
Genomic Landscape of Angiosarcoma: A Targeted and Immunotherapy Biomarker Analysis
 , , , , , , ,
, , , , , , , 

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
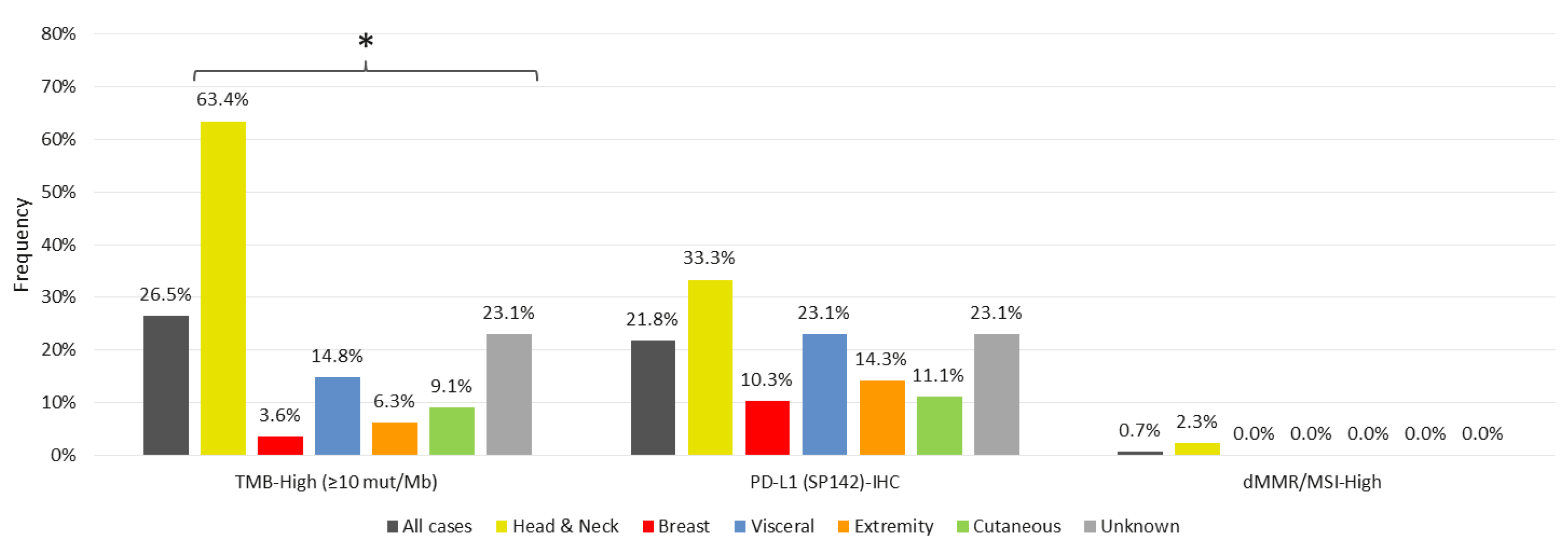
3.1. Markers of Immunotherapy Response
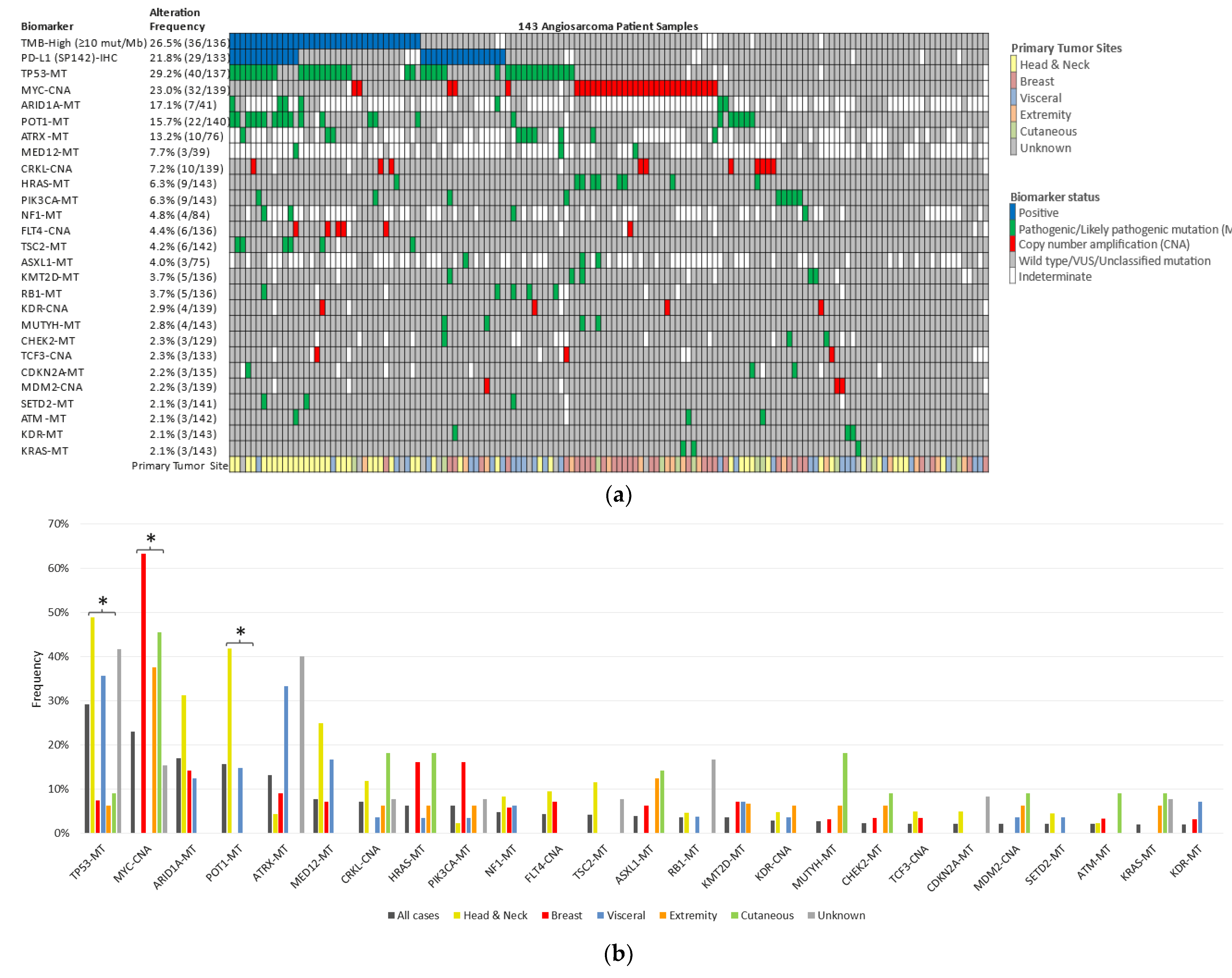
3.2. Genetic Alterations
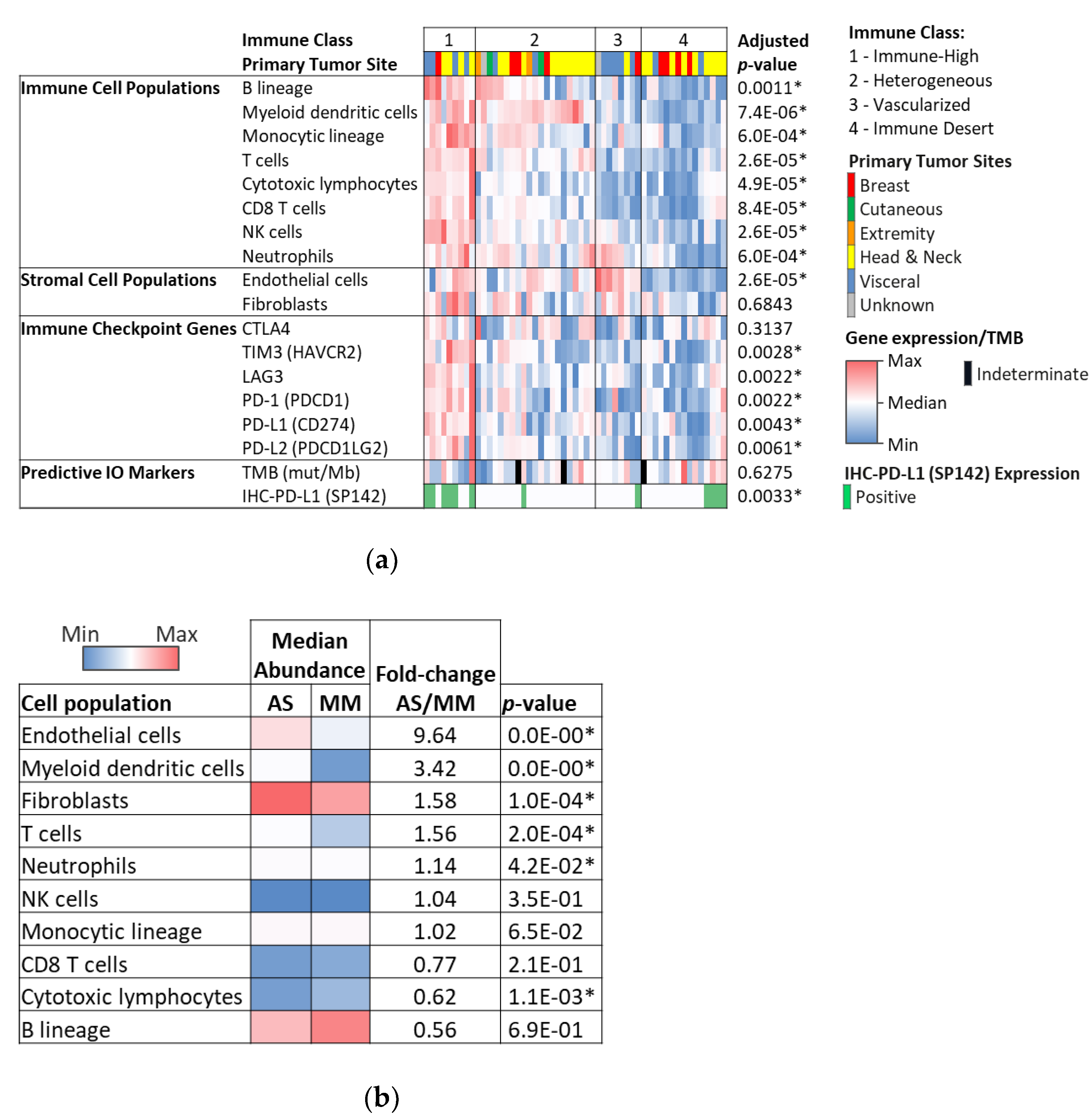
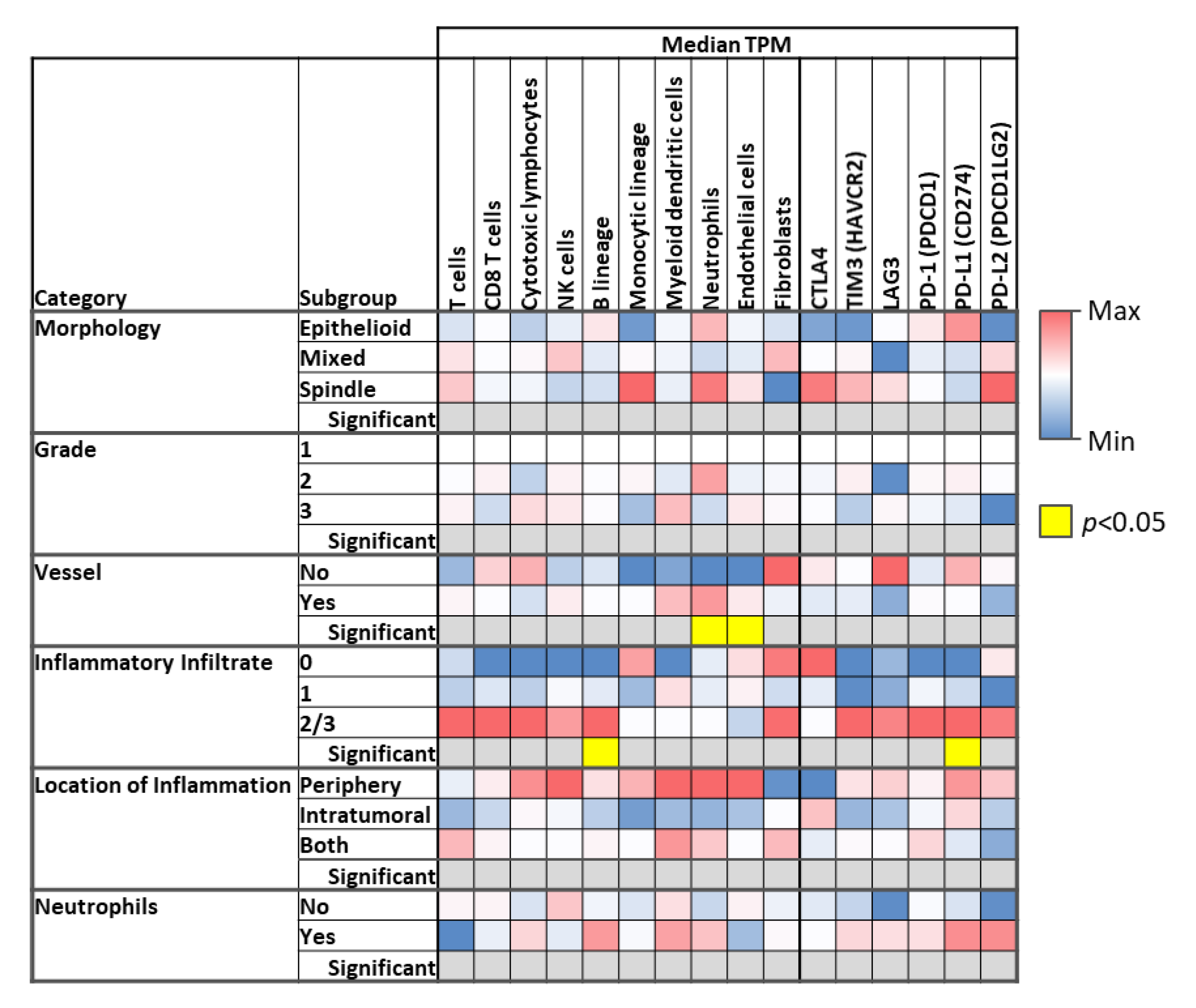
3.3. Microenvironment Phenotype
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ducimetière, F.; Lurkin, A.; Ranchère-Vince, D.; Decouvelaere, A.-V.; Peoc’H, M.; Istier, L.; Chalabreysse, P.; Muller, C.; Alberti, L.; Bringuier, P.-P.; et al. Incidence of Sarcoma Histotypes and Molecular Subtypes in a Prospective Epidemiological Study with Central Pathology Review and Molecular Testing. PLoS ONE 2011, 6, e20294. [Google Scholar] [CrossRef]
- Florou, V.; Wilky, B.A. Current and Future Directions for Angiosarcoma Therapy. Curr. Treat. Options Oncol. 2018, 19, 14. [Google Scholar] [CrossRef]
- Mito, J.K.; Mitra, D.; Barysauskas, C.M.; Mariño-Enriquez, A.; Morgan, E.A.; Fletcher, C.D.; Raut, C.P.; Baldini, E.H.; Doyle, L.A. A Comparison of Outcomes and Prognostic Features for Radiation-Associated Angiosarcoma of the Breast and Other Radiation-Associated Sarcomas. Int. J. Radiat. Oncol. 2019, 104, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Antonescu, C.R.; Van Zee, K.; Brennan, M.; Maki, R.G. A 14-Year Retrospective Review of Angiosarcoma. Cancer J. 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Agulnik, M.; Yarber, J.; Okuno, S.; von Mehren, M.; Jovanovic, B.; Brockstein, B.; Evens, A.; Benjamin, R. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann. Oncol. 2013, 24, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kollár, A.; Jones, R.L.; Stacchiotti, S.; Gelderblom, H.; Guida, M.; Grignani, G.; Steeghs, N.; Safwat, A.; Katz, D.; Duffaud, F.; et al. Pazopanib in advanced vascular sarcomas: An EORTC Soft Tissue and Bone Sarcoma Group (STBSG) retrospective analysis. Acta Oncol. 2016, 56, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G.; D’Adamo, D.R.; Keohan, M.L.; Saulle, M.; Schuetze, S.M.; Undevia, S.D.; Livingston, M.B.; Cooney, M.M.; Hensley, M.L.; Mita, M.M.; et al. Phase II Study of Sorafenib in Patients With Metastatic or Recurrent Sarcomas. J. Clin. Oncol. 2009, 27, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.L.; Domont, J.; Tresch-Bruneel, E.; Bompas, E.; Cassier, P.A.; Mir, O.; Piperno-Neumann, S.; Italiano, A.; Chevreau, C.; Cupissol, D.; et al. Paclitaxel Given Once Per Week With or Without Bevacizumab in Patients With Advanced Angiosarcoma: A Randomized Phase II Trial. J. Clin. Oncol. 2015, 33, 2797–2802. [Google Scholar] [CrossRef]
- Agulnik, M.; Schulte, B.; Robinson, S.; Hirbe, A.C.; Kozak, K.; Chawla, S.P.; Attia, S.; Rademaker, A.; Zhang, H.; Abbinanti, S.; et al. An open-label single-arm phase II study of regorafenib for the treatment of angiosarcoma. Eur. J. Cancer 2021, 154, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Florou, V.; Rosenberg, A.E.; Wieder, E.; Komanduri, K.V.; Kolonias, D.; Uduman, M.; Castle, J.C.; Buell, J.S.; Trent, J.C.; Wilky, B.A. Angiosarcoma patients treated with immune checkpoint inhibitors: A case series of seven patients from a single institution. J. Immunother. Cancer 2019, 7, 213. [Google Scholar] [CrossRef] [PubMed]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Zañudo, J.G.T.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef]
- Young, R.J.; Brown, N.; Reed, M.W.; Hughes, D.J.; Woll, P. Angiosarcoma. Lancet Oncol. 2010, 11, 983–991. [Google Scholar] [CrossRef]
- Fayette, J.; Martin, E.; Piperno-Neumann, S.; Le Cesne, A.; Robert, C.; Bonvalot, S.; Ranchère, D.; Pouillart, P.; Coindre, J.M.; Blay, J.Y. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: A retrospective study of 161 cases. Ann. Oncol. 2007, 18, 2030–2036. [Google Scholar] [CrossRef]
- Wang, L.; Lao, I.W.; Yu, L.; Wang, J. Clinicopathological features and prognostic factors in angiosarcoma: A retrospective analysis of 200 patients from a single Chinese medical institute. Oncol. Lett. 2017, 14, 5370–5378. [Google Scholar] [CrossRef]
- Yin, M.; Wang, W.; Drabick, J.J.; Harold, H.A. Prognosis and treatment of non-metastatic primary and secondary breast angiosarcoma: A comparative study. BMC Cancer 2017, 17, 295. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Yoshida, A.; Guo, T.; Chang, N.-E.; Zhang, L.; Agaram, N.P.; Qin, L.-X.; Brennan, M.; Singer, S.; Maki, R.G. KDR Activating Mutations in Human Angiosarcomas Are Sensitive to Specific Kinase Inhibitors. Cancer Res. 2009, 69, 7175–7179. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Sheldon, H.; Martincorena, I.; Van Loo, P.; Gundem, G.; Wedge, D.; Ramakrishna, M.; Cooke, S.L.; Pillay, N.; et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46, 376–379. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, L.; Chang, N.-E.; Maki, S.S.R.G.; Antonescu, C.R. Consistent MYC and FLT4 Gene Amplification in Radiation-Induced Angiosarcoma But Not in Other Radiation-Associated Atypical Vascular Lesions Tianhua. Cancer 2011, 396, 389–396. [Google Scholar]
- Motaparthi, K.; Lauer, S.R.; Patel, R.M.; Vidal, C.I.; Linos, K. MYC gene amplification by fluorescence in situ hybridization and MYC protein expression by immunohistochemistry in the diagnosis of cutaneous angiosarcoma: Systematic review and appropriate use criteria. J. Cutan. Pathol. 2020, 48, 578–586. [Google Scholar] [CrossRef]
- Manner, J.; Radlwimmer, B.; Hohenberger, P.; Mössinger, K.; Küffer, S.; Sauer, C.; Belharazem, D.; Zettl, A.; Coindre, J.-M.; Hallermann, C.; et al. MYC High Level Gene Amplification Is a Distinctive Feature of Angiosarcomas after Irradiation or Chronic Lymphedema. Am. J. Pathol. 2010, 176, 34–39. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Mahoney, M.R.; A Van Tine, B.; Atkins, J.; Milhem, M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Petitprez, F.; De Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 1–20. [Google Scholar] [CrossRef]
- Alexandrov, L.; Initiative, A.P.C.G.; Nik-Zainal, S.; Wedge, D.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.; Bignell, G.R.; Bolli, N.; Borg, A.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Boichard, A.; Wagner, M.J.; Kurzrock, R. Angiosarcoma heterogeneity and potential therapeutic vulnerability to immune checkpoint blockade: Insights from genomic sequencing. Genome Med. 2020, 12, 1–6. [Google Scholar] [CrossRef]
- Chan, J.Y.; Lim, J.Q.; Yeong, J.; Ravi, V.; Guan, P.; Boot, A.; Tay, T.K.Y.; Selvarajan, S.; Nasir, N.D.M.; Loh, J.H.; et al. Multiomic analysis and immunoprofiling reveal distinct subtypes of human angiosarcoma. J. Clin. Investig. 2020, 130, 5833–5846. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2017, 18, 89–102. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Walter-Petrich, A.; Che, J.L.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. APR-246 Combined with Azacitidine (AZA) in TP53 Mutated Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). a Phase 2 Study By the Groupe Francophone Des Myélodysplasies (GFM). Blood 2019, 134, 677. [Google Scholar] [CrossRef]
- Makita, S.; Tobinai, K. Targeting EZH2 with tazemetostat. Lancet Oncol. 2018, 19, 586–587. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Schoffski, P.; Jones, R.; Agulnik, M.; Villalobos, V.M.; Jahan, T.M.; Chen, T.W.-W.; Italiano, A.; Demetri, G.D.; Cote, G.M.; et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES) (NCT02601950). J. Clin. Oncol. 2019, 37, 11003. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Toulmonde, M.; Penel, N.; Mir, O.; Chevreau, C.; Anract, P.; Bompas, E.; Rios, M.; Firmin, N.; Italiano, A.; et al. Natural history of sarcomas and impact of reference centers in the nationwide NETSARC study on 35,784 patients (pts) from 2010 to 2017. Ann. Oncol. 2018, 29, viii576. [Google Scholar] [CrossRef]
- Alldredge, J.K.; Eskander, R.N. EZH2 inhibition in ARID1A mutated clear cell and endometrioid ovarian and endometrioid endometrial cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 1–9. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- Wu, Y.; Poulos, R.C.; Reddel, R.R. Role of POT1 in Human Cancer. Cancers 2020, 12, 2739. [Google Scholar] [CrossRef]
- Delehouze, C.; Godl, K.; Loaec, N.; Bruyere, C.; Desban, N.; Oumata, N.; Galons, H.; Roumeliotis, T.I.; Giannopoulou, E.G.; Grenet, J.; et al. CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene 2013, 33, 5675–5687. [Google Scholar] [CrossRef]
- Frame, S.; Saladino, C.; Mackay, C.; Atrash, B.; Sheldrake, P.; McDonald, E.; Clarke, P.A.; Workman, P.; Blake, D.; Zheleva, D. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS ONE 2020, 15, e0234103. [Google Scholar] [CrossRef]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef]
- Bolin, S.; Borgenvik, A.; Persson, C.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gupta, S.K.; Han, W.; Kundson, R.A.; Nelson, S.; Knutson, D.; Greipp, P.T.; Elsawa, S.F.; Sotomayor, E.M.; Gupta, M. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Liu, C.; Tao, Z.; Wang, M.; Jia, Y.; Sang, X.; Shen, L.; Xue, Y.; Jiang, K.; Luo, F.; et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019, 43, 225–237. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Megquier, K.; Turner-Maier, J.; Swofford, R.; Kim, J.-H.; Sarver, A.L.; Wang, C.; Sakthikumar, S.; Johnson, J.; Koltookian, M.; Lewellen, M.; et al. Comparative Genomics Reveals Shared Mutational Landscape in Canine Hemangiosarcoma and Human Angiosarcoma. Mol. Cancer Res. 2019, 17, 2410–2421. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Angiosarcoma Subgroup | All | Head and Neck | Breast | Visceral | Extremity | Cutaneous | Unknown | p-Value |
|---|---|---|---|---|---|---|---|---|
| Sample size, N (%) | 143 (100%) | 44 (30.8%) | 31 (21.7%) | 28 (19.6%) | 16 (11.2%) | 11 (7.7%) | 13 (9.1%) | |
| Morphology | ||||||||
| Epithelioid | 46 (32.9%) | 19 (43.2%) | 8 (26.7%) | 9 (32.1%) | 3 (18.8%) | 3 (30.0%) | 4 (36.4%) | 0.16 |
| Spindle | 9 (6.4%) | 0 (0.0%) | 3 (10.0%) | 4 (14.3%) | 0 (0.0%) | 1 (10.0%) | 1 (9.1%) | |
| Mixed | 85 (60.7%) | 25 (56.8%) | 19 (63.3%) | 15 (53.6%) | 13 (81.3%) | 6 (60.0%) | 6 (54.5%) | |
| Grade | ||||||||
| 1 | 2 (1.4%) | 0 (0.0%) | 1 (3.3%) | 1 (3.6%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0.52 |
| 2 | 78 (55.7%) | 21 (47.7%) | 21 (70.0%) | 15 (53.6%) | 8 (50.0%) | 5 (50.0%) | 8 (72.7%) | |
| 3 | 60 (42.9%) | 23 (52.3%) | 8 (26.7%) | 12 (42.9%) | 8 (50.0%) | 5 (50.0%) | 3 (27.3%) | |
| Vessel formation | ||||||||
| Yes | 117 (83.6%) | 35 (79.5%) | 28 (93.3%) | 23 (82.1%) | 12 (75.0%) | 9 (90.0%) | 9 (81.8%) | 0.43 |
| No | 23 (16.4%) | 9 (20.5%) | 2 (6.7%) | 5 (17.9%) | 4 (25.0%) | 1 (10.0%) | 2 (18.2%) | |
| Inflammatory infiltrate | ||||||||
| 0 | 8 (5.7%) | 1 (2.3%) | 2 (6.7%) | 3 (10.7%) | 2 (12.5%) | 0 (0.0%) | 0 (0.0%) | 0.11 |
| 1 | 105 (75.0%) | 31 (70.5%) | 28 (93.3%) | 19 (67.9%) | 11 (68.8%) | 8 (80.0%) | 7 (63.6%) | |
| 2 | 25 (17.9%) | 10 (22.7%) | 0 (0.0%) | 6 (21.4%) | 3 (18.8%) | 2 (20.0%) | 4 (36.4%) | |
| 3 | 2 (1.4%) | 2 (4.5%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Location of infiltrate | ||||||||
| Periphery | 8 (6.1%) | 2 (4.7%) | 1 (3.6%) | 3 (12.0%) | 1 (7.1%) | 0 (0.0%) | 1 (9.1%) | 0.73 |
| Intratumoral | 31 (23.5%) | 11 (25.6%) | 4 (14.3%) | 6 (24.0%) | 4 (28.6%) | 3 (30.0%) | 3 (27.3%) | |
| Both | 92 (69.7%) | 30 (69.8%) | 23 (82.1%) | 16 (64.0%) | 9 (64.3%) | 7 (70.0%) | 7 (63.6%) | |
| Neutrophils present | ||||||||
| Yes | 30 (22.7%) | 11 (25.6%) | 3 (10.7%) | 8 (32.0%) | 5 (35.7%) | 1 (10.0%) | 2 (18.2%) | 0.29 |
| No | 102 (77.3%) | 32 (74.4%) | 25 (89.3%) | 17 (68.0%) | 9 (64.3%) | 9 (90.0%) | 9 (81.8%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espejo Freire, A.; Elliott, A.; Rosenberg, A.; Costa, P.A.; Barreto-Coelho, P.; Jonczak, E.; D’Amato, G.; Subhawong, T.; Arshad, J.; Diaz-Perez, J.A.; et al. Genomic Landscape of Angiosarcoma: A Targeted and Immunotherapy Biomarker Analysis. Cancers 2021, 13, 4816. https://doi.org/10.3390/cancers13194816
Espejo Freire A, Elliott A, Rosenberg A, Costa PA, Barreto-Coelho P, Jonczak E, D’Amato G, Subhawong T, Arshad J, Diaz-Perez JA, et al. Genomic Landscape of Angiosarcoma: A Targeted and Immunotherapy Biomarker Analysis. Cancers. 2021; 13(19):4816. https://doi.org/10.3390/cancers13194816
Chicago/Turabian StyleEspejo Freire, Andrea, Andrew Elliott, Andrew Rosenberg, Philippos Apolinario Costa, Priscila Barreto-Coelho, Emily Jonczak, Gina D’Amato, Ty Subhawong, Junaid Arshad, Julio A. Diaz-Perez, and et al. 2021. "Genomic Landscape of Angiosarcoma: A Targeted and Immunotherapy Biomarker Analysis" Cancers 13, no. 19: 4816. https://doi.org/10.3390/cancers13194816
APA StyleEspejo Freire, A., Elliott, A., Rosenberg, A., Costa, P. A., Barreto-Coelho, P., Jonczak, E., D’Amato, G., Subhawong, T., Arshad, J., Diaz-Perez, J. A., Korn, W. M., Oberley, M. J., Magee, D., Dizon, D., von Mehren, M., Khushman, M. M., Hussein, A. M., Leu, K., & Trent, J. C. (2021). Genomic Landscape of Angiosarcoma: A Targeted and Immunotherapy Biomarker Analysis. Cancers, 13(19), 4816. https://doi.org/10.3390/cancers13194816