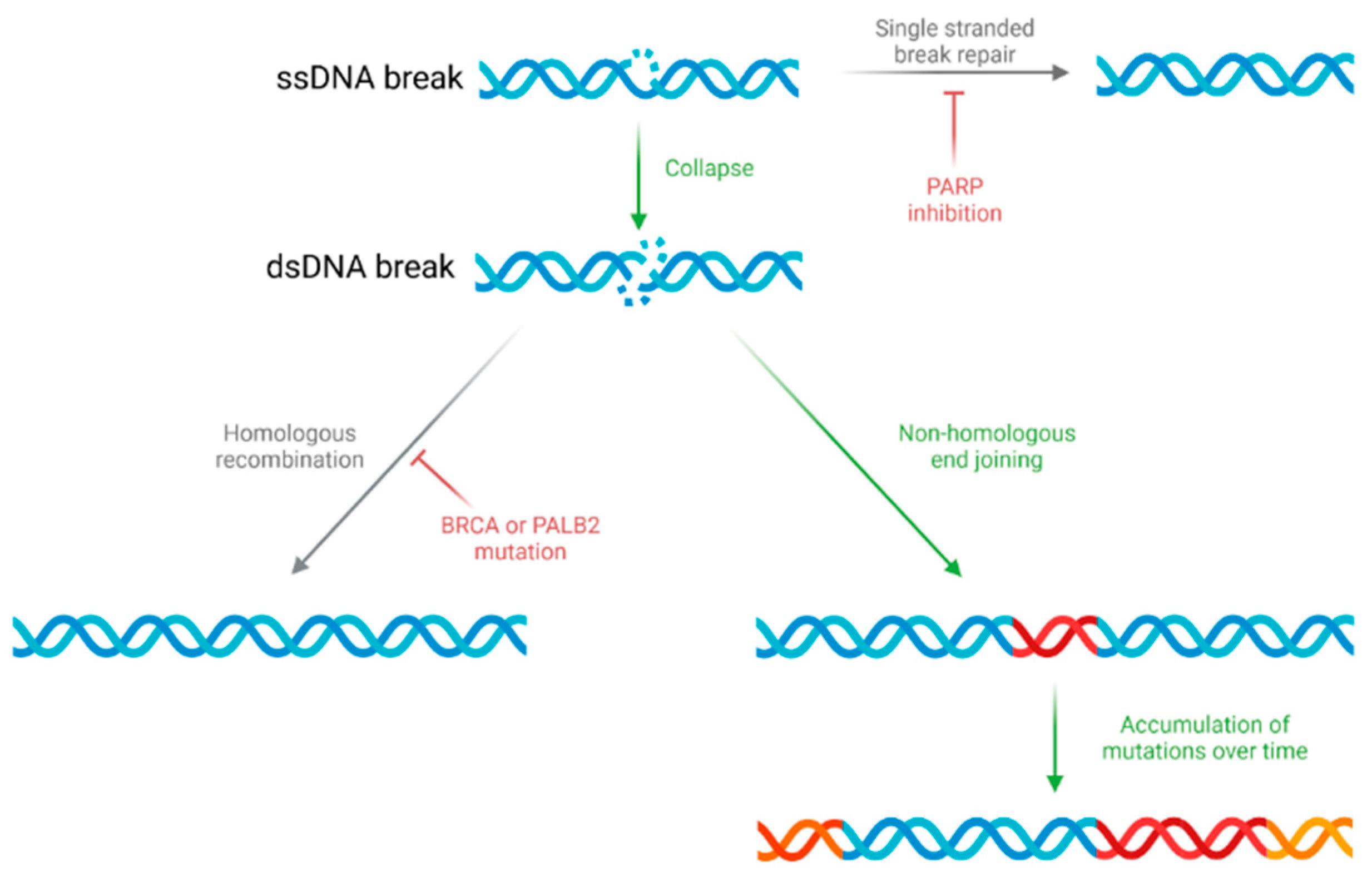
4. Genome Instability and DNA Damage
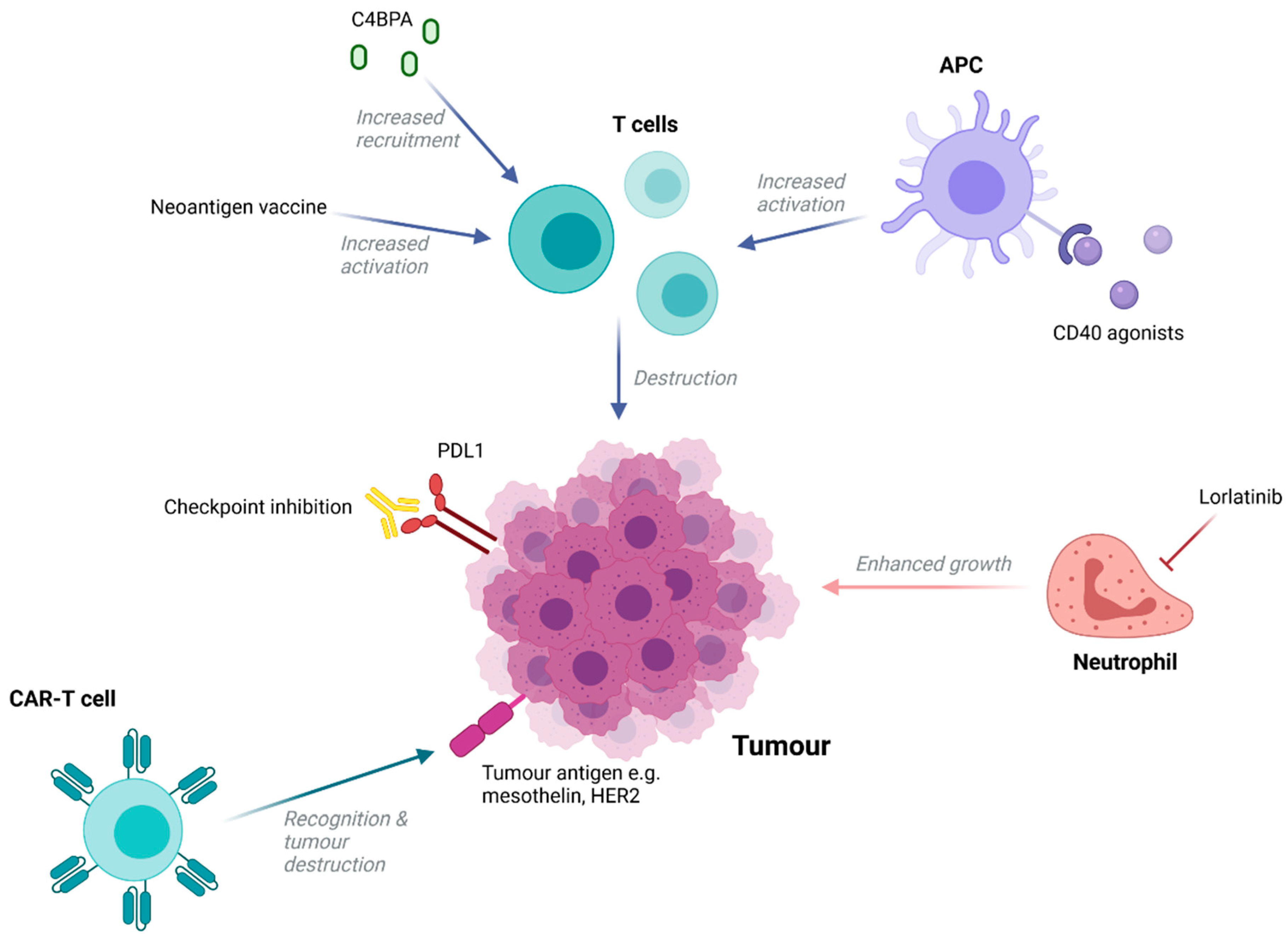
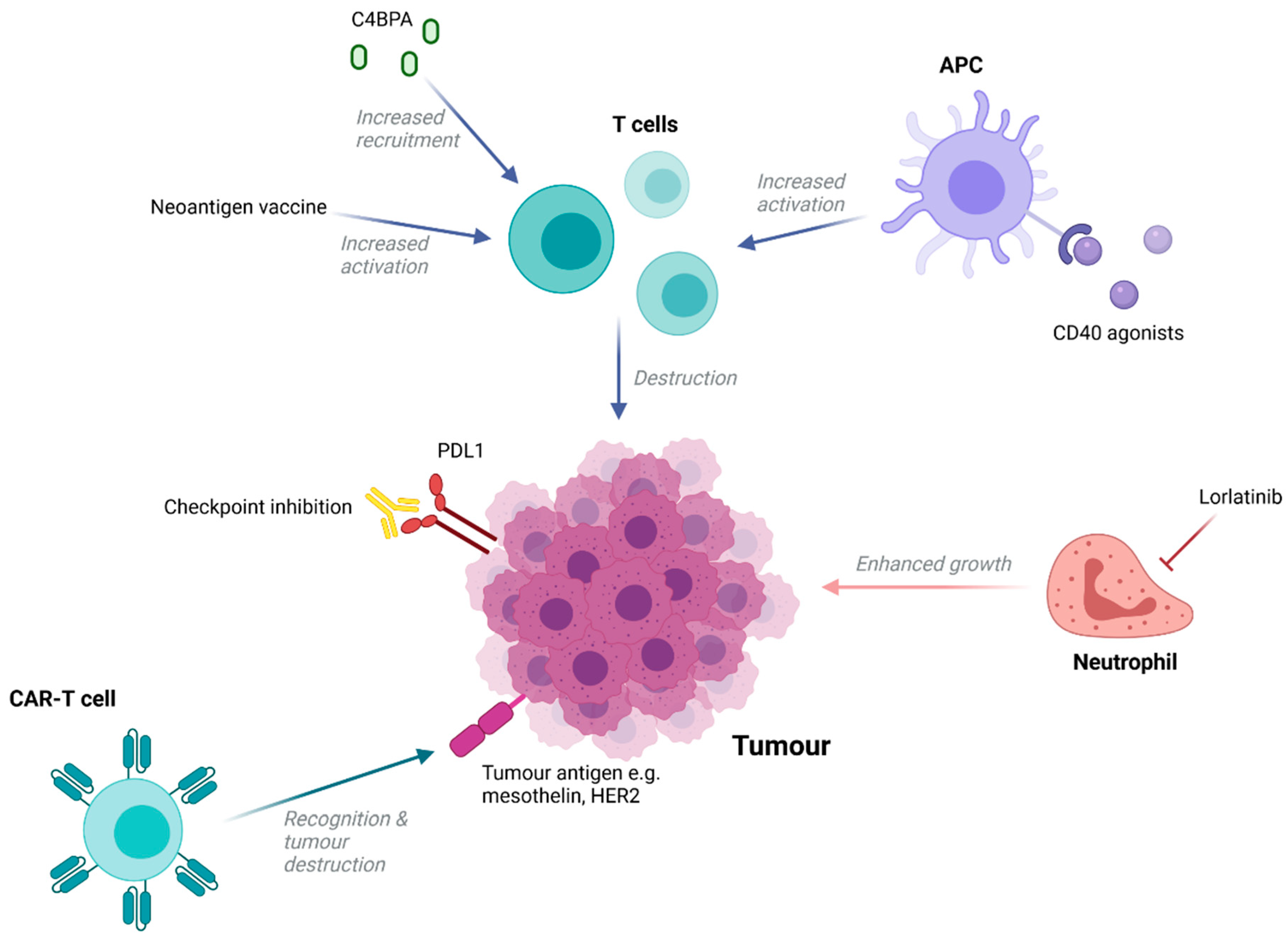
5. Immunotherapies
6. Extracellular Tumour Microenvironment Modification
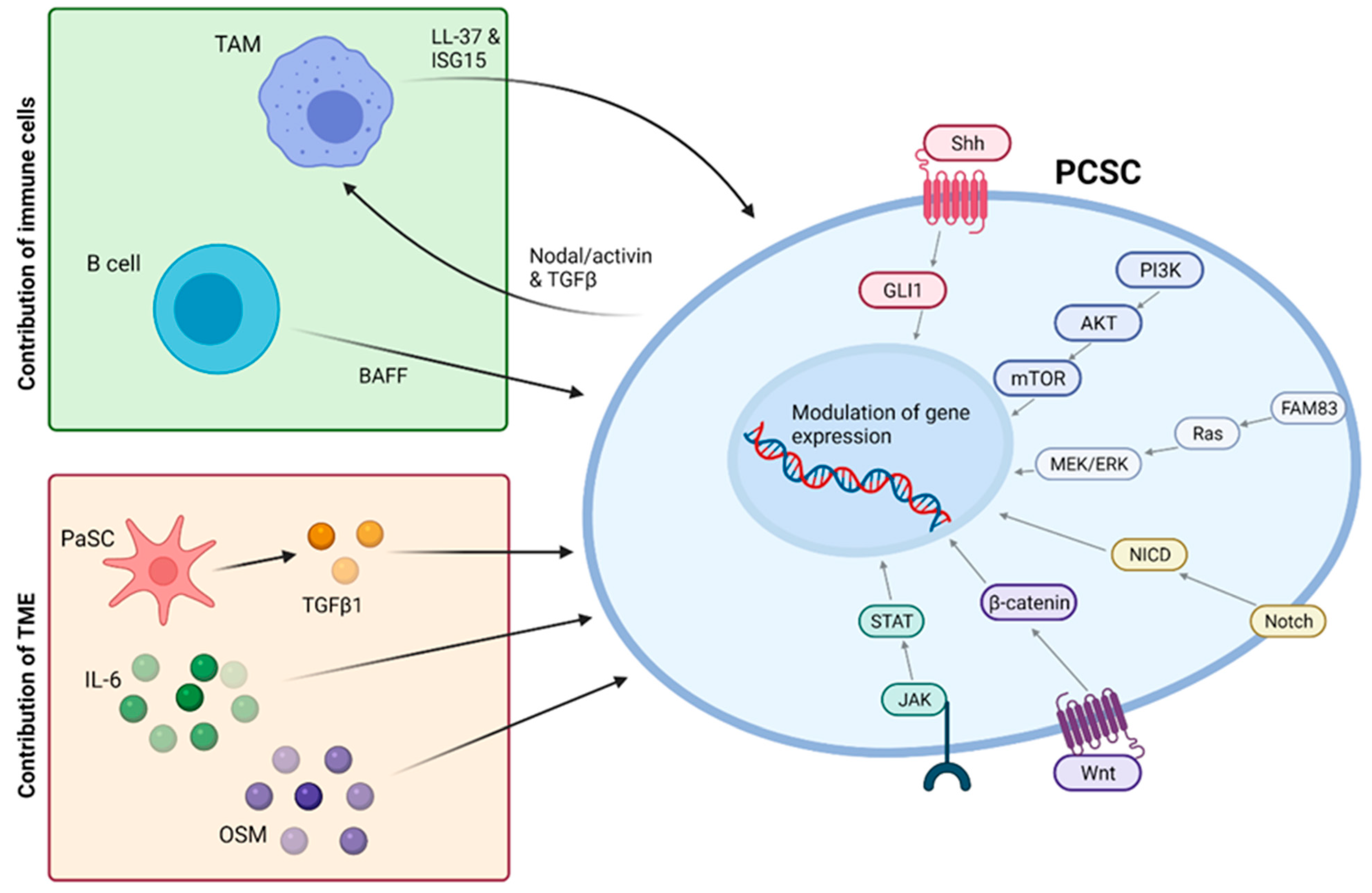
7. Elimination of Cancer Stem Cells
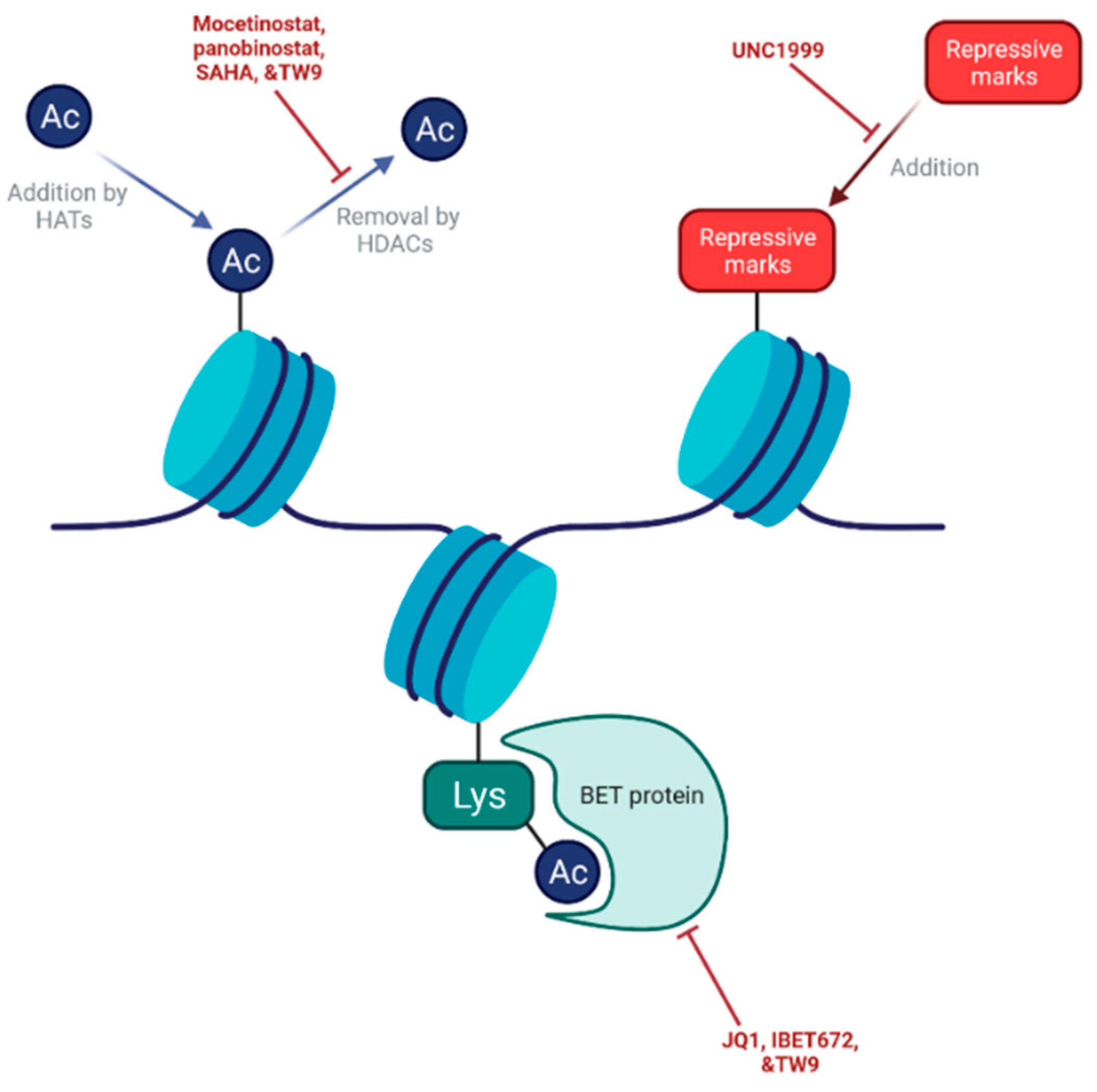
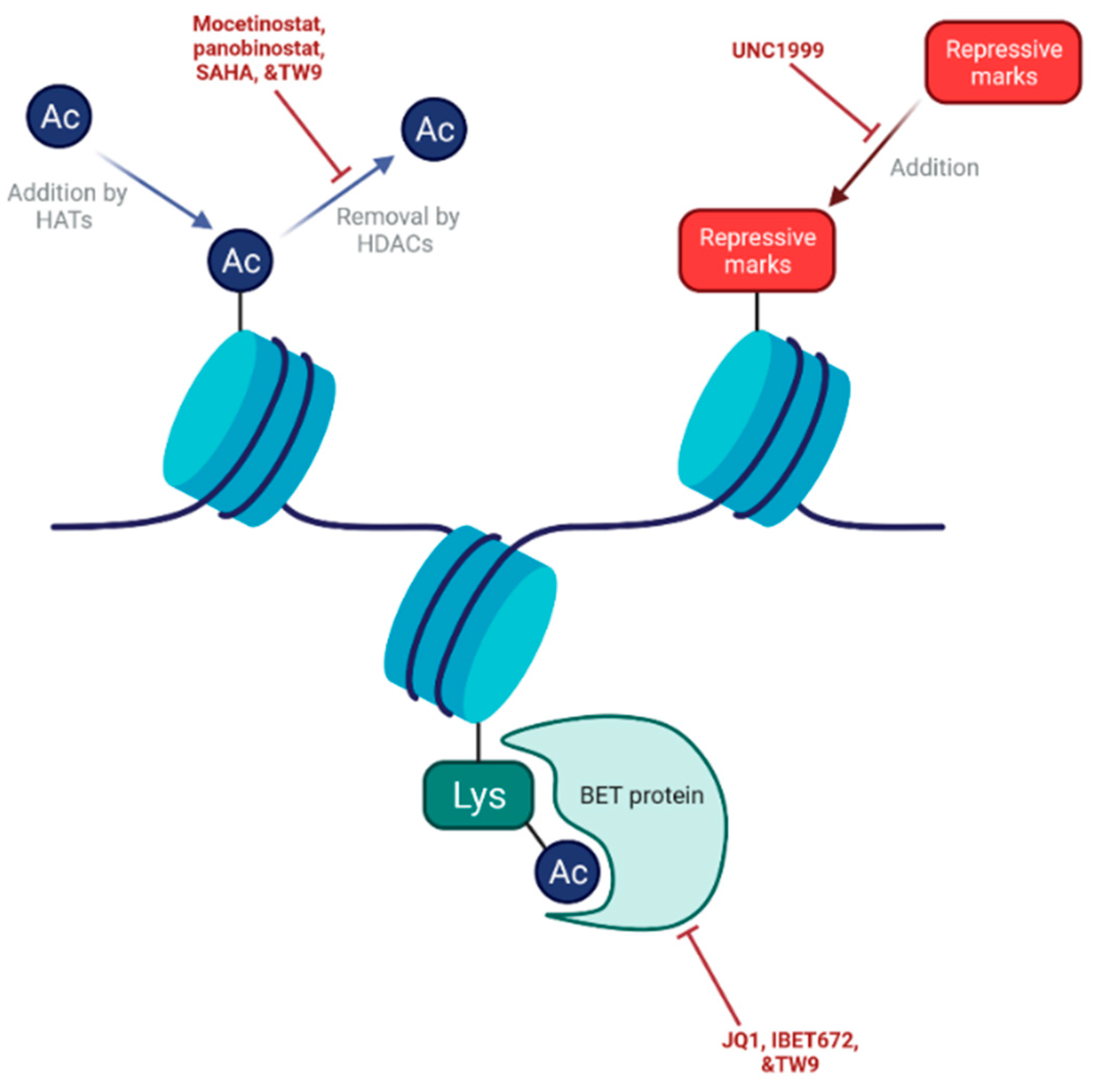
8. Targeting Epigenetic Mechanisms
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [Green Version]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 1–22. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Roser, M.; Ritchie, H. Cancer. Our World in Data. Available online: https://ourworldindata.org/cancer (accessed on 6 August 2021).
- Hill, A.; Chung, V. Pancreatic Cancer. Oncol. Precis. Med. Era 2020, 97–109. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wientjes, M.G.; Au, J.L.-S. Pancreatic Cancer: Pathobiology, Treatment Options, and Drug Delivery. AAPS J. 2010, 12, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy with Gemcitabine Plus Erlotinib versus Gemcitabine Alone in Patients after R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef]
- NICE. Pancreatic Cancer in Adults: Diagnosis and Management. NICE Guideline [NG85]. Available online: https://www.nice.org.uk/guidance/ng85/chapter/Recommendations#ftn.footnote_5 (accessed on 8 August 2021).
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, S.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin. Cancer Res. 2017, 23, 1638–1646. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Waters, A.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J. Refining Classification of Pancreatic Cancer Subtypes to Improve Clinical Care. Gastroenterology 2018, 155, 1689–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, S.; Lefesvre, P.; Nicolle, R.; Biankin, A.; Puleo, F.; Van Laethem, J.; Rooman, I. Different shades of pancreatic ductal adenocarcinoma, different paths towards precision therapeutic applications. Ann. Oncol. 2019, 30, 1428–1436. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Stoica, A.-F.; Chang, C.-H.; Pauklin, S. Molecular Therapeutics of Pancreatic Ductal Adenocarcinoma: Targeted Pathways and the Role of Cancer Stem Cells. Trends Pharmacol. Sci. 2020, 41, 977–993. [Google Scholar] [CrossRef]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.L.; Lertpiriyapong, K.; Cocco, L.; Martelli, A.M.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Steelman, L.; Abrams, S.L.; et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv. Biol. Regul. 2015, 59, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.T.; Navas, C.; Martín-Serrano, G.; Graña-Castro, O.; Lechuga, C.G.; Martín-Díaz, L.; Djurec, M.; Li, J.; Morales-Cacho, L.; Esteban-Burgos, L.; et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer Cell 2019, 35, 573–587.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Vena, F.; Causi, E.L.; Rodriguez-Justo, M.; Goodstal, S.; Hagemann, T.; Hartley, J.A.; Hochhauser, D. The MEK1/2 Inhibitor Pimasertib Enhances Gemcitabine Efficacy in Pancreatic Cancer Models by Altering Ribonucleotide Reductase Subunit-1 (RRM1). Clin. Cancer Res. 2015, 21, 5563–5577. [Google Scholar] [CrossRef] [Green Version]
- Kheder, E.S.; Hong, D.S. Emerging Targeted Therapy for Tumors with NTRK Fusion Proteins. Clin. Cancer Res. 2018, 24, 5807–5814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Rolfo, C.D.; Liu, S.V.; Multani, P.S.; Maneval, E.C.; Garrido-Laguna, I. Clinical benefit of entrectinib for patients with metastatic pancreatic cancer who harbor NTRK and ROS1 fusions. J. Clin. Oncol. 2018, 36, 521. [Google Scholar] [CrossRef]
- Laffranchi, B.; De Jonge, M.J.; Bajetta, E.; Hendlisz, A.; Giuliani, R.; Barone, C.; Endlicher, E.; Jannuzzo, M.G.; Spinelli, R.; Santoro, A. Phase II study of danusertib (D) in advanced/metastatic colorectal and pancreatic cancers (CRC, PC). J. Clin. Oncol. 2010, 28, e13558. [Google Scholar] [CrossRef]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; del Camino Menéndez, M.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353.e6. [Google Scholar] [CrossRef] [PubMed]
- García-Reyes, B.; Kretz, A.-L.; Ruff, J.-P.; Von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The Emerging Role of Cyclin-Dependent Kinases (CDKs) in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef] [Green Version]
- Annese, T.; Tamma, R.; Ruggieri, S.; Ribatti, D. Angiogenesis in Pancreatic Cancer: Pre-Clinical and Clinical Studies. Cancers 2019, 11, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isacoff, W.H.; Reber, H.A.; Bedford, R.; Hoos, W.; Rahib, L.; Upfill-Brown, A.; Donahue, T.; Hines, O.J. Low-Dose Continuous 5-Fluorouracil Combined with Leucovorin, nab-Paclitaxel, Oxaliplatin, and Bevacizumab for Patients with Advanced Pancreatic Cancer: A Retrospective Analysis. Target. Oncol. 2018, 13, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Pauklin, S. ROS and TGFβ: From pancreatic tumour growth to metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 1–11. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. A phase II, double-blind study of galunisertib+gemcitabine (GG) vs. gemcitabine+placebo (GP) in patients (pts) with unresectable pancreatic cancer (PC). J. Clin. Oncol. 2016, 34, 4019. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling intumorigenesis and the development ofcancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, R.; Varadhachary, G.R.; Bhosale, P.R.; Wang, X.; Fogelman, D.R.; Shroff, R.T.; Overman, M.J.; Wolff, R.A.; Javle, M. Randomized, phase I/II study of gemcitabine plus IGF-1R antagonist (MK-0646) versus gemcitabine plus erlotinib with and without MK-0646 for advanced pancreatic adenocarcinoma. J. Hematol. Oncol. 2018, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, A. Highly bioavailable curcumin (Theracurmin): Its development and clinical application. PharmaNutrition 2015, 3, 123–130. [Google Scholar] [CrossRef]
- Kanai, M.; Yoshimura, K.; Asada, M.; Imaizumi, A.; Suzuki, C.; Matsumoto, S.; Nishimura, T.; Mori, Y.; Masui, T.; Kawaguchi, Y.; et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother. Pharmacol. 2011, 68, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Hu, W. Progress of JAK/STAT 3 and its inhibitors in the treatment of cancer. In AIP Conference Proceedings; AIP Publishing LLC.: Melville, NY, USA, 2020; Volume 2208, p. 020045. [Google Scholar]
- Kim, S.-Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.; Van Cutsem, E.; Bendell, J.; Hidalgo, M.; Li, C.-P.; Salvo, M.G.; Macarulla, T.; Sahai, V.; Sama, A.; Greeno, E.; et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. New Drugs 2018, 36, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Hendifar, A.; Starodub, A.; Chaves, J.; Yang, Y.; Koh, B.; Barbie, D.; Hahn, W.C.; Fuchs, C.S. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2019, 37, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Qureshy, Z.; Johnson, D.E.; Grandis, J.R. Targeting the JAK/STAT pathway in solid tumors. J. Cancer Metastasis Treat. 2020, 2020, 27. [Google Scholar] [CrossRef]
- Yu, E.Y.; Wilding, G.; Posadas, E.; Gross, M.; Culine, S.; Massard, C.; Morris, M.J.; Hudes, G.; Calabrò, F.; Cheng, S.; et al. Phase II Study of Dasatinib in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2009, 15, 7421–7428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, T.; Van Cutsem, E.; Moore, M.; Bazin, I.; Rosemurgy, A.; Bodoky, G.; Deplanque, G.; Harrison, M.; Melichar, B.; Pezet, D.; et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann. Oncol. 2017, 28, 354–361. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempero, M.; Oh, D.; Macarulla, T.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.; Starling, N.; Bachet, J.; Chang, H.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine as first-line treatment for patients with metastatic pancreatic adenocarcinoma: Results from the phase 3 RESOLVE study. Ann. Oncol. 2019, 30, iv126. [Google Scholar] [CrossRef]
- Overman, M.; Javle, M.; Davis, R.E.; Vats, P.; Kumar-Sinha, C.; Xiao, L.; Mettu, N.B.; Parra, E.R.; Benson, A.B.; Lopez, C.D.; et al. Randomized phase II study of the Bruton tyrosine kinase inhibitor acalabrutinib, alone or with pembrolizumab in patients with advanced pancreatic cancer. J. Immunother. Cancer 2020, 8, e000587. [Google Scholar] [CrossRef] [Green Version]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial Metabolism in PDAC: From Better Knowledge to New Targeting Strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [Green Version]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Hadi, N.A.; Gicquel, T.; El Kaoutari, A.; Diémé, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients with Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel with or without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- M.D. Anderson Cancer Center. Binimetinib and Hydroxychloroquine in Treating Patients with KRAS Mutant Metastatic Pancreatic Cancer. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04132505 (accessed on 9 August 2021).
- Duong, H.-Q.; Yi, Y.W.; Kang, H.J.; Bin Hong, Y.; Tang, W.; Wang, A.; Seong, Y.-S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; et al. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1136–1146. [Google Scholar] [CrossRef] [Green Version]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Gruber, J.J.; Gross, W.; McMillan, A.; Ford, J.M.; Telli, M.L. A phase II clinical trial of talazoparib monotherapy for PALB2 mutation-associated advanced breast cancer. J. Clin. Oncol. 2021, 39, TPS1109. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib monotherapy for previously treated pancreatic cancer with DNA damage repair genetic alterations other than germline BRCA variants findings from 2 phase 2 nonrandomized clinical trials. JAMA Oncol. 2021, 7, 693–699. [Google Scholar] [CrossRef]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, I.H. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A.; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef] [PubMed]
- NCT03404960. Niraparib + Ipilimumab or Nivolumab in Progression Free Pancreatic Adenocarcinoma After Platinum-Based Chemotherapy. 2017. Available online: https://clinicaltrials.gov/show/nct03404960 (accessed on 11 August 2021).
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Br. Dent. J. 2014, 217, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Güthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial–mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Palii, S.S.; Innes, C.L.; Paules, R.S. Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents. Cell Cycle 2014, 13, 3541–3550. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti–PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Upadhrasta, S.; Zheng, L. Strategies in Developing Immunotherapy for Pancreatic Cancer: Recognizing and Correcting Multiple Immune “Defects” in the Tumor Microenvironment. J. Clin. Med. 2019, 8, 1472. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6Clow F4/80+ Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, M.H.; O’Reilly, E.M.; Rosemarie, M.; Varadhachary, G.; Wainberg, Z.A.; Ko, A.; Fisher, G.A.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. Cancer Res. 2019, 79, CT004. [Google Scholar]
- Sasaki, K.; Takano, S.; Tomizawa, S.; Miyahara, Y.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Takada, M.; Ohtsuka, M. C4b-binding protein α-chain enhances antitumor immunity by facilitating the accumulation of tumor-infiltrating lymphocytes in the tumor microenvironment in pancreatic cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–16. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Konduri, V.; Joseph, S.K.; Byrd, T.T.; Nawas, Z.; Vazquez-Perez, J.; Hofferek, C.J.; Halpert, M.M.; Liu, D.; Liang, Z.; Baig, Y.; et al. A subset of cytotoxic effector memory T cells enhances CAR T cell efficacy in a model of pancreatic ductal adenocarcinoma. Sci. Transl. Med. 2021, 13, eabc3196. [Google Scholar] [CrossRef]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.E.; Morse, M.A.; Zeh, H.J.; Weekes, C.D.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Lianyuan, T.; Gang, L.; Ming, T.; Dianrong, X.; Chunhui, Y.; Zhaolai, M.; Bin, J. Tumor associated neutrophils promote the metastasis of pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2020, 21, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.R.; Strøbech, J.E.; Horton, E.R.; Jackstadt, R.; Laitala, A.; Bravo, M.C.; Maltese, G.; Jensen, A.R.D.; Reuten, R.; Rafaeva, M.; et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.; Nywening, T.M.; Hawkins, T.M.N.W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennewith, K.L.; Huang, X.; Ham, C.M.; Graves, E.E.; Erler, J.; Kambham, N.; Feazell, J.; Yang, G.P.; Koong, A.; Giaccia, A.J. The Role of Tumor Cell–Derived Connective Tissue Growth Factor (CTGF/CCN2) in Pancreatic Tumor Growth. Cancer Res. 2009, 69, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Picozzi, V.J.; Pishvaian, M.J.; Mody, K.; Winter, J.M.; Glaspy, J.A.; Larson, T.; Matrana, M.R.; Saikali, K.; Carney, M.; Porter, S.; et al. Effect of anti-CTGF human recombinant monoclonal antibody pamrevlumab on resectability and resection rate when combined with gemcitabine/nab-paclitaxel in phase 1/2 clinical study for the treatment of locally advanced pancreatic cancer patients. J. Clin. Oncol. 2018, 36, 4016. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.M.; Silva, M.A.; D’Costa, Z.; Bockelmann, R.; Soonawalla, Z.; Liu, S.; O’Neill, E.; Mukherjee, S.; McKenna, W.G.; Muschel, R.; et al. The prognostic role of desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 4183–4194. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.-B.; Sato, N.; Kohi, S.; Yamaguchi, K. Prognostic Impact of Hyaluronan and Its Regulators in Pancreatic Ductal Adenocarcinoma. PLoS ONE 2013, 8, e80765. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.-M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.-M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007, 110, 587–595. [Google Scholar] [CrossRef]
- Tahkola, K.; Ahtiainen, M.; Mecklin, J.-P.; Kellokumpu, I.; Laukkarinen, J.; Tammi, M.; Tammi, R.; Väyrynen, J.P.; Böhm, J. Stromal hyaluronan accumulation is associated with low immune response and poor prognosis in pancreatic cancer. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Van Cutsem, E.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20) + nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinom. J. Clin. Oncol. 2020, 38, 638. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.R.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. A phase IB/II randomized study of mFOLFIRINOX (mFFOX) + pegylated recombinant human hyaluronidase (PEGPH20) versus mFFOX alone in patients with good performance status metastatic pancreatic adenocarcinoma (mPC): SWOG S1313 (NCT #01959139). J. Clin. Oncol. 2018, 36, 208. [Google Scholar] [CrossRef]
- Hakim, N.; Patel, R.; DeVoe, C.; Saif, M.W. Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas 2019, 3, e1–e4. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askan, G.; Sahin, I.H.; Chou, J.F.; Yavas, A.; Capanu, M.; Iacobuzio-Donahue, C.A.; Basturk, O.; O’Reilly, E.M. Pancreatic cancer stem cells may define tumor stroma characteristics and recurrence patterns in pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 385. [Google Scholar] [CrossRef]
- Mueller, M.; Hermann, P.C.; Witthauer, J.; Rubio–Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined Targeted Treatment to Eliminate Tumorigenic Cancer Stem Cells in Human Pancreatic Cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Nature 2020, 5, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics. Cancers 2018, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, C.; Sousa, B.C.; Manfredi, M.; Brandi, J.; Dalla Pozza, E.; Marengo, E.; Palmieri, M.; Dando, I.; Wakelam, M.J.; Lopez-Clavijo, A.F.; et al. Integrated lipidomics and proteomics reveal cardiolipin alterations, upregulation of HADHA and long chain fatty acids in pancreatic cancer stem cells. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sainz, B. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Biol. 2018, 53, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Q.; Dudek, A.Z. Phase II study of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy. Anticancer. Res. 2012, 32, 1027–1031. [Google Scholar] [PubMed]
- Leal, A.S.M.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef]
- Xie, F.; Huang, M.; Lin, X.; Liu, C.; Liu, Z.; Meng, F.; Wang, C.; Huang, Q. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET Bromodomain Inhibitors Block Growth of Pancreatic Cancer Cells in Three-Dimensional Collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Fei, Q.; Xiong, P.; Yang, J.; Zhang, Z.; Lin, X.; Pan, M.; Lu, F.; Huang, H. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. OncoImmunology 2019, 8, e1581529. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sánchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zegar, T.; Weiser, T.; Hamdan, F.; Berger, B.; Lucas, R.; Balourdas, D.-I.; Ladigan, S.; Cheung, P.F.; Liffers, S.; et al. Characterization of a dual BET/HDAC inhibitor for treatment of pancreatic ductal adenocarcinoma. Int. J. Cancer 2020, 147, 2847–2861. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, M.C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Bian, B.; Bigonnet, M.; Gayet, O.; Loncle, C.; Maignan, A.; Gilabert, M.; Moutardier, V.; Garcia, S.; Turrini, O.; Delpero, J.R.; et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ 1: Implications for individualized medicine efforts. EMBO Mol. Med. 2017, 9, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Juiz, N.A.; Gayet, O.; Bigonnet, M.; Brandone, N.; Roques, J.; Cros, J.; Wang, N.; Dusetti, N.; Iovanna, J. Pancreatic Cancer Organoids for Determining Sensitivity to Bromodomain and Extra-Terminal Inhibitors (BETi). Front. Oncol. 2019, 9, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Subtype | Characteristics |
|---|---|---|
| Collison | Classical | Better prognosis than QM subtype following resection High expression of adhesion-associated and epithelial genes, high GATA6 expression Increased dependence on KRAS and increased sensitivity to erlotinib compared to QM subtype |
| Quasi-mesenchymal (QM) | High expression of mesenchyme associated genes Increased sensitivity to gemcitabine compared to classical subtype | |
| Exocrine-like | Relatively high expression of tumour cell-derived digestive enzyme genes | |
| Moffitt tumour-specific subtypes | Classical | 20/22 genes shared with Collisson classical subtype |
| Basal-like | Worse median survival and 1-year survival compared to basal-like subtype Better response to adjuvant therapy compared to classical subtype High expression of laminins and keratins | |
| Moffitt stromal-specific subtypes | Normal | High expression of markers for pancreatic stellate cells, smooth muscle actin, vimentin, and desmin |
| Activated | Worse median and 1-year survival compared to normal subtype More diverse gene expression including genes associated with macrophages (e.g., ITGAM, CCL13, and CCL18) and genes involved in tumour promotion (e.g., SPARC, WNT2, WNT5A, MMP9) | |
| Bailey | Squamous | Poor prognosis compared to other subtypes High expression of TP53 and KDM6A, and upregulation of TP63∆N transcriptional network Hypermethylation of pancreatic endodermal cell-fate determining genes |
| Pancreatic progenitor | High expression of genes involved in early pancreatic development (FOXA2/3, PDX1, and MNX1) | |
| Immunogenic | Significant immune infiltrate Upregulation of immune networks, including pathways associated with acquired immune suppression | |
| Aberrantly differentiated endocrine exocrine | Subclass of pancreatic progenitor tumours High expression of genes involved in regulating KRAS activation, exocrine (NR5A2 and RBPJL), and endocrine differentiation (NEUROD1 and NKX2-2) |
| Pathway | Function | Relevant Results |
|---|---|---|
| Growth factor receptors and associated pathways | ||
| Epidermal growth factor (EGF) pathway | Important regulator of cell survival, able to activate multiple signalling pathways including PI3K/Akt, Ras/Raf/MEK/ERK, and Jak-STAT. EGF receptor (EGFR) is essential in PDAC tumourigenesis as its activity is needed for KRAS activation of ERK [24]. | Addition of the EGFR inhibitor erlotinib to gemcitabine significantly prolonged OS compared to gemcitabine alone in a phase III trial [25]. More recently, combined inhibition of EGFR and c-RAF induced complete tumour regression in genetically engineered PDAC mouse models and blocked tumour progression in patient-derived xenografts harbouring KRAS and TP53 mutations [26]. |
| KRAS | KRAS mutations are present in >90% of PDACs [20] and are thought to drive PDAC initiation [27]. | KRASG12C mutant inhibitors were able to inhibit tumour growth in vivo in pre-clinical models [13]. In phase I trials in patients with lung adenocarcinoma or colorectal cancer, the KRASG12C inhibitor AMG-510 was well tolerated and able to induce stable disease or partial response [14,28]. |
| Ras/Raf/MEK/ERK | Mediates cellular response to several growth factors. | Gemcitabine and pimasertib (MEK inhibitor) showed synergistic inhibition of tumour growth in PDAC mouse models [29]. |
| NRTK | Fusion of TRKA, TRKB, or TRKC proteins with a variety of partners leads to constitutive activation of Ras/Raf/MEK/ERK pathway and PI3K/AKR pathway to aid in tumourigenesis [30]. | In the NAVIGATE trial, selective TRK inhibitor Larotrectinib was able to produce a 75% response rate in patients with NRTK-fusion positive tumours, including one patient with PDAC who had a partial response [31] The STARTRK-2 trial investigated the use of entrectinib (an inhibitor of TRKA/B/C and Ros-1 or Alk-containing gene fusions) in 3 patients with PDAC and demonstrated clinical improvement in all 3 [32]. |
| Evading growth suppressors | ||
| Aurora kinase | Aurora kinase proteins play important roles in cell division and are overexpressed in a number of tumours. | Danusertib (pan-Aurora kinase inhibitor) showed limited clinical activity as a second line treatment in PDAC with just 3 out of 31 patients remaining stable for 6–8.5 months [33]. |
| Cyclin dependent kinase (CDK) | CDK proteins, overactivated in cancer, are vital cell cycle regulators and play a role in controlling RNA transcription. Loss of CDKN2A is frequently seen in PDAC and detected in 47% of patients in one study [21]. | One mouse xenograft study showed that CDK4/6 inhibitors prevent PDAC recovery following taxane-based chemotherapy [34]. However, clinical trials of several CDK inhibitors have failed to show the desired results in PDAC and solid tumours as a whole [35]. |
| Angiogenesis inhibitors | ||
| Vascular endothelial growth factor (VEGF) | VEGF pathway is a key regulator of angiogenesis and vital in enabling the development of a blood supply to support tumour growth. | Numerous clinical trials of VEGF inhibitors in PDAC have failed to show clinical benefit, with phase III trials combining gemcitabine with bevacizumab or axitinib failing to reach their primary endpoints of OS [36]. However, one study assessing the combination of the VEGF inhibitor bevacizumab with 5-FU, leucovorin, nab-paclitaxel, and oxaliplatin did report an impressive OS of >17 months [37]. |
| Invasion and metastasis inhibitors | ||
| TGF beta | TGF-beta pathway plays a role in cell proliferation, differentiation, migration, and apoptosis. Recently, it has been shown to switch from an initial anti-tumourigenic role to a pro-tumourigenic role during the course of cancer progression [38]. | The TGF beta inhibitor galunisertib has shown promising results in combination with gemcitabine in PDAC, improving OS and PFS compared to gemcitabine alone [39,40]. |
| Inhibitors of apoptosis resistance | ||
| Insulin-like growth factor (IGF) | The IGF pathway stimulates cell proliferation, suppresses programmed cell death, and promotes cell differentiation. It has also been shown to aid in CSC maintenance and self-renewal in PDAC [41]. | Dalotuzumab (an IGF-1R antagonist) improved OS but not PFS in combination with gemcitabine and erlotinib [42]. |
| NF-KB | NF-KB suppresses apoptosis, downregulates p53 expression, and induces matrix metalloprotease expression to contribute to cancer growth and metastasis. | Curcumin and Theracurmin (NF-KB inhibitors) have been highlighted as promising options in PDAC [43] and combined Theracurmin and gemcitabine was shown to be well tolerated in one phase I trial [44]. |
| Anti-inflammatory drugs | ||
| JAK-STAT | This pathway is known to play a key role in cell proliferation, angiogenesis, and apoptosis [45], while STAT3 has been linked to the emergence of CSCs from non-stem cancer cells [46]. | A phase III trial investigating the use of combined ruxolitinib (a specific JAK1/2 inhibitor) and capecitabine in patients with advanced/metastatic PDAC was terminated due to failure to improve OS [47]. This was similarly true for a phase I trial administering the JAK1 inhibitor momelotenib to patients with metastatic PDAC [48]. However, clinical trials into alternative JAK/STAT inhibitors are ongoing [49]. |
| Src | Src deregulation is thought to enhance tumour growth and invasion. | The Src inhibitor Dasatinib failed in phase II trials for PDAC, both as a monotherapy [50] and in combination with gemcitabine [51]. |
| Bruton tyrosine kinase (BTK) | BTK is a non-receptor tyrosine kinase of the B cell receptor signalling pathway. | BTK inhibition with ibrutinib was shown to restore the T-cell-driven anti-tumour response in mouse models of PDAC and enhance responses to chemotherapy [52]. However, in the RESOLVE trial, a phase III study investigating the addition of ibrutinib to nab-paclitaxel and gemcitabine, no benefit in OS or PFS was seen in patients with advanced-stage PDAC [53]. Acalabrutinib, an alternative BTK inhibitor, also showed limited clinical activity as a monotherapy or in combination with pembrolizumab [54]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pook, H.; Pauklin, S. Mechanisms of Cancer Cell Death: Therapeutic Implications for Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 4834. https://doi.org/10.3390/cancers13194834
Pook H, Pauklin S. Mechanisms of Cancer Cell Death: Therapeutic Implications for Pancreatic Ductal Adenocarcinoma. Cancers. 2021; 13(19):4834. https://doi.org/10.3390/cancers13194834
Chicago/Turabian StylePook, Hannah, and Siim Pauklin. 2021. "Mechanisms of Cancer Cell Death: Therapeutic Implications for Pancreatic Ductal Adenocarcinoma" Cancers 13, no. 19: 4834. https://doi.org/10.3390/cancers13194834
APA StylePook, H., & Pauklin, S. (2021). Mechanisms of Cancer Cell Death: Therapeutic Implications for Pancreatic Ductal Adenocarcinoma. Cancers, 13(19), 4834. https://doi.org/10.3390/cancers13194834