
Gene Expression Profiling as a Potential Tool for Precision Oncology in Non-Small Cell Lung Cancer

 ,
,  ,
, 
 and
and
Simple Summary
Abstract
1. Introduction
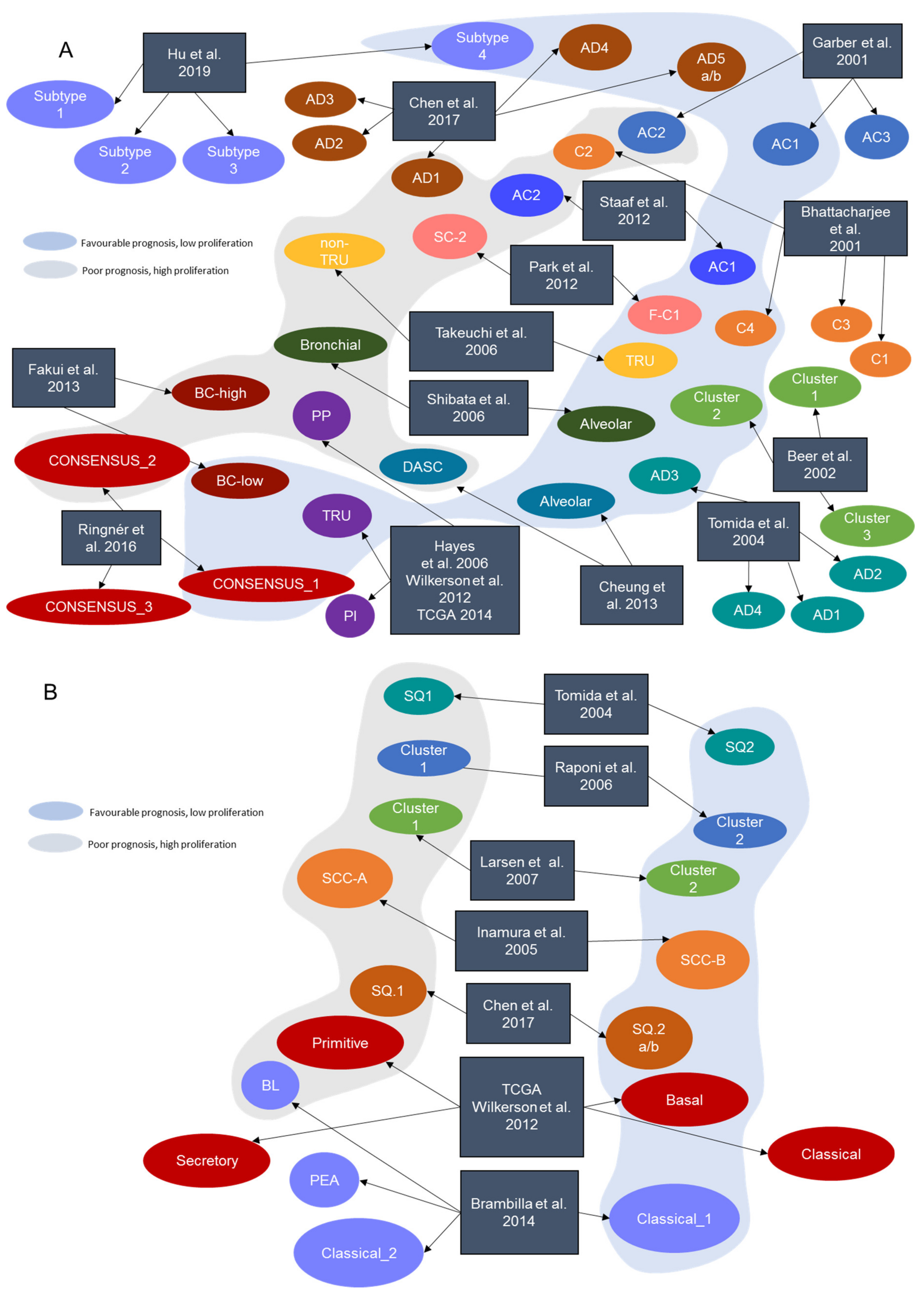
2. NSCLC Intrinsic Molecular Subtypes
2.1. Transcriptional Subtypes in Lung LUAD
2.2. Transcriptional Subtypes in Lung LUSC
2.3. Current Clinical Applicability of NSCLC Gene Expression Signatures
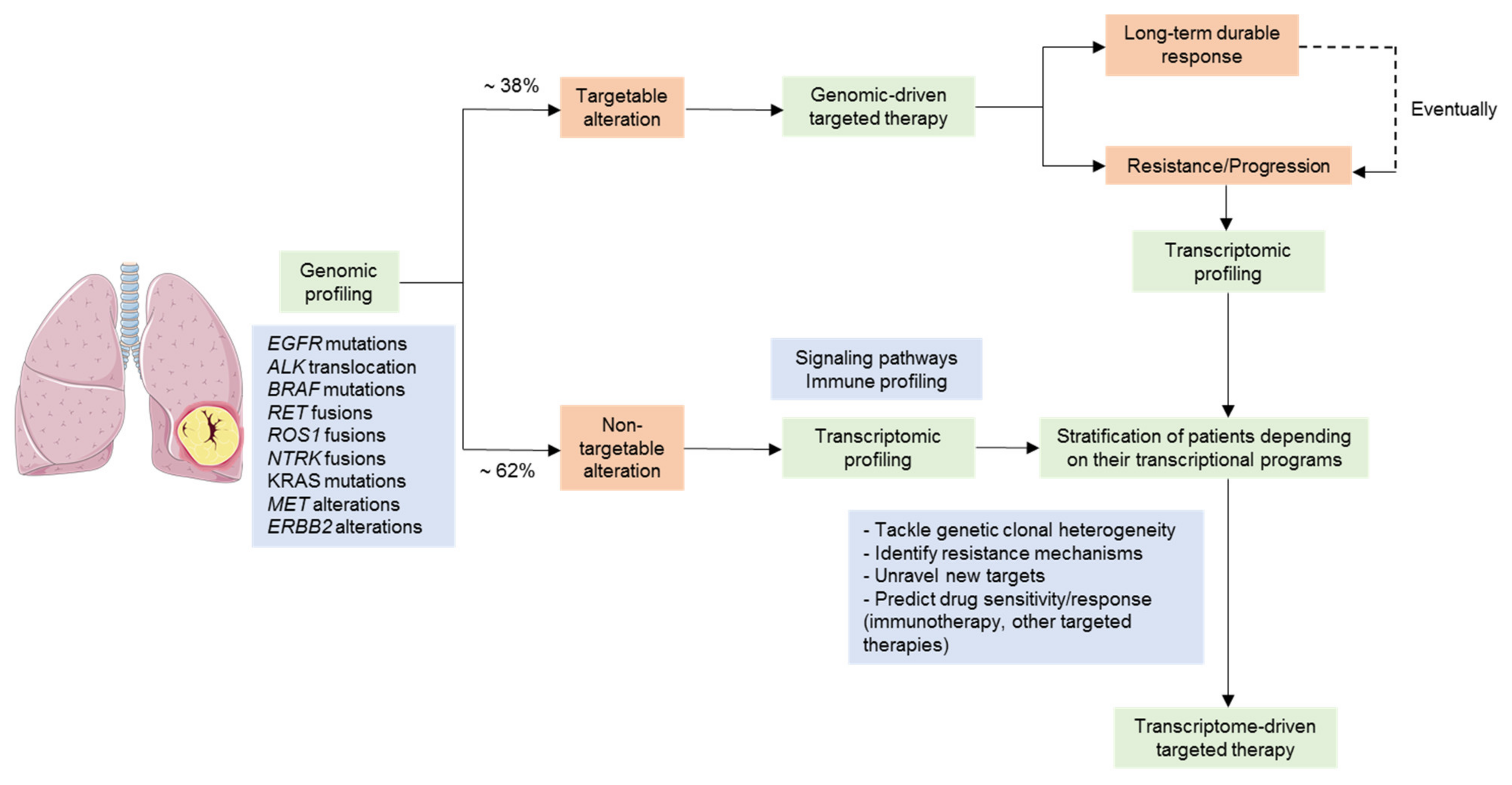
3. Gene Expression Profiling in the Context of Targeted Therapies in NSCLC
3.1. Gene Expression Profiling as a Tool for the Stratification of Driver-Positive Patients
3.2. NSCLC Tumors Lacking a Tractable Oncogenic Driver
3.3. Overcoming Targeted Therapy Resistance Mechanisms
4. Gene Expression Profiling in the Context of Immunotherapy in NSCLC
4.1. Gene Expression Signatures as Predictors of Immunotherapy Response
4.2. Linking Lung Cancer Molecular Subtypes with Immune Phenotype
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Cancer of the Lung and Bronchus—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/lungb.html (accessed on 19 August 2021).
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Uramoto, H.; Tanaka, F. Recurrence after Surgery in Patients with NSCLC. Transl. Lung Cancer Res. 2014, 3, 242–249. [Google Scholar] [CrossRef]
- Santos, C.; Sanz-Pamplona, R.; Nadal, E.; Grasselli, J.; Pernas, S.; Dienstmann, R.; Moreno, V.; Tabernero, J.; Salazar, R. Intrinsic Cancer Subtypes--next Steps into Personalized Medicine. Cell. Oncol. Dordr. 2015, 38, 3–16. [Google Scholar] [CrossRef]
- Garber, M.E.; Troyanskaya, O.G.; Schluens, K.; Petersen, S.; Thaesler, Z.; Pacyna-Gengelbach, M.; van de Rijn, M.; Rosen, G.D.; Perou, C.M.; Whyte, R.I.; et al. Diversity of Gene Expression in Adenocarcinoma of the Lung. Proc. Natl. Acad. Sci. USA 2001, 98, 13784–13789. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of Human Lung Carcinomas by MRNA Expression Profiling Reveals Distinct Adenocarcinoma Subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.G.; Kardia, S.L.R.; Huang, C.-C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-Expression Profiles Predict Survival of Patients with Lung Adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Tomida, S.; Koshikawa, K.; Yatabe, Y.; Harano, T.; Ogura, N.; Mitsudomi, T.; Some, M.; Yanagisawa, K.; Takahashi, T.; Osada, H.; et al. Gene Expression-Based, Individualized Outcome Prediction for Surgically Treated Lung Cancer Patients. Oncogene 2004, 23, 5360–5370. [Google Scholar] [CrossRef]
- Inamura, K.; Fujiwara, T.; Hoshida, Y.; Isagawa, T.; Jones, M.H.; Virtanen, C.; Shimane, M.; Satoh, Y.; Okumura, S.; Nakagawa, K.; et al. Two Subclasses of Lung Squamous Cell Carcinoma with Different Gene Expression Profiles and Prognosis Identified by Hierarchical Clustering and Non-Negative Matrix Factorization. Oncogene 2005, 24, 7105–7113. [Google Scholar] [CrossRef]
- Hayes, D.N.; Monti, S.; Parmigiani, G.; Gilks, C.B.; Naoki, K.; Bhattacharjee, A.; Socinski, M.A.; Perou, C.; Meyerson, M. Gene Expression Profiling Reveals Reproducible Human Lung Adenocarcinoma Subtypes in Multiple Independent Patient Cohorts. J. Clin. Oncol. 2006, 24, 5079–5090. [Google Scholar] [CrossRef]
- Raponi, M.; Zhang, Y.; Yu, J.; Chen, G.; Lee, G.; Taylor, J.M.G.; Macdonald, J.; Thomas, D.; Moskaluk, C.; Wang, Y.; et al. Gene Expression Signatures for Predicting Prognosis of Squamous Cell and Adenocarcinomas of the Lung. Cancer Res. 2006, 66, 7466–7472. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tomida, S.; Yatabe, Y.; Kosaka, T.; Osada, H.; Yanagisawa, K.; Mitsudomi, T.; Takahashi, T. Expression Profile–Defined Classification of Lung Adenocarcinoma Shows Close Relationship with Underlying Major Genetic Changes and Clinicopathologic Behaviors. J. Clin. Oncol. 2006, 24, 1679–1688. [Google Scholar] [CrossRef]
- Larsen, J.E.; Pavey, S.J.; Passmore, L.H.; Bowman, R.; Clarke, B.E.; Hayward, N.K.; Fong, K.M. Expression Profiling Defines a Recurrence Signature in Lung Squamous Cell Carcinoma. Carcinogenesis 2007, 28, 760–766. [Google Scholar] [CrossRef]
- Shibata, T.; Hanada, S.; Kokubu, A.; Matsuno, Y.; Asamura, H.; Ohta, T.; Sakamoto, M.; Hirohashi, S. Gene Expression Profiling of Epidermal Growth Factor Receptor/KRAS Pathway Activation in Lung Adenocarcinoma. Cancer Sci. 2007, 98, 985–991. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Yin, X.; Hoadley, K.A.; Liu, Y.; Hayward, M.C.; Cabanski, C.R.; Muldrew, K.; Miller, C.R.; Randell, S.H.; Socinski, M.A.; et al. Lung Squamous Cell Carcinoma MRNA Expression Subtypes Are Reproducible, Clinically Important, and Correspond to Normal Cell Types. Clin. Cancer Res. 2010, 16, 4864–4875. [Google Scholar] [CrossRef]
- Park, Y.-Y.; Park, E.S.; Kim, S.B.; Kim, S.C.; Sohn, B.H.; Chu, I.-S.; Jeong, W.; Mills, G.B.; Byers, L.A.; Lee, J.-S. Development and Validation of a Prognostic Gene-Expression Signature for Lung Adenocarcinoma. PLoS ONE 2012, 7, e44225. [Google Scholar] [CrossRef]
- Staaf, J.; Jönsson, G.; Jönsson, M.; Karlsson, A.; Isaksson, S.; Salomonsson, A.; Pettersson, H.M.; Soller, M.; Ewers, S.-B.; Johansson, L.; et al. Relation between Smoking History and Gene Expression Profiles in Lung Adenocarcinomas. BMC Med. Genom. 2012, 5, 22. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef]
- Cheung, W.K.C.; Zhao, M.; Liu, Z.; Stevens, L.E.; Cao, P.D.; Fang, J.E.; Westbrook, T.F.; Nguyen, D.X. Control of Alveolar Differentiation by the Lineage Transcription Factors GATA6 and HOPX Inhibits Lung Adenocarcinoma Metastasis. Cancer Cell 2013, 23, 725–738. [Google Scholar] [CrossRef]
- Fukui, T.; Shaykhiev, R.; Agosto-Perez, F.; Mezey, J.G.; Downey, R.J.; Travis, W.D.; Crystal, R.G. Lung Adenocarcinoma Subtypes Based on Expression of Human Airway Basal Cell Genes. Eur. Respir. J. 2013, 42, 1332–1344. [Google Scholar] [CrossRef]
- Brambilla, C.; Laffaire, J.; Lantuejoul, S.; Moro-Sibilot, D.; Mignotte, H.; Arbib, F.; Toffart, A.-C.; Petel, F.; Hainaut, P.; Rousseaux, S.; et al. Lung Squamous Cell Carcinomas with Basaloid Histology Represent a Specific Molecular Entity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5777–5786. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Ringnér, M.; Staaf, J. Consensus of Gene Expression Phenotypes and Prognostic Risk Predictors in Primary Lung Adenocarcinoma. Oncotarget 2016, 7, 52957–52973. [Google Scholar] [CrossRef][Green Version]
- Chen, F.; Zhang, Y.; Parra, E.; Rodriguez, J.; Behrens, C.; Akbani, R.; Lu, Y.; Kurie, J.M.; Gibbons, D.L.; Mills, G.B.; et al. Multiplatform-Based Molecular Subtypes of Non-Small-Cell Lung Cancer. Oncogene 2017, 36, 1384–1393. [Google Scholar] [CrossRef]
- Hu, F.; Zhou, Y.; Wang, Q.; Yang, Z.; Shi, Y.; Chi, Q. Gene Expression Classification of Lung Adenocarcinoma into Molecular Subtypes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 1187–1197. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Tang, H.; Wang, S.; Xiao, G.; Schiller, J.; Papadimitrakopoulou, V.; Minna, J.; Wistuba, I.I.; Xie, Y. Comprehensive Evaluation of Published Gene Expression Prognostic Signatures for Biomarker-Based Lung Cancer Clinical Studies. Ann. Oncol. 2017, 28, 733–740. [Google Scholar] [CrossRef]
- Subramanian, J.; Simon, R. Gene Expression-Based Prognostic Signatures in Lung Cancer: Ready for Clinical Use? JNCI J. Natl. Cancer Inst. 2010, 102, 464–474. [Google Scholar] [CrossRef]
- Kratz, J.R.; Jablons, D.M. Genomic Prognostic Models in Early-Stage Lung Cancer. Clin. Lung Cancer 2009, 10, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kim, S.; Taylor, J.M.G.; Wang, Z.; Lee, O.; Ramnath, N.; Reddy, R.M.; Lin, J.; Chang, A.C.; Orringer, M.B.; et al. Development and Validation of a Quantitative Real-Time Polymerase Chain Reaction Classifier for Lung Cancer Prognosis. J. Thorac. Oncol. 2011, 6, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Richards, W.G.; Harpole, D.H.; Ballman, K.V.; Tsao, M.-S.; Chen, Z.; Wang, X.; Chen, G.; Chirieac, L.R.; Chui, M.H.; et al. Multi-Institutional Prospective Validation of Prognostic MRNA Signatures in Early Stage Squamous Lung Cancer (Alliance). J. Thorac. Oncol. 2020, 15, 1748–1757. [Google Scholar] [CrossRef]
- Kratz, J.R.; He, J.; Van Den Eeden, S.K.; Zhu, Z.-H.; Gao, W.; Pham, P.T.; Mulvihill, M.S.; Ziaei, F.; Zhang, H.; Su, B.; et al. A Practical Molecular Assay to Predict Survival in Resected Non-Squamous, Non-Small-Cell Lung Cancer: Development and International Validation Studies. Lancet Lond. Engl. 2012, 379, 823–832. [Google Scholar] [CrossRef]
- Brant, R.; Sharpe, A.; Liptrot, T.; Dry, J.R.; Harrington, E.A.; Barrett, J.C.; Whalley, N.; Womack, C.; Smith, P.; Hodgson, D.R. Clinically Viable Gene Expression Assays with Potential for Predicting Benefit from MEK Inhibitors. Clin. Cancer Res. 2017, 23, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H. The Genomics of Lung Adenocarcinoma: Opportunities for Targeted Therapies. Genes Cancer 2010, 1, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Dacic, S.; Nikiforova, M.N. Present and Future Molecular Testing of Lung Carcinoma. Adv. Anat. Pathol. 2014, 21, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Gazdar, A.F. Personalized Medicine and Inhibition of EGFR Signaling in Lung Cancer. N. Engl. J. Med. 2009, 361, 1018–1020. [Google Scholar] [CrossRef]
- Casadio, C.; Guarize, J.; Donghi, S.; Di Tonno, C.; Fumagalli, C.; Vacirca, D.; Dell’Orto, P.; De Marinis, F.; Spaggiari, L.; Viale, G.; et al. Molecular Testing for Targeted Therapy in Advanced Non-Small Cell Lung Cancer: Suitability of Endobronchial Ultrasound Transbronchial Needle Aspiration. Am. J. Clin. Pathol. 2015, 144, 629–634. [Google Scholar] [CrossRef]
- Janku, F.; Garrido-Laguna, I.; Petruzelka, L.B.; Stewart, D.J.; Kurzrock, R. Novel Therapeutic Targets in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 1601–1612. [Google Scholar] [CrossRef]
- Savas, P.; Hughes, B.; Solomon, B. Targeted Therapy in Lung Cancer: IPASS and beyond, Keeping Abreast of the Explosion of Targeted Therapies for Lung Cancer. J. Thorac. Dis. 2013, 5 (Suppl. 5), S579–S592. [Google Scholar] [CrossRef]
- Pakkala, S.; Ramalingam, S.S. Personalized Therapy for Lung Cancer: Striking a Moving Target. JCI Insight 2018, 3, e120858. [Google Scholar] [CrossRef]
- Guo, L.; Chen, Z.; Xu, C.; Zhang, X.; Yan, H.; Su, J.; Yang, J.; Xie, Z.; Guo, W.; Li, F.; et al. Intratumoral Heterogeneity of EGFR-Activating Mutations in Advanced NSCLC Patients at the Single-Cell Level. BMC Cancer 2019, 19, 369. [Google Scholar] [CrossRef]
- Angulo, B.; Suarez-Gauthier, A.; Lopez-Rios, F.; Medina, P.P.; Conde, E.; Tang, M.; Soler, G.; Lopez-Encuentra, A.; Cigudosa, J.C.; Sanchez-Cespedes, M. Expression Signatures in Lung Cancer Reveal a Profile for EGFR-Mutant Tumours and Identify Selective PIK3CA Overexpression by Gene Amplification. J. Pathol. 2008, 214, 347–356. [Google Scholar] [CrossRef]
- Planck, M.; Isaksson, S.; Veerla, S.; Staaf, J. Identification of Transcriptional Subgroups in EGFR-Mutated and EGFR/KRAS Wild-Type Lung Adenocarcinoma Reveals Gene Signatures Associated with Patient Outcome. Clin. Cancer Res. 2013, 19, 5116–5126. [Google Scholar] [CrossRef]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of Genes Upregulated in ALK -Positive and EGFR/KRAS/ALK -Negative Lung Adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef]
- Sweet-Cordero, A.; Mukherjee, S.; Subramanian, A.; You, H.; Roix, J.J.; Ladd-Acosta, C.; Mesirov, J.; Golub, T.R.; Jacks, T. An Oncogenic KRAS2 Expression Signature Identified by Cross-Species Gene-Expression Analysis. Nat. Genet. 2005, 37, 48–55. [Google Scholar] [CrossRef]
- Ghimessy, A.; Radeczky, P.; Laszlo, V.; Hegedus, B.; Renyi-Vamos, F.; Fillinger, J.; Klepetko, W.; Lang, C.; Dome, B.; Megyesfalvi, Z. Current Therapy of KRAS-Mutant Lung Cancer. Cancer Metastasis Rev. 2020, 39, 1159–1177. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. JMD 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Lang, U.E.; Yeh, I.; McCalmont, T.H. Molecular Melanoma Diagnosis Update: Gene Fusion, Genomic Hybridization, and Massively Parallel Short-Read Sequencing. Clin. Lab. Med. 2017, 37, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative Clinical Genomics of Metastatic Cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and Transcriptomic Profiling Expands Precision Cancer Medicine: The WINTHER Trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Tuxen, I.V.; Rohrberg, K.S.; Oestrup, O.; Ahlborn, L.B.; Schmidt, A.Y.; Spanggaard, I.; Hasselby, J.P.; Santoni-Rugiu, E.; Yde, C.W.; Mau-Sørensen, M.; et al. Copenhagen Prospective Personalized Oncology (CoPPO)—Clinical Utility of Using Molecular Profiling to Select Patients to Phase I Trials. Clin. Cancer Res. 2019, 25, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.; Bivona, T.G. Understanding and Targeting Resistance Mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sakao, Y.; Ito, S.; Park, J.; Kuroda, H.; Sakakura, N.; Usami, N.; Mitsudomi, T.; Yatabe, Y. Transformation to Sarcomatoid Carcinoma in ALK-Rearranged Adenocarcinoma, Which Developed Acquired Resistance to Crizotinib and Received Subsequent Chemotherapies. J. Thorac. Oncol. 2013, 8, e75–e78. [Google Scholar] [CrossRef]
- Cha, Y.J.; Cho, B.C.; Kim, H.R.; Lee, H.-J.; Shim, H.S. A Case of ALK-Rearranged Adenocarcinoma with Small Cell Carcinoma-Like Transformation and Resistance to Crizotinib. J. Thorac. Oncol. 2016, 11, e55–e58. [Google Scholar] [CrossRef]
- Park, J.W.; Han, J.-W. Targeting Epigenetics for Cancer Therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef]
- Chung, F.-T.; Lee, K.-Y.; Wang, C.-W.; Heh, C.-C.; Chan, Y.-F.; Chen, H.-W.; Kuo, C.-H.; Feng, P.-H.; Lin, T.-Y.; Wang, C.-H.; et al. Tumor-Associated Macrophages Correlate with Response to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Advanced Non-Small Cell Lung Cancer. Int. J. Cancer 2012, 131, E227–E235. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Tan, C.-S.; Gilligan, D.; Pacey, S. Treatment Approaches for EGFR-Inhibitor-Resistant Patients with Non-Small-Cell Lung Cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.-L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung Cancers with Acquired Resistance to EGFR Inhibitors Occasionally Harbor BRAF Gene Mutations but Lack Mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef]
- Bruin, D.E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 Expression Confers Resistance to EGFR Inhibition in Lung Cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK Signaling Causes Resistance to EGFR Kinase Inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef]
- Ho, C.-C.; Liao, W.-Y.; Lin, C.-A.; Shih, J.-Y.; Yu, C.-J.; Chih-Hsin Yang, J. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK Dependence Underlies a Rational Polytherapy Strategy in EML4-ALK-Positive Lung Cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef]
- Fumarola, C.; Bonelli, M.A.; Petronini, P.G.; Alfieri, R.R. Targeting PI3K/AKT/MTOR Pathway in Non Small Cell Lung Cancer. Biochem. Pharmacol. 2014, 90, 197–207. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt Signalling Pathway and Cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Ono, N.; Yamazaki, T.; Tsukaguchi, T.; Fujii, T.; Sakata, K.; Suda, A.; Tsukuda, T.; Mio, T.; Ishii, N.; Kondoh, O.; et al. Enhanced Antitumor Activity of Erlotinib in Combination with the Hsp90 Inhibitor CH5164840 against Non-Small-Cell Lung Cancer. Cancer Sci. 2013, 104, 1346–1352. [Google Scholar] [CrossRef]
- Chen, M.; Shao, W.; He, J.; Wang, D. Role of Pemetrexed and Platinums Combination in Patients with Non-Small Cell Lung Cancer. Curr. Drug Targets 2010, 11, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hussmann, D.; Madsen, A.T.; Jakobsen, K.R.; Luo, Y.; Sorensen, B.S.; Nielsen, A.L. IGF1R Depletion Facilitates MET-Amplification as Mechanism of Acquired Resistance to Erlotinib in HCC827 NSCLC Cells. Oncotarget 2017, 8, 33300–33315. [Google Scholar] [CrossRef] [PubMed]
- Sasada, T.; Azuma, K.; Ohtake, J.; Fujimoto, Y. Immune Responses to Epidermal Growth Factor Receptor (EGFR) and Their Application for Cancer Treatment. Front. Pharmacol. 2016, 7, 405. [Google Scholar] [CrossRef] [PubMed]
- Coldren, C.D.; Helfrich, B.A.; Witta, S.E.; Sugita, M.; Lapadat, R.; Zeng, C.; Barón, A.; Franklin, W.A.; Hirsch, F.R.; Geraci, M.W.; et al. Baseline Gene Expression Predicts Sensitivity to Gefitinib in Non–Small Cell Lung Cancer Cell Lines. Mol. Cancer Res. 2006, 4, 521–528. [Google Scholar] [CrossRef]
- Balko, J.M.; Potti, A.; Saunders, C.; Stromberg, A.; Haura, E.B.; Black, E.P. Gene Expression Patterns That Predict Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Lung Cancer Cell Lines and Human Lung Tumors. BMC Genom. 2006, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL Kinase Causes Resistance to EGFR-Targeted Therapy in Lung Cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 Autocrine Pathway: A Novel Mechanism of Acquired Resistance to Gefitinib in NSCLC. Mol. Cancer Res. 2013, 11, 759–767. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.J.; Huang, R. Clinical Drug Response Can Be Predicted Using Baseline Gene Expression Levels and in Vitro Drug Sensitivity in Cell Lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Chang, T.-H.; Tsai, M.-F.; Wu, S.-G.; Tsai, T.-H.; Chen, H.-Y.; Yu, S.-L.; Yang, J.C.-H.; Shih, J.-Y. IL-8 Confers Resistance to EGFR Inhibitors by Inducing Stem Cell Properties in Lung Cancer. Oncotarget 2015, 6, 10415–10431. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; Concannon, K.; Cullen, S.; Boulay, G.; Turke, A.B.; Faber, A.C.; Lockerman, E.L.; Rivera, M.N.; Engelman, J.A.; Maheswaran, S.; et al. Inhibition of Mutant EGFR in Lung Cancer Cells Triggers SOX2-FOXO6-Dependent Survival Pathways. eLife 2015, 4, e06132. [Google Scholar] [CrossRef]
- Mojtabavi Naeini, M.; Tavassoli, M.; Ghaedi, K. Systematic Bioinformatic Approaches Reveal Novel Gene Expression Signatures Associated with Acquired Resistance to EGFR Targeted Therapy in Lung Cancer. Gene 2018, 667, 62–69. [Google Scholar] [CrossRef]
- Cheng, C.; Zhao, Y.; Schaafsma, E.; Weng, Y.; Amos, C. An EGFR Signature Predicts Cell Line and Patient Sensitivity to Multiple Tyrosine Kinase Inhibitors. Int. J. Cancer 2020, 147, 2621–2633. [Google Scholar] [CrossRef]
- Novello, S.; Barlesi, F.; Califano, R.; Cufer, T.; Ekman, S.; Levra, M.G.; Kerr, K.; Popat, S.; Reck, M.; Senan, S.; et al. Metastatic Non-Small-Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, v1–v27. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 807–821. [Google Scholar] [CrossRef]
- Lagos, G.G.; Izar, B.; Rizvi, N.A. Beyond Tumor PD-L1: Emerging Genomic Biomarkers for Checkpoint Inhibitor Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2020, 40, 1–11. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Navarro, A.; Paré, L.; Reguart, N.; Galván, P.; Pascual, T.; Martínez, A.; Nuciforo, P.; Comerma, L.; Alos, L.; et al. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non–Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res. 2017, 77, 3540–3550. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–Related MRNA Profile Predicts Clinical Response to PD-1 Blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-Cancer Adaptive Immune Resistance as Defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-Tumor Genomic Biomarkers for PD-1 Checkpoint Blockade–Based Immunotherapy. Science 2018, 362, 6411. [Google Scholar] [CrossRef]
- Wallden, B.; Pekker, I.; Popa, S.; Dowidar, N.; Sullivan, A.; Hood, T.; Danaher, P.; Mashadi-Hossein, A.; Lunceford, J.K.; Marton, M.J.; et al. Development and Analytical Performance of a Molecular Diagnostic for Anti-PD1 Response on the NCounter Dx Analysis System. J. Clin. Oncol. 2016, 34, 3034. [Google Scholar] [CrossRef]
- Hwang, S.; Kwon, A.-Y.; Jeong, J.-Y.; Kim, S.; Kang, H.; Park, J.; Kim, J.-H.; Han, O.J.; Lim, S.M.; An, H.J. Immune Gene Signatures for Predicting Durable Clinical Benefit of Anti-PD-1 Immunotherapy in Patients with Non-Small Cell Lung Cancer. Sci. Rep. 2020, 10, 643. [Google Scholar] [CrossRef]
- Faruki, H.; Mayhew, G.M.; Serody, J.S.; Hayes, D.N.; Perou, C.M.; Lai-Goldman, M. Lung Adenocarcinoma and Squamous Cell Carcinoma Gene Expression Subtypes Demonstrate Significant Differences in Tumor Immune Landscape. J. Thorac. Oncol. 2017, 12, 943–953. [Google Scholar] [CrossRef]
- Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma; Shedden, K.; Taylor, J.M.G.; Enkemann, S.A.; Tsao, M.-S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene Expression-Based Survival Prediction in Lung Adenocarcinoma: A Multi-Site, Blinded Validation Study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.-J.; Song, I.H.; Park, I.A.; Kang, J.; Yu, J.H.; Ahn, J.-H.; Gong, G. Prognostic and Predictive Value of NanoString-Based Immune-Related Gene Signatures in a Neoadjuvant Setting of Triple-Negative Breast Cancer: Relationship to Tumor-Infiltrating Lymphocytes. Breast Cancer Res. Treat. 2015, 151, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Signature | Year | Journal | Reference | Tissue of Origin | Number of Genes | Identified Subtypes | Technology |
|---|---|---|---|---|---|---|---|
| Bhattacharjee et al. | 2001 | Proc. Natl. Acad. Sci. USA | [7] | LUAD | 100 | C1, C2, C3, C4 | Microarray |
| Garber et al. | 2001 | Proc. Natl. Acad. Sci. USA | [6] | LUAD | 146 | AC1, AC2, AC3 | Microarray |
| Beer et al. | 2002 | Nat. Med. | [8] | LUAD | 4966 | Cluster 1, Cluster 2, Cluster 3 | Microarray |
| Tomida et al. | 2004 | Oncogene | [9] | LUAD, LUSC | 829 | AD1, AD2, AD3, AD4, SQ1, SQ2 | Microarray |
| Inamura et al. | 2005 | Oncogene | [10] | LUSC | 432 | SCC-A, SCC-B | Microarray |
| Hayes et al. | 2006 | J. Clin. Onc. | [11] | LUAD | 2553 | Bronchioid, Squamoid, Magnoid | Microarray |
| Raponi et al. | 2006 | Cancer Res. | [12] | LUSC | 11,101 | Cluster 1, Cluster 2 | Microarray |
| Takeuchi et al. | 2006 | J. Clin. Onc. | [13] | LUAD | 293 | TRU, non-TRU | Microarray |
| Larsen et al. | 2007 | Carcinogenesis | [14] | LUSC | 6748 | Cluster 1, Cluster 2 | Microarray |
| Shibata et al. | 2007 | Cancer Sci. | [15] | LUAD | 78 | Alveolar, Bronchiolar | Microarray |
| Wilkerson et al. | 2010 | Clin. Cancer Res. Off. J. Am. Assoc Cancer Res | [16] | LUSC | 208 | Primitive, Basal, Secretory, Classical | Microarray |
| Park et al. | 2012 | PloS ONE | [17] | LUAD | 191 | S_C1, F_C2 | Microarray |
| Staaf et al. | 2012 | BMC Med. Genomics | [18] | LUAD | 176 | AC1, AC2 | Microarray |
| TCGA-LUSC | 2012 | Nature | [19] | LUSC | 208 | Primitive, Basal, Secretory, Classical | RNA-Seq |
| Wilkerson et al. | 2012 | PloS ONE | [20] | LUAD | 506 | Bronchioid, Squamoid, Magnoid | Microarray |
| Cheung et al. | 2013 | Cancer Cell | [21] | LUAD | 249 | Alveolar, Distal airway stem cell-like (DASC) | Microarray |
| Fukui et al. | 2013 | Eur. Respir. J. | [22] | LUAD | 1829 | BC-low, BC-high | Microarray |
| Brambilla et al. | 2014 | Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res | [23] | LUSC | 139 | Classical_1, Classical_2, PEA, BL | Microarray |
| TCGA-LUAD | 2014 | Nature | [24] | LUAD | 506 | TRU, PI, PP | RNA-Seq |
| Ringnér et al. | 2016 | Oncotarget | [25] | LUAD | Consensus classification | CONSENSUS_1, CONSENSUS_2, CONSENSUS_3 | Consensus |
| Chen et al. | 2017 | Oncogene | [26] | LUAD, LUSC | 700 | AD.1, AD.2, AD.3, AD.4, AD.5a, AD.5b, SQ.1, SQ.2a, SQ.2b | RNA-Seq + Other omics |
| Hu et al. | 2019 | Trans. Comput. Biol. Bioinform. | [27] | LUAD | 30 | AD.1, AD.2, AD.3, AD.4, AD.5a, AD.5b | RNA-Seq |
| Signature | Histology | Evaluated Genomic Alterations | Associated Subtype |
|---|---|---|---|
| Takeuchi et al. [13] 2006 | LUAD | EGFR | TRU-type |
| KRAS | - | ||
| TP53 | - | ||
| Shibata et al. [15] 2007 | LUAD | KRAS | Bronchial |
| EGFR | Alveolar | ||
| Angulo et al. [46] 2008 | NSCLC | PI3K3CA | - |
| BRAF | - | ||
| LKB1 | - | ||
| KRAS | - | ||
| TP53 | - | ||
| EGFR | EGFR mutated LUAD transcriptional cluster | ||
| Wilkerson et al. [20] 2012 | LUAD | EGFR | Bronchioid |
| KRAS | Magnoid | ||
| TP53 | Magnoid | ||
| STK11 | Magnoid | ||
| LRP1B | - | ||
| BRAF | - | ||
| PTEN | - | ||
| Okayama et al. [48] 2012 | LUAD | EGFR | - |
| KRAS | - | ||
| ALK | ALK positive cluster | ||
| Hammerman et al. * [19] 2012 | LUSC | KEAP1 | Classical |
| NFE2L2 | Classical | ||
| PTEN | Classical, Primitive | ||
| RB1 | Primitive | ||
| Planck et al. [47] 2013 | LUAD | EGFR | EGFR-1 and EGFR-2 subtypes |
| KRAS | - | ||
| Collison et al. * [24] 2014 | LUAD | KRAS | PP |
| STK11 | PP | ||
| NF1 | PI | ||
| TP53 | PI | ||
| EGFR | TRU | ||
| Rignér et al. [25] 2016 | LUAD | EGFR | CONSENSUS_1 |
| KRAS | CONSENSUS_2 | ||
| Chen et al. * [26] 2017 | NSCLC | TP53 | SQ.2a, SQ.2b, AD.1, AD.2, AD.3 |
| RASA1 | - | ||
| PTEN | - | ||
| SMARCA4 | - | ||
| CDKN2A | - | ||
| NFE2L2 | - | ||
| STK11 | AD.1, AD.5b | ||
| KRAS | AD.2, AD.5b | ||
| KEAP1 | - | ||
| EGFR | - | ||
| Hu et al. * [27] 2019 | LUAD | TP53 | Subtype 1, Subtype 2 |
| EGFR | Subtype 4 |
| Study | Targeted Therapy | Target Gene | Sensitivity or Resistance | N Signature Genes | Main Proposed Mechanism |
|---|---|---|---|---|---|
| Coldren et al. [77] 2006 | Gefitinib | EGFR | Both | Sensitivity: 305 Resistance: 105 | E-cadherin upregulation in sensitive cell lines |
| Balko et al. [78] 2006 | Erlotinib | EGFR | Sensitivity | 180 | Overexpression of MAPK and PI3K pathways |
| Zhang et al. [79] 2012 | Erlotinib | EGFR | Resistance | 21 | AXL overexpression |
| Byers et al. [80] 2013 | Erlotinib PI3K-i | EGFR PI3K | Resistance | 76 | AXL overexpression and epithelial–mesenchymal transition activation |
| Terai et al. [81] 2013 | Gefitinib | EGFR | Resistance | - | FDF2-FGFR pathway activation |
| Geeleher et al. [82] 2014 | Erlotinib | EGFR | Sensitivity | 1000 | Not highlighted |
| Liu et al. [83] 2015 | Gefitinib | EGFR | Resistance | - | IL-8 overexpression and enriched stemness properties |
| Rothenberg et al. [84] 2015 | Erlotinib | EGFR | Resistance | 35 | SOX2 overexpression |
| Naeini et al. [85] 2018 | TKIs | EGFR | Resistance | 1286 | - Glycolysis and cell cycle upregulation - Immune response downregulation - Apoptosis downregulation - P53 pathway downregulation - TNF-alpha pathway downregulation - Xenobiotic metabolism downregulation |
| Cheng et al. [86] 2020 | Erlotinib Gefitinib Other TKIs | EGFR | Sensitivity | 11,431 | Not highlighted |
| Study | Year | Description |
|---|---|---|
| Liberzon et al. [90] | 2011 | Immunologic signatures of MSigDB. |
| Bindea et al. [91] | 2013 | 28 transcriptional signatures to quantify the degree of infiltration of different immune cell subpopulations. Both innate immune cells and adaptative immune cells are included. |
| Rooney et al. [92] | 2015 | CYT score based on the expression of the effector molecules that drive cytolysis. |
| Prat et al. [93] | 2017 | 23 immune-related signatures linked to response and progression-free survival after treatment with anti-PD1 therapy. |
| Ayers et al. [94] | 2017 | IFN-gamma pathway gene signature related to antigen presentation, chemokine expression, cytotoxic activity, and adaptative immune resistance. It has been associated with clinical benefit upon anti-PD1 pembrolizumab treatment. |
| Thorsson et al. [95] | 2018 | Identification of 6 immune subtypes/signatures characterized by differences in tumor microenvironment and prognosis. Specific mutations correlated with lower (CTNNB1, NRAS, IDH1) or higher (BRAF, TP53, CASP8) lymphocytes presence. |
| Danaher et al. [96] | 2018 | Tumor Inflammation Signature (TIS) takes into account genes related to antigen presentation, cytotoxic activity, and adaptative immune resistance. The TIS has been shown to enrich for patients who respond to the anti-PD1 treatment pembrolizumab. The TIS has been applied retrospectively in multiple immuno-oncology trials. |
| Critescu et al. [97] | 2018 | T-Cell inflamed gene expression signature and TMB potential to jointly predict response to pembrolizumab in more than 300 patients with advanced solid tumors and melanoma from KEYNOTE trials. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hijazo-Pechero, S.; Alay, A.; Marín, R.; Vilariño, N.; Muñoz-Pinedo, C.; Villanueva, A.; Santamaría, D.; Nadal, E.; Solé, X. Gene Expression Profiling as a Potential Tool for Precision Oncology in Non-Small Cell Lung Cancer. Cancers 2021, 13, 4734. https://doi.org/10.3390/cancers13194734
Hijazo-Pechero S, Alay A, Marín R, Vilariño N, Muñoz-Pinedo C, Villanueva A, Santamaría D, Nadal E, Solé X. Gene Expression Profiling as a Potential Tool for Precision Oncology in Non-Small Cell Lung Cancer. Cancers. 2021; 13(19):4734. https://doi.org/10.3390/cancers13194734
Chicago/Turabian StyleHijazo-Pechero, Sara, Ania Alay, Raúl Marín, Noelia Vilariño, Cristina Muñoz-Pinedo, Alberto Villanueva, David Santamaría, Ernest Nadal, and Xavier Solé. 2021. "Gene Expression Profiling as a Potential Tool for Precision Oncology in Non-Small Cell Lung Cancer" Cancers 13, no. 19: 4734. https://doi.org/10.3390/cancers13194734
APA StyleHijazo-Pechero, S., Alay, A., Marín, R., Vilariño, N., Muñoz-Pinedo, C., Villanueva, A., Santamaría, D., Nadal, E., & Solé, X. (2021). Gene Expression Profiling as a Potential Tool for Precision Oncology in Non-Small Cell Lung Cancer. Cancers, 13(19), 4734. https://doi.org/10.3390/cancers13194734