
The Role of the Hedgehog Pathway in Cholangiocarcinoma



Simple Summary
Abstract
1. Introduction
2. Cholangiocarcinoma Overview
3. Current Therapeutic Intervention in Cholangiocarcinoma
3.1. Surgery
3.2. Systemic Chemotherapy
3.3. Other Specific in Loco Therapies
3.4. Molecular Targeted Therapy
3.5. FGFR Inhibitors
3.6. IDH1/2 Inhibitors
3.7. BRAF-Directed Therapy
3.8. Other Molecular Targets
4. The Hedgehog Signaling Pathway
4.1. The Hedgehog Signaling in the Liver
4.2. Hedgehog and Cholangiocarcinoma
4.2.1. Hedgehog in CCA Patients
4.2.2. Hedgehog Aberrant Activation in CCA Cells
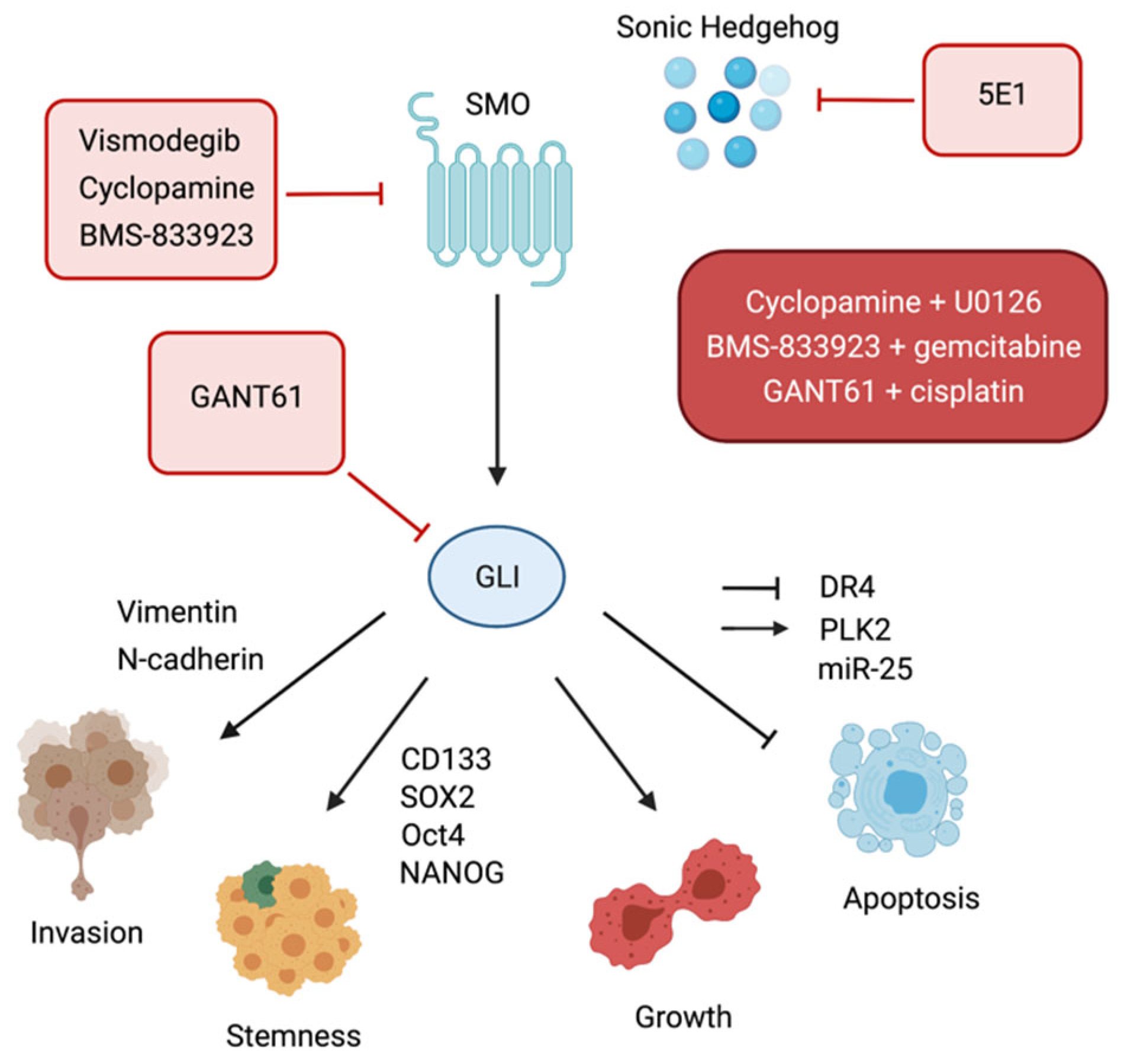
4.2.3. Hedgehog Targeting Strategies in CCA
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Komuta, M.; Roskams, T.; Gores, G.J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 512–522. [Google Scholar] [CrossRef]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: Pathogenesis and rationale for molecular therapies. Oncogene 2013, 32, 4861–4870. [Google Scholar] [CrossRef]
- Cardinale, V.; Carpino, G.; Reid, L.; Gaudio, E.; Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest. Oncol. 2012, 4, 94–102. [Google Scholar] [CrossRef]
- Raggi, C.; Invernizzi, P.; Andersen, J.B. Impact of microenvironment and stem-like plasticity in cholangiocarcinoma: Molecular networks and biological concepts. J. Hepatol. 2015, 62, 198–207. [Google Scholar] [CrossRef]
- Rizvi, S.; Borad, M.J.; Patel, T.; Gores, G.J. Cholangiocarcinoma: Molecular pathways and therapeutic opportunities. Semin. Liver Dis. 2014, 34, 456–464. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin. Cancer Res. 2006, 12, 1680–1685. [Google Scholar] [CrossRef]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Corrigendum: Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2015, 528, 152. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Cillo, U.; Fondevila, C.; Donadon, M.; Gringeri, E.; Mocchegiani, F.; Schlitt, H.J.; Ijzermans, J.N.M.; Vivarelli, M.; Zieniewicz, K.; Damink, S.O.; et al. Surgery for cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. S1), 143–155. [Google Scholar] [CrossRef]
- Kelley, R.K.; Bridgewater, J.; Gores, G.J.; Zhu, A.X. Systemic therapies for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Gringeri, E.; Gambato, M.; Sapisochin, G.; Ivanics, T.; Lynch, E.N.; Mescoli, C.; Burra, P.; Cillo, U.; Russo, F.P. Cholangiocarcinoma as an Indication for Liver Transplantation in the Era of Transplant Oncology. J. Clin. Med. 2020, 9, 1353. [Google Scholar] [CrossRef]
- Lehrke, H.D.; Heimbach, J.K.; Wu, T.T.; Jenkins, S.M.; Gores, G.J.; Rosen, C.B.; Mounajjed, T. Prognostic Significance of the Histologic Response of Perihilar Cholangiocarcinoma to Preoperative Neoadjuvant Chemoradiation in Liver Explants. Am. J. Surg. Pathol. 2016, 40, 510–518. [Google Scholar] [CrossRef]
- Sapisochin, G.; Facciuto, M.; Rubbia-Brandt, L.; Marti, J.; Mehta, N.; Yao, F.Y.; Vibert, E.; Cherqui, D.; Grant, D.R.; Hernandez-Alejandro, R.; et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016, 64, 1178–1188. [Google Scholar] [CrossRef]
- Sapisochin, G.; de Lope, C.R.; Gastaca, M.; de Urbina, J.O.; López-Andujar, R.; Palacios, F.; Ramos, E.; Fabregat, J.; Castroagudín, J.F.; Varo, E.; et al. Intrahepatic cholangiocarcinoma or mixed hepatocellular-cholangiocarcinoma in patients undergoing liver transplantation: A Spanish matched cohort multicenter study. Ann. Surg. 2014, 259, 944–952. [Google Scholar] [CrossRef]
- Valle, J.W.; Furuse, J.; Jitlal, M.; Beare, S.; Mizuno, N.; Wasan, H.; Bridgewater, J.; Okusaka, T. Cisplatin and gemcitabine for advanced biliary tract cancer: A meta-analysis of two randomised trials. Ann. Oncol. 2014, 25, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. ABC-06|A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin / 5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced / metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. J. Clin. Oncol. 2019, 37, 4003. [Google Scholar] [CrossRef]
- Shroff, R.T.; Javle, M.M.; Xiao, L.; Kaseb, A.O.; Varadhachary, G.R.; Wolff, R.A.; Raghav, K.P.S.; Iwasaki, M.; Masci, P.; Ramanathan, R.K.; et al. Gemcitabine, Cisplatin, and nab-Paclitaxel for the Treatment of Advanced Biliary Tract Cancers: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 824–830. [Google Scholar] [CrossRef]
- Seidensticker, R.; Ricke, J.; Seidensticker, M. Integration of chemoembolization and radioembolization into multimodal treatment of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Boerner, T.; Tan, B.R.; Chou, J.F.; Gönen, M.; Boucher, T.M.; Hauser, H.F.; Do, R.K.G.; Lowery, M.A.; Harding, J.J.; et al. Assessment of Hepatic Arterial Infusion of Floxuridine in Combination with Systemic Gemcitabine and Oxaliplatin in Patients with Unresectable Intrahepatic Cholangiocarcinoma: A Phase 2 Clinical Trial. JAMA Oncol. 2020, 6, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B. Cholangiocarcinoma: Current Knowledge and New Developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef]
- Verlingue, L.; Malka, D.; Allorant, A.; Massard, C.; Ferté, C.; Lacroix, L.; Rouleau, E.; Auger, N.; Ngo, M.; Nicotra, C.; et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial. Eur. J. Cancer 2017, 87, 122–130. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet. Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; He, Y.; Soni, N.; et al. FOENIX-CCA2: A phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. J. Clin. Oncol. 2020, 38, 108. [Google Scholar] [CrossRef]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef]
- Infigratinib Approved for Cholangiocarcinoma. Cancer Discov. 2021, 11, Of5. [CrossRef]
- Bekaii-Saab, T.S.; Valle, J.W.; Van Cutsem, E.; Rimassa, L.; Furuse, J.; Ioka, T.; Melisi, D.; Macarulla, T.; Bridgewater, J.; Wasan, H.; et al. FIGHT-302: First-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncol. 2020, 16, 2385–2399. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D.; Brandi, G. IDH inhibitors in advanced cholangiocarcinoma: Another arrow in the quiver? Cancer Treat. Res. Commun. 2021, 27, 100356. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet. Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla Mercade, T.; Javle, M.; Kelley, R.K.; Lubner, S.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.A.; et al. LBA10_PR—ClarIDHy: A global, phase III, randomized, double-blind study of ivosidenib (IVO) vs placebo in patients with advanced cholangiocarcinoma (CC) with an isocitrate dehydrogenase 1 (IDH1) mutation. Ann. Oncol. 2019, 30, v872–v873. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- IDH-Mutant Tumors Vulnerable to PARP Inhibition. Cancer Discov. 2017, 7, Of4. [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): A phase 2, open-label, single-arm, multicentre basket trial. Lancet. Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.P.; Jeong, J.H.; Chang, H.M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Bekaii-Saab, T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: Exploiting synthetic lethality with PARP inhibitors. Chin. Clin. Oncol. 2020, 9, 6. [Google Scholar] [CrossRef]
- Nam, A.R.; Jin, M.H.; Park, J.E.; Bang, J.H.; Oh, D.Y.; Bang, Y.J. Therapeutic Targeting of the DNA Damage Response Using an ATR Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2019, 51, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Nam, A.R.; Jin, M.H.; Bang, J.H.; Oh, K.S.; Seo, H.R.; Oh, D.Y.; Bang, Y.J. Inhibition of ATR Increases the Sensitivity to WEE1 Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2020, 52, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, X.; Wang, H.; Wang, J.; Qiao, Y.; Cigliano, A.; Utpatel, K.; Ribback, S.; Pilo, M.G.; Serra, M.; et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2019, 25, 403–413. [Google Scholar] [CrossRef]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef]
- Kheder, E.S.; Hong, D.S. Emerging Targeted Therapy for Tumors with NTRK Fusion Proteins. Clin. Cancer Res. 2018, 24, 5807–5814. [Google Scholar] [CrossRef]
- Law, L.Y. Dramatic response to trastuzumab and paclitaxel in a patient with human epidermal growth factor receptor 2-positive metastatic cholangiocarcinoma. J. Clin. Oncol. 2012, 30, e271–e273. [Google Scholar] [CrossRef]
- Riedlinger, D.; Bahra, M.; Boas-Knoop, S.; Lippert, S.; Bradtmöller, M.; Guse, K.; Seehofer, D.; Bova, R.; Sauer, I.M.; Neuhaus, P.; et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. J. Hepato-Biliary-Pancreat. Sci. 2014, 21, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar] [CrossRef] [PubMed]
- Sagai, T.; Hosoya, M.; Mizushina, Y.; Tamura, M.; Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 2005, 132, 797–803. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef]
- Dyer, M.A.; Farrington, S.M.; Mohn, D.; Munday, J.R.; Baron, M.H. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development 2001, 128, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef]
- Yao, H.H.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef]
- Wijgerde, M.; Ooms, M.; Hoogerbrugge, J.W.; Grootegoed, J.A. Hedgehog signaling in mouse ovary: Indian hedgehog and desert hedgehog from granulosa cells induce target gene expression in developing theca cells. Endocrinology 2005, 146, 3558–3566. [Google Scholar] [CrossRef]
- Denef, N.; Neubüser, D.; Perez, L.; Cohen, S.M. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 2000, 102, 521–531. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y. Evidence for the direct involvement of {β}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. β-arrestin-mediated localization of smoothened to the primary cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.; Curran, T. The Hedgehog’s tale: Developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Stecca, B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells 2018, 7, 272. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-β promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Swarbrick, A.; Sutherland, R.L. The Hedgehog signalling pathway as a therapeutic target in early breast cancer development. Expert Opin. Ther. Targets 2009, 13, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Matsui, W. Molecular pathways: The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2012, 18, 4883–4888. [Google Scholar] [CrossRef]
- Hirose, Y.; Itoh, T.; Miyajima, A. Hedgehog signal activation coordinates proliferation and differentiation of fetal liver progenitor cells. Exp. Cell Res. 2009, 315, 2648–2657. [Google Scholar] [CrossRef]
- Jeng, K.S.; Chang, C.F.; Lin, S.-S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Li, Y.X.; Melhem, A.; Schmelzer, E.; Zdanowicz, M.; Huang, J.; Caballero, M.; Fair, J.H.; Ludlow, J.W.; McClelland, R.E.; et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G859–G870. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Hedgehog signalling in liver pathophysiology. J. Hepatol. 2018, 68, 550–562. [Google Scholar] [CrossRef]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Omenetti, A.; Popov, Y.; Jung, Y.; Choi, S.S.; Witek, R.P.; Yang, L.; Brown, K.D.; Schuppan, D.; Diehl, A.M. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut 2008, 57, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Yang, L.; Li, Y.X.; McCall, S.J.; Jung, Y.; Sicklick, J.K.; Huang, J.; Choi, S.; Suzuki, A.; Diehl, A.M. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; McCall, S.J.; Li, Y.X.; Diehl, A.M. Bile ductules and stromal cells express hedgehog ligands and/or hedgehog target genes in primary biliary cirrhosis. Hepatology 2007, 45, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Omenetti, A.; Witek, R.P.; Moylan, C.A.; Syn, W.K.; Jung, Y.; Yang, L.; Sudan, D.L.; Sicklick, J.K.; Michelotti, G.A.; et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1093–G1106. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Witek, R.P.; Yang, L.; Omenetti, A.; Syn, W.K.; Moylan, C.A.; Jung, Y.; Karaca, G.F.; Teaberry, V.S.; Pereira, T.A.; et al. Activation of Rac1 promotes hedgehog-mediated acquisition of the myofibroblastic phenotype in rat and human hepatic stellate cells. Hepatology 2010, 52, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.X.; Choi, S.S.; Qi, Y.; Chen, W.; Bustamante, M.; Huang, J.; Zdanowicz, M.; Camp, T.; Torbenson, M.S.; et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Mao, H.; Fleig, S.; Omenetti, A.; Brown, K.D.; Sicklick, J.K.; Li, Y.-X.; Diehl, A.M. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 2008, 48, 98–106. [Google Scholar] [CrossRef]
- Shackel, N.A.; McGuinness, P.H.; Abbott, C.A.; Gorrell, M.D.; McCaughan, G.W. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut 2001, 49, 565–576. [Google Scholar] [CrossRef]
- Omenetti, A.; Diehl, A.M. The adventures of sonic hedgehog in development and repair. II. Sonic hedgehog and liver development, inflammation, and cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G595–G598. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, M.O.; Shin, J.S.; Park, S.H.; Kim, S.B.; Kim, J.; Park, S.C.; Han, C.J.; Ryu, J.K.; Yoon, Y.B.; et al. Hedgehog signaling between cancer cells and hepatic stellate cells in promoting cholangiocarcinoma. Ann. Surg. Oncol. 2014, 21, 2684–2698. [Google Scholar] [CrossRef]
- Tang, L.; Tan, Y.X.; Jiang, B.G.; Pan, Y.F.; Li, S.X.; Yang, G.Z.; Wang, M.; Wang, Q.; Zhang, J.; Zhou, W.P.; et al. The prognostic significance and therapeutic potential of hedgehog signaling in intrahepatic cholangiocellular carcinoma. Clin. Cancer Res. 2013, 19, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Eichenmüller, M.; Gruner, I.; Hagl, B.; Häberle, B.; Müller-Höcker, J.; von Schweinitz, D.; Kappler, R. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009, 49, 482–490. [Google Scholar] [CrossRef]
- Li, Y.-C.; Deng, Y.-H.; Guo, Z.-H.; Zhang, M.-M.; Zhu, J.; Pu, C.-L.; Xiang, C.-P.; Guo, C.-B.; Li, Y.-C.; Deng, Y.-H.; et al. Prognostic value of hedgehog signal component expressions in hepatoblastoma patients. Eur. J. Med. Res. 2010, 15, 468–474. [Google Scholar] [CrossRef]
- Matsushita, S.; Onishi, H.; Nakano, K.; Nagamatsu, I.; Imaizumi, A.; Hattori, M.; Oda, Y.; Tanaka, M.; Katano, M. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. Cancer Sci. 2014, 105, 272–280. [Google Scholar] [CrossRef]
- Takebe, N.; Yang, S.X. Sonic hedgehog signaling pathway and gallbladder cancer: Targeting with precision medicine approach. Chin. Clin. Oncol. 2016, 5, 1. [Google Scholar] [CrossRef]
- Dixit, R.; Pandey, M.; Tripathi, S.K.; Dwivedi, A.N.; Shukla, V.K. Comparative Analysis of Mutational Profile of Sonic hedgehog Gene in Gallbladder Cancer. Dig. Dis. Sci. 2017, 62, 708–714. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Ohta, H.; Aoyagi, K.; Fukaya, M.; Danjoh, I.; Ohta, A.; Isohata, N.; Saeki, N.; Taniguchi, H.; Sakamoto, H.; Shimoda, T.; et al. Cross talk between hedgehog and epithelial-mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br. J. Cancer 2009, 100, 389–398. [Google Scholar] [CrossRef]
- Riaz, S.K.; Khan, J.S.; Shah, S.T.A.; Wang, F.; Ye, L.; Jiang, W.G.; Malik, M.F.A. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun. Signal. CCS 2018, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Jäger, W.; Thomas, C.; Fazli, L.; Hurtado-Coll, A.; Li, E.; Janssen, C.; Gust, K.M.; So, A.I.; Hainz, M.; Schmidtmann, I.; et al. DHH is an independent prognosticator of oncologic outcome of clear cell renal cell carcinoma. J. Urol. 2014, 192, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Togni, M.; Astolfi, A.; Pigazzi, M.; Manara, E.; Indio, V.; Rizzari, C.; Rutella, S.; Basso, G.; Pession, A.; et al. DHH-RHEBL1 fusion transcript: A novel recurrent feature in the new landscape of pediatric CBFA2T3-GLIS2-positive acute myeloid leukemia. Oncotarget 2013, 4, 1712–1720. [Google Scholar] [CrossRef]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef]
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Ruiz i Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881. [Google Scholar] [CrossRef]
- McGarvey, T.W.; Maruta, Y.; Tomaszewski, J.E.; Linnenbach, A.J.; Malkowicz, S.B. PTCH gene mutations in invasive transitional cell carcinoma of the bladder. Oncogene 1998, 17, 1167–1172. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Kool, M.; Jones, D.T.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef]
- El Khatib, M.; Kalnytska, A.; Palagani, V.; Kossatz, U.; Manns, M.P.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 2013, 57, 1035–1045. [Google Scholar] [CrossRef]
- Kiesslich, T.; Mayr, C.; Wachter, J.; Bach, D.; Fuereder, J.; Wagner, A.; Alinger, B.; Pichler, M.; Di Fazio, P.; Ocker, M.; et al. Activated hedgehog pathway is a potential target for pharmacological intervention in biliary tract cancer. Mol. Cell. Biochem. 2014, 396, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Al-Bahrani, R.; Nagamori, S.; Leng, R.; Petryk, A.; Sergi, C. Differential Expression of Sonic Hedgehog Protein in Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Pathol. Oncol. Res. 2015, 21, 901–908. [Google Scholar] [CrossRef]
- Nanashima, A.; Hatachi, G.; Tsuchiya, T.; Matsumoto, H.; Arai, J.; Abo, T.; Murakami, G.; Tominaga, T.; Takagi, K.; Nagayasu, T. Clinical significances of cancer stem cells markers in patients with intrahepatic cholangiocarcinoma who underwent hepatectomy. Anticancer Res. 2013, 33, 2107–2114. [Google Scholar]
- Peng, Y.; Meng, G.; Sheng, X.; Gao, H. Transcriptome and DNA methylation analysis reveals molecular mechanisms underlying intrahepatic cholangiocarcinoma progression. J. Cell. Mol. Med. 2021, 25, 6373–6387. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Guo, L.; Zhou, Y.; Chen, Y.; Sun, H.; Wang, Y.; Qu, Y. LncRNA ASAP1-IT1 positively modulates the development of cholangiocarcinoma via hedgehog signaling pathway. Biomed. Pharmacother. 2018, 103, 167–173. [Google Scholar] [CrossRef]
- Kurita, S.; Mott, J.L.; Almada, L.L.; Bronk, S.F.; Werneburg, N.W.; Sun, S.Y.; Roberts, L.R.; Fernandez-Zapico, M.E.; Gores, G.J. GLI3-dependent repression of DR4 mediates hedgehog antagonism of TRAIL-induced apoptosis. Oncogene 2010, 29, 4848–4858. [Google Scholar] [CrossRef]
- Razumilava, N.; Bronk, S.F.; Smoot, R.L.; Fingas, C.D.; Werneburg, N.W.; Roberts, L.R.; Mott, J.L. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012, 55, 465–475. [Google Scholar] [CrossRef]
- Mansini, A.P.; Peixoto, E.; Thelen, K.M.; Gaspari, C.; Jin, S.; Gradilone, S.A. The cholangiocyte primary cilium in health and disease. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 1245–1253. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Wong, S.Y.; Seol, A.D.; So, P.L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H., Jr.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 2009, 15, 1062–1065. [Google Scholar] [CrossRef]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef]
- Moeller, B.J.; Richardson, R.A.; Dewhirst, M.W. Hypoxia and radiotherapy: Opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007, 26, 241–248. [Google Scholar] [CrossRef]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- Ellis, L.M.; Hicklin, D.J. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, e5990–e5999. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, M.; Xing, L.; Wang, Y.; Xiao, Y.; Wu, Y. HIF-1α contributes to proliferation and invasiveness of neuroblastoma cells via SHH signaling. PLoS ONE 2015, 10, e0121115. [Google Scholar] [CrossRef]
- Vanichapol, T.; Leelawat, K.; Hongeng, S. Hypoxia enhances cholangiocarcinoma invasion through activation of hepatocyte growth factor receptor and the extracellular signal-regulated kinase signaling pathway. Mol. Med. Rep. 2015, 12, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The Role of Stroma in Cholangiocarcinoma: The Intriguing Interplay between Fibroblastic Component, Immune Cell Subsets and Tumor Epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Guicciardi, M.E.; Cazanave, S.C.; Mertens, J.C.; Sirica, A.E.; Gores, G.J. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 2011, 54, 2076–2088. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Mertens, J.C.; Razumilava, N.; Sydor, S.; Bronk, S.F.; Christensen, J.D.; Rizvi, S.H.; Canbay, A.; Treckmann, J.W.; Paul, A.; et al. Polo-like kinase 2 is a mediator of hedgehog survival signaling in cholangiocarcinoma. Hepatology 2013, 58, 1362–1374. [Google Scholar] [CrossRef]
- Milenkovic, L.; Scott, M.P. Not lost in space: Trafficking in the hedgehog signaling pathway. Sci. Signal. 2010, 3, pe14. [Google Scholar] [CrossRef][Green Version]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef]
- Theunissen, J.W.; de Sauvage, F.J. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Investig. 2015, 125, 1269–1285. [Google Scholar] [CrossRef] [PubMed]
- Jinawath, A.; Akiyama, Y.; Sripa, B.; Yuasa, Y. Dual blockade of the Hedgehog and ERK1/2 pathways coordinately decreases proliferation and survival of cholangiocarcinoma cells. J. Cancer Res. Clin. Oncol. 2007, 133, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, C.; Chen, Z.; Zhao, W. Overcoming the resistance mechanisms of Smoothened inhibitors. Drug Discov. Today 2018, 23, 704–710. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef]
- Tao, H.; Jin, Q.; Koo, D.I.; Liao, X.; Englund, N.P.; Wang, Y.; Ramamurthy, A.; Schultz, P.G.; Dorsch, M.; Kelleher, J.; et al. Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of smoothened. Chem. Biol. 2011, 18, 432–437. [Google Scholar] [CrossRef]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Trang, V.; Lee, A.; Williams, N.S.; Wilson, A.N.; Epstein, E.H., Jr.; Tang, J.Y.; Kim, J. Posaconazole, a Second-Generation Triazole Antifungal Drug, Inhibits the Hedgehog Signaling Pathway and Progression of Basal Cell Carcinoma. Mol. Cancer Ther. 2016, 15, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mook, R.A., Jr.; Lu, J.; Gooden, D.M.; Ribeiro, A.; Guo, A.; Barak, L.S.; Lyerly, H.K.; Chen, W. Identification of a novel Smoothened antagonist that potently suppresses Hedgehog signaling. Bioorgan. Med. Chem. 2012, 20, 6751–6757. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chaudhary, A.K.; Dong, Y.; Zhong, H.A.; Mondal, G.; Lin, F.; Kumar, V.; Mahato, R.I. Design, Synthesis and Biological Evaluation of novel Hedgehog Inhibitors for treating Pancreatic Cancer. Sci. Rep. 2017, 7, 1665. [Google Scholar] [CrossRef]
- Lin, P.; He, Y.; Chen, G.; Ma, H.; Zheng, J.; Zhang, Z.; Cao, B.; Zhang, H.; Zhang, X.; Mao, X. A novel hedgehog inhibitor for the treatment of hematological malignancies. Anti-Cancer Drugs 2018, 29, 995–1003. [Google Scholar] [CrossRef]
- Manetti, F.; Faure, H.; Roudaut, H.; Gorojankina, T.; Traiffort, E.; Schoenfelder, A.; Mann, A.; Solinas, A.; Taddei, M.; Ruat, M. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as smoothened antagonists. Mol. Pharmacol. 2010, 78, 658–665. [Google Scholar] [CrossRef]
- Pietrobono, S.; Santini, R.; Gagliardi, S.; Dapporto, F.; Colecchia, D.; Chiariello, M.; Leone, C.; Valoti, M.; Manetti, F.; Petricci, E.; et al. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 2018, 9, 142. [Google Scholar] [CrossRef]
- Chiarenza, A.; Manetti, F.; Petricci, E.; Ruat, M.; Naldini, A.; Taddei, M.; Carraro, F. Novel Acylguanidine Derivatives Targeting Smoothened Induce Antiproliferative and Pro-Apoptotic Effects in Chronic Myeloid Leukemia Cells. PLoS ONE 2016, 11, e0149919. [Google Scholar] [CrossRef]
- Bernardini, G.; Geminiani, M.; Gambassi, S.; Orlandini, M.; Petricci, E.; Marzocchi, B.; Laschi, M.; Taddei, M.; Manetti, F.; Santucci, A. Novel smoothened antagonists as anti-neoplastic agents for the treatment of osteosarcoma. J. Cell. Physiol. 2018, 233, 4961–4971. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Missense | Nonsense | In/Del | Splicing | CNA | Frequency (%) |
|---|---|---|---|---|---|---|
| GLI1 | Q867K, A108D, A941T, P821S, S89L, A670S, P842S, E1015D | G274Afs*6 | AMP | 2.5 | ||
| GLI2 | P386L | E431 * | 2.7 | |||
| GLI3 | P575T, D848G, Y1223F, N1203K, R875C, S1025I, G1001S, E729K, L364I, G221=, R1010W, R220P, V778I, R1189H, R1010Q, G371D, P791L, A1005D, L690I, I254V, N1203K, D428N, T167M, E1014K, E551D | R667 * | X158_splice | 4.6 | ||
| SMO | S785L, G402V, T307I, A631S | L23dup | 0.7 | |||
| PTCH1 | G880V, D301N, L775M, E405K, P1315L, A412T, D878H | 1.1 | ||||
| PTCH2 | A396V | 2.9 | ||||
| SUFU | V323I, L68S | 0.4 | ||||
| DHH | S384P | AMP | 5.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anichini, G.; Carrassa, L.; Stecca, B.; Marra, F.; Raggi, C. The Role of the Hedgehog Pathway in Cholangiocarcinoma. Cancers 2021, 13, 4774. https://doi.org/10.3390/cancers13194774
Anichini G, Carrassa L, Stecca B, Marra F, Raggi C. The Role of the Hedgehog Pathway in Cholangiocarcinoma. Cancers. 2021; 13(19):4774. https://doi.org/10.3390/cancers13194774
Chicago/Turabian StyleAnichini, Giulia, Laura Carrassa, Barbara Stecca, Fabio Marra, and Chiara Raggi. 2021. "The Role of the Hedgehog Pathway in Cholangiocarcinoma" Cancers 13, no. 19: 4774. https://doi.org/10.3390/cancers13194774
APA StyleAnichini, G., Carrassa, L., Stecca, B., Marra, F., & Raggi, C. (2021). The Role of the Hedgehog Pathway in Cholangiocarcinoma. Cancers, 13(19), 4774. https://doi.org/10.3390/cancers13194774