
Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges

 , , , , ,
, , , , ,  ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Acute Myeloid Leukemia (AML)
3. Chronic Myeloid Leukemia (CML)
4. Acute Lymphoblastic Leukemia (ALL)
5. Chronic Lymphocytic Leukemia (CLL)
6. Non-Hodgkin Lymphomas (NHLs)
7. Multiple Myeloma (MM)
8. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wood, B.L.; Arroz, M.; Barnett, D.; DiGiuseppe, J.; Greig, B.; Kussick, S.J.; Oldaker, T.; Shenkin, M.; Stone, E.; Wallace, P. 2006 Bethesda International Consensus Recommendations on the Immunophenotypic Analysis of Hematolymphoid Neoplasia by Flow Cytometry: Optimal Reagents and Reporting for the Flow Cytometric Diagnosis of Hematopoietic Neoplasia. Cytometry 2007, 72B, S14–S22. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; van der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow Antibody Panels for Standardized N-Dimensional Flow Cytometric Immunophenotyping of Normal, Reactive and Malignant Leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/Measurable Residual Disease in AML: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Plesa, A.; Labussière-Wallet, H.; Hayette, S.; Salles, G.; Thomas, X.; Sujobert, P. Efficiency of Blinatumomab in a t(8;21) Acute Myeloid Leukemia Expressing CD19. Haematologica 2019, 104, e487–e488. [Google Scholar] [CrossRef] [PubMed]
- El Chaer, F.; Ali, O.M.; Sausville, E.A.; Law, J.Y.; Lee, S.T.; Duong, V.H.; Baer, M.R.; Koka, R.; Singh, Z.N.; Wong, J.; et al. Treatment of CD19-Positive Mixed Phenotype Acute Leukemia with Blinatumomab. Am. J. Hematol. 2019, 94, E7–E8. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Kelder, A.; Oussoren-Brockhoff, Y.J.M.; Scholten, W.J.; Snel, A.N.; Veldhuizen, D.; Cloos, J.; Ossenkoppele, G.J.; Schuurhuis, G.J. Peripheral Blood Minimal Residual Disease May Replace Bone Marrow Minimal Residual Disease as an Immunophenotypic Biomarker for Impending Relapse in Acute Myeloid Leukemia. Leukemia 2016, 30, 708–715. [Google Scholar] [CrossRef]
- Dillon, R.; Potter, N.; Freeman, S.; Russell, N. How We Use Molecular Minimal Residual Disease (MRD) Testing in Acute Myeloid Leukaemia (AML). Br. J. Haematol. 2021, 193, 231–244. [Google Scholar] [CrossRef]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Song, G.; Shurtleff, S.; Yeoh, A.E.-J.; Chng, W.J.; Chen, S.P.; Rubnitz, J.E.; Pui, C.-H.; Downing, J.R.; Campana, D. Universal Monitoring of Minimal Residual Disease in Acute Myeloid Leukemia. JCI Insight 2018, 3, e98561. [Google Scholar] [CrossRef]
- Kandeel, E.Z.; El Sharkawy, N.; Hanafi, M.; Samra, M.; Kamel, A. Tracing Leukemia Stem Cells and Their Influence on Clinical Course of Adult Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 383–393. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef]
- Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. [Google Scholar] [CrossRef]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The Interleukin-3 Receptor Alpha Chain is a Unique Marker for Human Acute Myelogenous Leukemia Stem Cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated Expression of IL-3Rα in Acute Myelogenous Leukemia is Associated with Enhanced Blast Proliferation, Increased Cellularity, and Poor Prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of Leukemic Stem Cells CD34+/CD38−/CD123+ Delineate FLT3/ITD-Positive Clones. J. Hematol. Oncol. 2016, 9, 61. [Google Scholar] [CrossRef]
- Montesinos, P.; Roboz, G.J.; Bulabois, C.-E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V.; et al. Safety and Efficacy of Talacotuzumab plus Decitabine or Decitabine Alone in Patients with Acute Myeloid Leukemia not Eligible for Chemotherapy: Results from a Multicenter, Randomized, Phase 2/3 Study. Leukemia 2021, 35, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Goswami, S.; Gopalakrishnan, B.; Ramaswamy, R.; Wasmuth, R.; Tran, M.; Mo, X.; Gordon, A.; Bucci, D.; Lucas, D.M.; et al. The Interleukin-3 Receptor CD123 Targeted SL-401 Mediates Potent Cytotoxic Activity against CD34+CD123+ Cells from Acute Myeloid Leukemia/Myelodysplastic Syndrome Patients and Healthy Donors. Haematologica 2018, 103, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A. Targeting CD123 in AML. Clin. Lymphoma Myeloma Leuk. 2020, 20 (Suppl. 1), S67–S68. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Alhabbab, R.Y. Targeting Cancer Stem Cells by Genetically Engineered Chimeric Antigen Receptor T Cells. Front. Genet. 2020, 11, 312. [Google Scholar] [CrossRef]
- Bueno, C.; Velasco-Hernandez, T.; Gutiérrez-Agüera, F.; Zanetti, S.R.; Baroni, M.L.; Sánchez-Martínez, D.; Molina, O.; Closa, A.; Agraz-Doblás, A.; Marín, P.; et al. CD133-Directed CAR T-Cells for MLL Leukemia: On-Target, off-Tumor Myeloablative Toxicity. Leukemia 2019, 33, 2090–2125. [Google Scholar] [CrossRef]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for Acute Myeloid Leukemia Therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef]
- Ngai, L.L.; Ma, C.Y.; Maguire, O.; Do, A.D.; Robert, A.; Logan, A.C.; Griffiths, E.A.; Nemeth, M.J.; Green, C.; Pourmohamad, T.; et al. Bimodal Expression of Potential Drug Target CLL-1 (CLEC12A) on CD34+ Blasts of AML Patients. Eur. J. Haematol. 2021. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, D.H.; Cortes, J.; Andreeff, M. The Elusive Chronic Myeloid Leukemia Stem Cell: Does it Matter and How do We Eliminate it? Semin. Hematol. 2010, 47, 362–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janssen, J.J.W.M.; Deenik, W.; Smolders, K.G.M.; van Kuijk, B.J.; Pouwels, W.; Kelder, A.; Cornelissen, J.J.; Schuurhuis, G.J.; Ossenkoppele, G.J. Residual Normal Stem Cells Can Be Detected in Newly Diagnosed Chronic Myeloid Leukemia Patients by a New Flow Cytometric Approach and Predict for Optimal Response to Imatinib. Leukemia 2012, 26, 977–984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raspadori, D.; Pacelli, P.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Cattaneo, D.; Gozzini, A.; Galimberti, S.; Baratè, C.; Pregno, P.; et al. Flow Cytometry Assessment of CD26+ Leukemic Stem Cells in Peripheral Blood: A Simple and Rapid New Diagnostic Tool for Chronic Myeloid Leukemia. Cytometry 2019, 96, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Rülicke, T.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of Target Expression Profiles in CD34+/CD38− and CD34+/CD38+ Stem and Progenitor Cells in AML and CML. Blood Adv. 2020, 4, 5118–5132. [Google Scholar] [CrossRef]
- Weerkamp, F.; Dekking, E.; Ng, Y.Y.; van der Velden, V.H.J.; Wai, H.; Böttcher, S.; Brüggemann, M.; van der Sluijs, A.J.; Koning, A.; Boeckx, N.; et al. Flow Cytometric Immunobead Assay for the Detection of BCR-ABL Fusion Proteins in Leukemia Patients. Leukemia 2009, 23, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Raponi, S.; De Propris, M.S.; Wai, H.; Intoppa, S.; Elia, L.; Diverio, D.; Vitale, A.; Foa, R.; Guarini, A. An Accurate and Rapid Flow Cytometric Diagnosis of BCR-ABL Positive Acute Lymphoblastic Leukemia. Haematologica 2009, 94, 1767–1770. [Google Scholar] [CrossRef][Green Version]
- Yujie, W.; Yu, Z.; Sixuan, Q.; Li, W.; Peng, L.; Zeng, G.; Sujiang, Z.; Jianyong, L. Detection of BCR-ABL Fusion Proteins in Patients with Leukemia Using a Cytometric Bead Array. Leuk. Lymphoma 2012, 53, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Löf, L.; Arngården, L.; Olsson-Strömberg, U.; Siart, B.; Jansson, M.; Dahlin, J.S.; Thörn, I.; Christiansson, L.; Hermansson, M.; Larsson, A.; et al. Flow Cytometric Measurement of Blood Cells with BCR-ABL1 Fusion Protein in Chronic Myeloid Leukemia. Sci. Rep. 2017, 7, 623. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, M.; Raff, T.; Kneba, M. Has MRD Monitoring Superseded Other Prognostic Factors in Adult ALL? Blood 2012, 120, 4470–4481. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Zhou, S.; Higley, H.; Mukundan, L.; Fu, S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease With Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-Analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrová, M.; Novakova, M.; Sonneveld, E.; et al. Standardized Flow Cytometry for Highly Sensitive MRD Measurements in B-Cell Acute Lymphoblastic Leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Chen, S.; Wang, C.; Brown, P.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Flow-Cytometric vs. -Morphologic Assessment of Remission in Childhood Acute Lymphoblastic Leukemia: A Report from the Children’s Oncology Group (COG). Leukemia 2018, 32, 1370–1379. [Google Scholar] [CrossRef]
- Burns, M.A.; Place, A.E.; Stevenson, K.E.; Gutiérrez, A.; Forrest, S.; Pikman, Y.; Vrooman, L.M.; Harris, M.H.; Hunt, S.K.; O’Brien, J.E.; et al. Identification of Prognostic Factors in Childhood T-Cell Acute Lymphoblastic Leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr. Blood Cancer 2021, 68, e28719. [Google Scholar] [CrossRef] [PubMed]
- Ciudad, J.; San Miguel, J.F.; López-Berges, M.C.; Vidriales, B.; Valverde, B.; Ocqueteau, M.; Mateos, G.; Caballero, M.D.; Hernández, J.; Moro, M.J.; et al. Prognostic Value of Immunophenotypic Detection of Minimal Residual Disease in Acute Lymphoblastic Leukemia. J. Clin. Oncol. 1998, 16, 3774–3781. [Google Scholar] [CrossRef]
- Malec, M.; Björklund, E.; Söderhäll, S.; Mazur, J.; Sjögren, A.M.; Pisa, P.; Björkholm, M.; Porwit-MacDonald, A. Flow Cytometry and Allele-Specific Oligonucleotide PCR Are Equally Effective in Detection of Minimal Residual Disease in ALL. Leukemia 2001, 15, 716–727. [Google Scholar] [CrossRef][Green Version]
- Vidriales, M.-B.; Pérez, J.J.; López-Berges, M.C.; Gutiérrez, N.; Ciudad, J.; Lucio, P.; Vazquez, L.; García-Sanz, R.; del Cañizo, M.C.; Fernández-Calvo, J.; et al. Minimal Residual Disease in Adolescent (Older than 14 Years) and Adult Acute Lymphoblastic Leukemias: Early Immunophenotypic Evaluation Has High Clinical Value. Blood 2003, 101, 4695–4700. [Google Scholar] [CrossRef]
- Björklund, E.; Mazur, J.; Söderhäll, S.; Porwit-MacDonald, A. Flow Cytometric Follow-up of Minimal Residual Disease in Bone Marrow Gives Prognostic Information in Children with Acute Lymphoblastic Leukemia. Leukemia 2003, 17, 138–148. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Gaipa, G.; Ratei, R.; Veltroni, M.; Schumich, A.; Maglia, O.; Karawajew, L.; Benetello, A.; Pötschger, U.; Husak, Z.; et al. Standardization of Flow Cytometric Minimal Residual Disease Evaluation in Acute Lymphoblastic Leukemia: Multicentric Assessment Is Feasible. Cytometry B Clin. Cytom. 2008, 74, 331–340. [Google Scholar] [CrossRef]
- Robillard, N.; Cavé, H.; Méchinaud, F.; Guidal, C.; Garnache-Ottou, F.; Rohrlich, P.S.; Avet-Loiseau, H.; Garand, R. Four-Color Flow Cytometry Bypasses Limitations of IG/TCR Polymerase Chain Reaction for Minimal Residual Disease Detection in Certain Subsets of Children with Acute Lymphoblastic Leukemia. Haematologica 2005, 90, 1516–1523. [Google Scholar]
- Garand, R.; Beldjord, K.; Cavé, H.; Fossat, C.; Arnoux, I.; Asnafi, V.; Bertrand, Y.; Boulland, M.-L.; Brouzes, C.; Clappier, E.; et al. Flow Cytometry and IG/TCR Quantitative PCR for Minimal Residual Disease Quantitation in Acute Lymphoblastic Leukemia: A French Multicenter Prospective Study on Behalf of the FRALLE, EORTC and GRAALL. Leukemia 2013, 27, 370–376. [Google Scholar] [CrossRef]
- Denys, B.; van der Sluijs-Gelling, A.J.; Homburg, C.; van der Schoot, C.E.; de Haas, V.; Philippé, J.; Pieters, R.; van Dongen, J.J.M.; van der Velden, V.H.J. Improved Flow Cytometric Detection of Minimal Residual Disease in Childhood Acute Lymphoblastic Leukemia. Leukemia 2013, 27, 635–641. [Google Scholar] [CrossRef]
- Modvig, S.; Hallböök, H.; Madsen, H.O.; Siitonen, S.; Rosthøj, S.; Tierens, A.; Juvonen, V.; Osnes, L.T.N.; Vålerhaugen, H.; Hultdin, M.; et al. Value of Flow Cytometry for MRD-Based Relapse Prediction in B-Cell Precursor ALL in a Multicenter Setting. Leukemia 2021, 35, 1894–1906. [Google Scholar] [CrossRef]
- Theunissen, P.M.J.; Sedek, L.; De Haas, V.; Szczepanski, T.; Van Der Sluijs, A.; Mejstrikova, E.; Nováková, M.; Kalina, T.; Lecrevisse, Q.; Orfao, A.; et al. Detailed Immunophenotyping of B-Cell Precursors in Regenerating Bone Marrow of Acute Lymphoblastic Leukaemia Patients: Implications for Minimal Residual Disease Detection. Br. J. Haematol. 2017, 178, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.A.; Sale, G.E.; Shulman, H.M.; Myerson, D.; Bryant, E.M.; Gooley, T.; Loken, M.R. Multidimensional Flow Cytometry of Marrow Can Differentiate Leukemic From Normal Lymphoblasts and Myeloblasts After Chemotherapy and Bone Marrow Transplantation. Am. J. Clin. Pathol. 1998, 110, 84–94. [Google Scholar] [CrossRef]
- DiGiuseppe, J.A.; Wood, B.L. Applications of Flow Cytometric Immunophenotyping in the Diagnosis and Posttreatment Monitoring of B and T Lymphoblastic Leukemia/Lymphoma. Cytometry B Clin. Cytom. 2019, 96, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Shi, C. Monitoring Minimal/Measurable Residual Disease in B-Cell Acute Lymphoblastic Leukemia by Flow Cytometry during Targeted Therapy. Int. J. Hematol. 2021, 113, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Basso, G.; Veltroni, M.; Valsecchi, M.G.; Dworzak, M.N.; Ratei, R.; Silvestri, D.; Benetello, A.; Buldini, B.; Maglia, O.; Masera, G.; et al. Risk of Relapse of Childhood Acute Lymphoblastic Leukemia Is Predicted by Flow Cytometric Measurement of Residual Disease on Day 15 Bone Marrow. J. Clin. Oncol. 2009, 27, 5168–5174. [Google Scholar] [CrossRef] [PubMed]
- Eveillard, M.; Robillard, N.; Arnoux, I.; Garand, R.; Rialland, F.; Thomas, C.; Strullu, M.; Michel, G.; Béné, M.C.; Fossat, C.; et al. Major Impact of an Early Bone Marrow Checkpoint (Day 21) for Minimal Residual Disease in Flow Cytometry in Childhood Acute Lymphoblastic Leukemia. Hematol. Oncol. 2017, 35, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Szczepański, T. Why and How to Quantify Minimal Residual Disease in Acute Lymphoblastic Leukemia? Leukemia 2007, 21, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Oberley, M.J.; Raikar, S.S.; Wertheim, G.B.; Malvar, J.; Sposto, R.; Rabin, K.R.; Punia, J.N.; Seif, A.E.; Cahen, V.C.; Schore, R.J.; et al. Significance of Minimal Residual Disease in Pediatric Mixed Phenotype Acute Leukemia: A Multicenter Cohort Study. Leukemia 2020, 34, 1741–1750. [Google Scholar] [CrossRef]
- Rezvani, K.; Yong, A.S.M.; Savani, B.N.; Mielke, S.; Keyvanfar, K.; Gostick, E.; Price, D.A.; Douek, D.C.; Barrett, A.J. Graft-versus-Leukemia Effects Associated with Detectable Wilms Tumor-1 Specific T Lymphocytes after Allogeneic Stem-Cell Transplantation for Acute Lymphoblastic Leukemia. Blood 2007, 110, 1924–1932. [Google Scholar] [CrossRef]
- Riva, G.; Luppi, M.; Barozzi, P.; Quadrelli, C.; Basso, S.; Vallerini, D.; Zanetti, E.; Morselli, M.; Forghieri, F.; Maccaferri, M.; et al. Emergence of BCR-ABL-Specific Cytotoxic T Cells in the Bone Marrow of Patients with Ph+ Acute Lymphoblastic Leukemia during Long-Term Imatinib Mesylate Treatment. Blood 2010, 115, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Riva, G.; Luppi, M.; Quadrelli, C.; Barozzi, P.; Basso, S.; Vallerini, D.; Zanetti, E.; Morselli, M.; Forghieri, F.; Maccaferri, M.; et al. BCR-ABL-Specific Cytotoxic T Cells in the Bone Marrow of Patients with Ph(+) Acute Lymphoblastic Leukemia during Second-Generation Tyrosine-Kinase Inhibitor Therapy. Blood Cancer J. 2011, 1, e30. [Google Scholar] [CrossRef]
- Krampera, M.; Vitale, A.; Vincenzi, C.; Perbellini, O.; Guarini, A.; Annino, L.; Todeschini, G.; Camera, A.; Fabbiano, F.; Fioritoni, G.; et al. Outcome Prediction by Immunophenotypic Minimal Residual Disease Detection in Adult T-Cell Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2003, 120, 74–79. [Google Scholar] [CrossRef]
- van der Velden, V.H.J.; Jacobs, D.C.H.; Wijkhuijs, A.J.M.; Comans-Bitter, W.M.; Willemse, M.J.; Hählen, K.; Kamps, W.A.; van Wering, E.R.; van Dongen, J.J.M. Minimal Residual Disease Levels in Bone Marrow and Peripheral Blood Are Comparable in Children with T Cell Acute Lymphoblastic Leukemia (ALL), but Not in Precursor-B-ALL. Leukemia 2002, 16, 1432–1436. [Google Scholar] [CrossRef]
- Keegan, A.; Charest, K.; Schmidt, R.; Briggs, D.; Deangelo, D.J.; Li, B.; Morgan, E.A.; Pozdnyakova, O. Flow Cytometric Minimal Residual Disease Assessment of Peripheral Blood in Acute Lymphoblastic Leukaemia Patients Has Potential for Early Detection of Relapsed Extramedullary Disease. J. Clin. Pathol. 2018, 71, 653–658. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Fröschl, G.; Printz, D.; Zen, L.D.; Gaipa, G.; Ratei, R.; Basso, G.; Biondi, A.; Ludwig, W.-D.; Gadner, H. CD99 Expression in T-Lineage ALL: Implications for Flow Cytometric Detection of Minimal Residual Disease. Leukemia 2004, 18, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Tembhare, P.R.; Sriram, H.; Khanka, T.; Chatterjee, G.; Panda, D.; Ghogale, S.; Badrinath, Y.; Deshpande, N.; Patkar, N.V.; Narula, G.; et al. Flow Cytometric Evaluation of CD38 Expression Levels in the Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia and the Effect of Chemotherapy on Its Expression in Measurable Residual Disease, Refractory Disease and Relapsed Disease: An Implication for Anti-CD38 Immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Vega-García, N.; Perez-Jaume, S.; Esperanza-Cebollada, E.; Vicente-Garcés, C.; Torrebadell, M.; Jiménez-Velasco, A.; Ortega, M.; Llop, M.; Abad, L.; Vagace, J.M.; et al. Measurable Residual Disease Assessed by Flow-Cytometry is a Stable Prognostic Factor for Pediatric T-Cell Acute Lymphoblastic Leukemia in Consecutive SEHOP Protocols Whereas the Impact of Oncogenetics Depends on Treatment. Front. Pediatr. 2020, 8, 614521. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Narula, G.; Khanka, T.; Ghogale, S.; Chatterjee, G.; Patkar, N.V.; Prasad, M.; Badrinath, Y.; Deshpande, N.; Gudapati, P.; et al. Post-Induction Measurable Residual Disease Using Multicolor Flow Cytometry is Strongly Predictive of Inferior Clinical Outcome in the Real-Life Management of Childhood T-Cell Acute Lymphoblastic Leukemia: A Study of 256 Patients. Front. Oncol. 2020, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, Y.; Huang, X.; Zhang, Y.; Qian, J.; Li, J.; Li, C.; Li, X.; Lou, Y.; Zhu, Q.; et al. Minimal Residual Disease Level Determined by Flow Cytometry Provides Reliable Risk Stratification in Adults with T-Cell Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2021, 193, 1096–1104. [Google Scholar] [CrossRef]
- Modvig, S.; Madsen, H.O.; Siitonen, S.M.; Rosthøj, S.; Tierens, A.; Juvonen, V.; Osnes, L.T.N.; Vålerhaugen, H.; Hultdin, M.; Thörn, I.; et al. Minimal Residual Disease Quantification by Flow Cytometry Provides Reliable Risk Stratification in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2019, 33, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Tembhare, P.R.; Chatterjee, G.; Khanka, T.; Ghogale, S.; Badrinath, Y.; Deshpande, N.; Panda, D.; Patkar, N.V.; Narula, G.; Girase, K.; et al. Eleven-Marker 10-Color Flow Cytometric Assessment of Measurable Residual Disease for T-Cell Acute Lymphoblastic Leukemia Using an Approach of Exclusion. Cytometry B Clin. Cytom. 2020, 100, 421–433. [Google Scholar] [CrossRef]
- Hallek, M. Chronic Lymphocytic Leukemia: 2020 Update on Diagnosis, Risk Stratification and Treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef]
- Robertson, L.; Huh, Y.; Butler, J.; Pugh, W.; Hirsch-Ginsberg, C.; Stass, S.; Kantarjian, H.; Keating, M. Response Assessment in Chronic Lymphocytic Leukemia after Fludarabine plus Prednisone: Clinical, Pathologic, Immunophenotypic, and Molecular Analysis [See Comments]. Blood 1992, 80, 29–36. [Google Scholar] [CrossRef]
- Vuillier, F.; Claisse, J.F.; Vandenvelde, C.; Travade, P.; Magnac, C.; Chevret, S.; Desablens, B.; Binet, J.L.; Dighiero, G. Evaluation of Residual Disease in B-Cell Chronic Lymphocytic Leukemia Patients in Clinical and Bone-Marrow Remission Using CD5-CD19 Markers and PCR Study of Gene Rearrangements. Leuk. Lymphoma. 1992, 7, 195–204. [Google Scholar] [CrossRef]
- European Medicines Agency. Appendix 4 to the Guidelines on the Evaluation of Anticancer Medicinal Products in Man. Available online: https://www.ema.europa.eu/en/appendix-4-guideline-evaluation-anticancer-medicinal-products-man-condition-specific-guidance (accessed on 27 June 2021).
- Thompson, P.A.; Wierda, W.G. Eliminating Minimal Residual Disease as a Therapeutic End Point: Working toward Cure for Patients with CLL. Blood 2016, 127, 279–286. [Google Scholar] [CrossRef]
- Dimier, N.; Delmar, P.; Ward, C.; Morariu-Zamfir, R.; Fingerle-Rowson, G.; Bahlo, J.; Fischer, K.; Eichhorst, B.; Goede, V.; van Dongen, J.J.M.; et al. A Model for Predicting Effect of Treatment on Progression-Free Survival Using MRD as a Surrogate End Point in CLL. Blood 2018, 131, 955–962. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. IwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Administration UFaD. Hematologic Malignancies: Regulatory Considerations for Use of Minimal Residual Disease in Development of Drug and Biological Products for Treatment. Guidance for Industry. 2018. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM623333.pdf (accessed on 27 June 2021).
- Wierda, W.G.; Rawstron, A.; Cymbalista, F.; Badoux, X.; Rossi, D.; Brown, J.R.; Egle, A.; Abello, V.; Cervera Ceballos, E.; Herishanu, Y.; et al. Measurable Residual Disease in Chronic Lymphocytic Leukemia: Expert Review and Consensus Recommendations. Leukemia 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Böttcher, S.; Letestu, R.; Villamor, N.; Fazi, C.; Kartsios, H.; de Tute, R.M.; Shingles, J.; Ritgen, M.; Moreno, C.; et al. Improving Efficiency and Sensitivity: European Research Initiative in CLL (ERIC) Update on the International Harmonised Approach for Flow Cytometric Residual Disease Monitoring in CLL. Leukemia 2013, 27, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Kennedy, B.; Evans, P.A.; Davies, F.E.; Richards, S.J.; Haynes, A.P.; Russell, N.H.; Hale, G.; Morgan, G.J.; Jack, A.S.; et al. Quantitation of Minimal Disease Levels in Chronic Lymphocytic Leukemia Using a Sensitive Flow Cytometric Assay Improves the Prediction of Outcome and Can Be Used to Optimize Therapy. Blood 2001, 98, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sorigue, M.; Juncà, J.; Sarrate, E.; Grau, J. Expression of CD43 in Chronic Lymphoproliferative Leukemias: CD43 IN LYMPHOPROLIFERATIVE LEUKEMIAS. Cytometry 2018, 94, 136–142. [Google Scholar] [CrossRef]
- Böttcher, S.; Ritgen, M.; Pott, C.; Brüggemann, M.; Raff, T.; Stilgenbauer, S.; Döhner, H.; Dreger, P.; Kneba, M. Comparative Analysis of Minimal Residual Disease Detection Using Four-Color Flow Cytometry, Consensus IgH-PCR, and Quantitative IgH PCR in CLL after Allogeneic and Autologous Stem Cell Transplantation. Leukemia 2004, 18, 1637–1645. [Google Scholar] [CrossRef]
- Rawstron, A.C.; de Tute, R.; Jack, A.S.; Hillmen, P. Flow Cytometric Protein Expression Profiling as a Systematic Approach for Developing Disease-Specific Assays: Identification of a Chronic Lymphocytic Leukaemia-Specific Assay for Use in Rituximab-Containing Regimens. Leukemia 2006, 20, 2102–2110. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Villamor, N.; Ritgen, M.; Böttcher, S.; Ghia, P.; Zehnder, J.L.; Lozanski, G.; Colomer, D.; Moreno, C.; Geuna, M.; et al. International Standardized Approach for Flow Cytometric Residual Disease Monitoring in Chronic Lymphocytic Leukaemia. Leukemia 2007, 21, 956–964. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Fazi, C.; Agathangelidis, A.; Villamor, N.; Letestu, R.; Nomdedeu, J.; Palacio, C.; Stehlikova, O.; Kreuzer, K.-A.; Liptrot, S.; et al. A Complementary Role of Multiparameter Flow Cytometry and High-Throughput Sequencing for Minimal Residual Disease Detection in Chronic Lymphocytic Leukemia: An European Research Initiative on CLL Study. Leukemia 2016, 30, 929–936. [Google Scholar] [CrossRef]
- Farren, T.W.; Giustiniani, J.; Fanous, M.; Liu, F.; Macey, M.G.; Wright, F.; Prentice, A.; Nathwani, A.; Agrawal, S.G. Minimal Residual Disease Detection with Tumor-Specific CD160 Correlates with Event-Free Survival in Chronic Lymphocytic Leukemia. Blood Cancer J. 2015, 5, e273. [Google Scholar] [CrossRef][Green Version]
- Köhnke, T.; Wittmann, V.K.; Bücklein, V.L.; Lichtenegger, F.; Pasalic, Z.; Hiddemann, W.; Spiekermann, K.; Subklewe, M. Diagnosis of CLL Revisited: Increased Specificity by a Modified Five-Marker Scoring System Including CD200. Br. J. Haematol. 2017, 179, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Baskar, S.; Kwong, K.Y.; Hofer, T.; Levy, J.M.; Kennedy, M.G.; Lee, E.; Staudt, L.M.; Wilson, W.H.; Wiestner, A.; Rader, C. Unique Cell Surface Expression of Receptor Tyrosine Kinase ROR1 in Human B-Cell Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2008, 14, 396–404. [Google Scholar] [CrossRef]
- Bewarder, M.; Stilgenbauer, S.; Thurner, L.; Kaddu-Mulindwa, D. Current Treatment Options in CLL. Cancers 2021, 13, 2468. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.J.; Danilov, A.V. The Evolving Role of Bruton’s Tyrosine Kinase Inhibitors in Chronic Lymphocytic Leukemia. Ther. Adv. Hematol. 2021, 12, 2040620721989588. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged Lymphocytosis during Ibrutinib Therapy Is Associated with Distinct Molecular Characteristics and Does Not Indicate a Suboptimal Response to Therapy. Blood 2014, 123, 1810–1817. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Niemann, C.U.; Farooqui, M.; Jones, J.; Mustafa, R.Z.; Lipsky, A.; Saba, N.; Martyr, S.; Soto, S.; Valdez, J.; et al. Ibrutinib-Induced Lymphocytosis in Patients with Chronic Lymphocytic Leukemia: Correlative Analyses from a Phase II Study. Leukemia 2014, 28, 2188–2196. [Google Scholar] [CrossRef]
- Perini, G.F.; Feres, C.C.P.; Teixeira, L.L.C.; Hamerschlak, N. BCL-2 Inhibition as Treatment for Chronic Lymphocytic Leukemia. Curr. Treat. Options Oncol. 2021, 22, 66. [Google Scholar] [CrossRef]
- Kater, A.P.; Seymour, J.F.; Hillmen, P.; Eichhorst, B.; Langerak, A.W.; Owen, C.; Verdugo, M.; Wu, J.; Punnoose, E.A.; Jiang, Y.; et al. Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J. Clin. Oncol. 2019, 37, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Genuardi, E.; Mazziotta, F.; Iovino, L.; Morabito, F.; Grassi, S.; Ciabatti, E.; Guerrini, F.; Petrini, M. The Minimal Residual Disease in Non-Hodgkin’s Lymphomas: From the Laboratory to the Clinical Practice. Front. Oncol. 2019, 9, 528. [Google Scholar] [CrossRef] [PubMed]
- Bayly, E.; Nguyen, V.; Binek, A.; Piggin, A.; Baldwin, K.; Westerman, D.; Came, N. Validation of a Modified Pre-lysis Sample Preparation Technique for Flow Cytometric Minimal Residual Disease Assessment in Multiple Myeloma, Chronic Lymphocytic Leukemia, and B-non Hodgkin Lymphoma. Cytometry 2020, 98, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.L.; Armand, P. Minimal Residual Disease in Non-Hodgkin Lymphoma—Current Applications and Future Directions. Br. J. Haematol. 2018, 180, 177–188. [Google Scholar] [CrossRef]
- Bottcher, S.; Ritgen, M.; Buske, S.; Gesk, S.; Klapper, W.; Hoster, E.; Hiddemann, W.; Unterhalt, M.; Dreyling, M.; Siebert, R.; et al. Minimal Residual Disease Detection in Mantle Cell Lymphoma: Methods and Significance of Four-Color Flow Cytometry Compared to Consensus IGH-Polymerase Chain Reaction at Initial Staging and for Follow-up Examinations. Haematologica 2008, 93, 551–559. [Google Scholar] [CrossRef]
- Chovancová, J.; Bernard, T.; Stehlíková, O.; Šálek, D.; Janíková, A.; Mayer, J.; Doubek, M. Detection of Minimal Residual Disease in Mantle Cell Lymphoma-Establishment of Novel Eight-Color Flow Cytometry Approach. Cytometry B Clin. Cytom. 2015, 88, 92–100. [Google Scholar] [CrossRef]
- Cheminant, M.; Derrieux, C.; Touzart, A.; Schmit, S.; Grenier, A.; Trinquand, A.; Delfau-Larue, M.-H.; Lhermitte, L.; Thieblemont, C.; Ribrag, V.; et al. Minimal Residual Disease Monitoring by 8-Color Flow Cytometry in Mantle Cell Lymphoma: An EU-MCL and LYSA Study. Haematologica 2016, 101, 336–345. [Google Scholar] [CrossRef]
- Noel, P. Definition of Remission, Minimal Residual Disease, and Relapse in Hairy Cell Leukemia Bone Marrow Biopsy Histology and Immunohistology Specimens. Leuk. Lymphoma 2011, 52 (Suppl. 2), 62–64. [Google Scholar] [CrossRef]
- Garnache Ottou, F.; Chandesris, M.-O.; Lhermitte, L.; Callens, C.; Beldjord, K.; Garrido, M.; Bedin, A.-S.; Brouzes, C.; Villemant, S.; Rubio, M.-T.; et al. Peripheral Blood 8 Colour Flow Cytometry Monitoring of Hairy Cell Leukaemia Allows Detection of High-Risk Patients. Br. J. Haematol. 2014, 166, 50–59. [Google Scholar] [CrossRef]
- Guerrini, F.; Paolicchi, M.; Ghio, F.; Ciabatti, E.; Grassi, S.; Salehzadeh, S.; Ercolano, G.; Metelli, M.R.; Del Re, M.; Iovino, L.; et al. The Droplet Digital PCR: A New Valid Molecular Approach for the Assessment of B-RAF V600E Mutation in Hairy Cell Leukemia. Front. Pharmacol. 2016, 7, 363. [Google Scholar] [CrossRef]
- Grever, M.R.; Abdel-Wahab, O.; Andritsos, L.A.; Banerji, V.; Barrientos, J.; Blachly, J.S.; Call, T.G.; Catovsky, D.; Dearden, C.; Demeter, J.; et al. Consensus Guidelines for the Diagnosis and Management of Patients with Classic Hairy Cell Leukemia. Blood 2017, 129, 553–560. [Google Scholar] [CrossRef]
- Lyu, R.; Wang, T.; Wang, Y.; Xiong, W.; Wang, H.; Yan, Y.; Wang, Q.; Liu, W.; An, G.; Huang, W.; et al. Undetectable Minimal Residual Disease Is an Independent Prognostic Factor in Splenic Marginal Zone Lymphoma. Br. J. Haematol. 2021, 194, 862–869. [Google Scholar] [CrossRef]
- van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple Myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- on behalf of the International Myeloma Working Group; Durie, B.G.M.; Harousseau, J.-L.; Miguel, J.S.; Bladé, J.; Barlogie, B.; Anderson, K.; Gertz, M.; Dimopoulos, M.; Westin, J.; et al. International Uniform Response Criteria for Multiple Myeloma. Leukemia 2006, 20, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Legarda, M.A.; Cejalvo, M.J.; de la Rubia, J. Recent Advances in the Treatment of Patients with Multiple Myeloma. Cancers 2020, 12, 3576. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Martinez-Lopez, J.; Vidriales, M.-B.; Mateos, M.-V.; Montalban, M.-A.; Fernandez-Redondo, E.; Alonso, L.; Oriol, A.; Teruel, A.-I.; de Paz, R.; et al. Comparison of Immunofixation, Serum Free Light Chain, and Immunophenotyping for Response Evaluation and Prognostication in Multiple Myeloma. J. Clin. Oncol. 2011, 29, 1627–1633. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-Analysis. JAMA Oncol. 2017, 3, 28. [Google Scholar] [CrossRef]
- Landgren, O.; Devlin, S.; Boulad, M.; Mailankody, S. Role of MRD Status in Relation to Clinical Outcomes in Newly Diagnosed Multiple Myeloma Patients: A Meta-Analysis. Bone Marrow Transpl. 2016, 51, 1565–1568. [Google Scholar] [CrossRef]
- Lahuerta, J.-J.; Paiva, B.; Vidriales, M.-B.; Cordón, L.; Cedena, M.-T.; Puig, N.; Martinez-Lopez, J.; Rosiñol, L.; Gutierrez, N.C.; Martín-Ramos, M.-L.; et al. Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. JCO 2017, 35, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; de Tute, R.; Paiva, B.; Perez, J.J.; Böttcher, S.; Wind, H.; Sanoja, L.; Puig, N.; Lecrevisse, Q.; Vidriales, M.B.; et al. Immunophenotype of Normal vs. Myeloma Plasma Cells: Toward Antibody Panel Specifications for MRD Detection in Multiple Myeloma: MM MRD antibody panels. Cytometry 2016, 90, 61–72. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Davies, F.E.; DasGupta, R.; Ashcroft, A.J.; Patmore, R.; Drayson, M.T.; Owen, R.G.; Jack, A.S.; Child, J.A.; Morgan, G.J. Flow Cytometric Disease Monitoring in Multiple Myeloma: The Relationship between Normal and Neoplastic Plasma Cells Predicts Outcome after Transplantation. Blood 2002, 100, 3095–3100. [Google Scholar] [CrossRef] [PubMed]
- San Miguel, J.F.; Almeida, J.; Mateo, G.; Bladé, J.; López-Berges, C.; Caballero, D.; Hernández, J.; Moro, M.J.; Fernández-Calvo, J.; Díaz-Mediavilla, J.; et al. Immunophenotypic Evaluation of the Plasma Cell Compartment in Multiple Myeloma: A Tool for Comparing the Efficacy of Different Treatment Strategies and Predicting Outcome. Blood 2002, 99, 1853–1856. [Google Scholar] [CrossRef]
- Arroz, M.; Came, N.; Lin, P.; Chen, W.; Yuan, C.; Lagoo, A.; Monreal, M.; de Tute, R.; Vergilio, J.-A.; Rawstron, A.C.; et al. Consensus Guidelines on Plasma Cell Myeloma Minimal Residual Disease Analysis and Reporting: Guidelines on Myeloma MRD Analysis and Reporting. Cytometry 2016, 90, 31–39. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Yuan, C.M.; Venzon, D.; Braylan, R.; Korde, N.; Manasanch, E.; Zuchlinsky, D.; Calvo, K.; Kurlander, R.; Bhutani, M.; et al. Flow Cytometric Differentiation of Abnormal and Normal Plasma Cells in the Bone Marrow in Patients with Multiple Myeloma and Its Precursor Diseases. Leuk. Res. 2014, 38, 371–376. [Google Scholar] [CrossRef]
- Liu, D.; Lin, P.; Hu, Y.; Zhou, Y.; Tang, G.; Powers, L.; Medeiros, L.J.; Jorgensen, J.L.; Wang, S.A. Immunophenotypic Heterogeneity of Normal Plasma Cells: Comparison with Minimal Residual Plasma Cell Myeloma. J. Clin. Pathol. 2012, 65, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Child, J.A.; de Tute, R.M.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Feyler, S.; et al. Minimal Residual Disease Assessed by Multiparameter Flow Cytometry in Multiple Myeloma: Impact on Outcome in the Medical Research Council Myeloma IX Study. J. Clin. Oncol. 2013, 31, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Goolsby, C.L.; Nelson, B.P.; Singhal, S.; Mehta, J.; Peterson, L.C. Instability of Immunophenotype in Plasma Cell Myeloma. Am. J. Clin. Pathol. 2008, 129, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Oberle, A.; Brandt, A.; Alawi, M.; Langebrake, C.; Janjetovic, S.; Wolschke, C.; Schütze, K.; Bannas, P.; Kröger, N.; Koch-Nolte, F.; et al. Long-Term CD38 Saturation by Daratumumab Interferes with Diagnostic Myeloma Cell Detection. Haematologica 2017, 102, e368–e370. [Google Scholar] [CrossRef]
- Paiva, B.; Almeida, J.; Pérez-Andrés, M.; Mateo, G.; López, A.; Rasillo, A.; Vídriales, M.-B.; López-Berges, M.-C.; Miguel, J.F.S.; Orfao, A. Utility of Flow Cytometry Immunophenotyping in Multiple Myeloma and Other Clonal Plasma Cell-Related Disorders. Cytometry B Clin. Cytom. 2010, 78, 239–252. [Google Scholar] [CrossRef]
- Mathis, S.; Chapuis, N.; Borgeot, J.; Maynadié, M.; Fontenay, M.; Béné, M.-C.; Guy, J.; Bardet, V. Comparison of Cross-Platform Flow Cytometry Minimal Residual Disease Evaluation in Multiple Myeloma Using a Common Antibody Combination and Analysis Strategy. Cytometry B Clin. Cytom. 2015, 88, 101–109. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Orfao, A.; Beksac, M.; Bezdickova, L.; Brooimans, R.A.; Bumbea, H.; Dalva, K.; Fuhler, G.; Gratama, J.; Hose, D.; et al. Report of the European Myeloma Network on Multiparametric Flow Cytometry in Multiple Myeloma and Related Disorders. Haematologica 2008, 93, 431–438. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Pérez-Morán, J.-J.; Vidriales, M.-B.; García-Sanz, R.; et al. Next Generation Flow for Highly Sensitive and Standardized Detection of Minimal Residual Disease in Multiple Myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef]
- Paiva, B.; Cedena, M.-T.; Puig, N.; Arana, P.; Vidriales, M.-B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.-L.; Martinez-Lopez, J.; et al. Minimal Residual Disease Monitoring and Immune Profiling in Multiple Myeloma in Elderly Patients. Blood 2016, 127, 3165–3174. [Google Scholar] [CrossRef]
- Paiva, B.; Puig, N.; Cedena, M.-T.; Rosiñol, L.; Cordón, L.; Vidriales, M.-B.; Burgos, L.; Flores-Montero, J.; Sanoja-Flores, L.; Lopez-Anglada, L.; et al. Measurable Residual Disease by Next-Generation Flow Cytometry in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 784–792. [Google Scholar] [CrossRef]
- Oliva, S.; op Bruinink, D.H.; Rihova, L.; D’Agostino, M.; Pantani, L.; Capra, A.; van der Holt, B.; Troia, R.; Petrucci, M.T.; Villanova, T.; et al. Minimal Residual Disease Assessment by Multiparameter Flow Cytometry in Transplant-Eligible Myeloma in the EMN02/HOVON 95 MM Trial. Blood Cancer J. 2021, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Bene, M.C.; Wuilleme, S.; Corre, J.; Attal, M.; Arnulf, B.; Garderet, L.; Macro, M.; Stoppa, A.-M.; Delforge, M.; et al. Concordance of Post-Consolidation Minimal Residual Disease Rates by Multiparametric Flow Cytometry and Next-Generation Sequencing in CASSIOPEIA. Clin. Lymphoma Myeloma Leuk. 2019, 19, e3–e4. [Google Scholar] [CrossRef]
- Oliva, S.; Genuardi, E.; Belotti, A.; Frascione, P.M.M.; Galli, M.; Capra, A.; Offidani, M.; Vozella, F.; Zambello, R.; Auclair, D.; et al. Minimal Residual Disease Evaluation By Multiparameter Flow Cytometry and Next Generation Sequencing in the Forte Trial for Newly Diagnosed Multiple Myeloma Patients. Blood 2019, 134, 4322. [Google Scholar] [CrossRef]
- Medina, A.; Puig, N.; Flores-Montero, J.; Jimenez, C.; Sarasquete, M.-E.; Garcia-Alvarez, M.; Prieto-Conde, I.; Chillon, C.; Alcoceba, M.; Gutierrez, N.C.; et al. Comparison of Next-Generation Sequencing (NGS) and next-Generation Flow (NGF) for Minimal Residual Disease (MRD) Assessment in Multiple Myeloma. Blood Cancer J. 2020, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Sanoja-Flores, L.; Flores-Montero, J.; Puig, N.; Contreras-Sanfeliciano, T.; Pontes, R.; Corral-Mateos, A.; García-Sánchez, O.; Díez-Campelo, M.; Pessoa de Magalhães, R.J.; García-Martín, L.; et al. Blood Monitoring of Circulating Tumor Plasma Cells by next Generation Flow in Multiple Myeloma after Therapy. Blood 2019, 134, 2218–2222. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.J.; Derman, B.A.; Bal, S.; Sidana, S.; Chhabra, S.; Silbermann, R.; Ye, J.C.; Cook, G.; Cornell, R.F.; Holstein, S.A.; et al. International Harmonization in Performing and Reporting Minimal Residual Disease Assessment in Multiple Myeloma Trials. Leukemia 2021, 35, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Seymour, J.F.; Kater, A.P.; Fischer, K. Should Undetectable Minimal Residual Disease Be the Goal of Chronic Lymphocytic Leukemia Therapy? Hematol Oncol. Clin. North Am. 2021, 35, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Should Minimal Residual Disease Negativity Be the End Point of Myeloma Therapy? Blood Adv. 2017, 1, 517–521. [Google Scholar] [CrossRef]
- Comoli, P.; Basso, S.; Riva, G.; Barozzi, P.; Guido, I.; Gurrado, A.; Quartuccio, G.; Rubert, L.; Lagreca, I.; Vallerini, D.; et al. BCR-ABL-Specific T-Cell Therapy in Ph+ ALL Patients on Tyrosine-Kinase Inhibitors. Blood 2017, 129, 582–586. [Google Scholar] [CrossRef]
- Greiner, J.; Ono, Y.; Hofmann, S.; Schmitt, A.; Mehring, E.; Götz, M.; Guillaume, P.; Döhner, K.; Mytilineos, J.; Döhner, H.; et al. Mutated Regions of Nucleophosmin 1 Elicit Both CD4(+) and CD8(+) T-Cell Responses in Patients with Acute Myeloid Leukemia. Blood 2012, 120, 1282–1289. [Google Scholar] [CrossRef]
- Forghieri, F.; Riva, G.; Lagreca, I.; Barozzi, P.; Vallerini, D.; Morselli, M.; Paolini, A.; Bresciani, P.; Colaci, E.; Maccaferri, M.; et al. Characterization and Dynamics of Specific T Cells against Nucleophosmin-1 (NPM1)-Mutated Peptides in Patients with NPM1-Mutated Acute Myeloid Leukemia. Oncotarget 2019, 10, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Riva, G.; Lagreca, I.; Barozzi, P.; Bettelli, F.; Paolini, A.; Nasillo, V.; Lusenti, B.; Pioli, V.; Giusti, D.; et al. Neoantigen-Specific T-Cell Immune Responses: The Paradigm of NPM1-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 9159. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Ho, P.-C.; Verdeil, G. Regulatory Circuits of T Cell Function in Cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T Lymphocyte Interplay in Controlling Tumor Growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef]



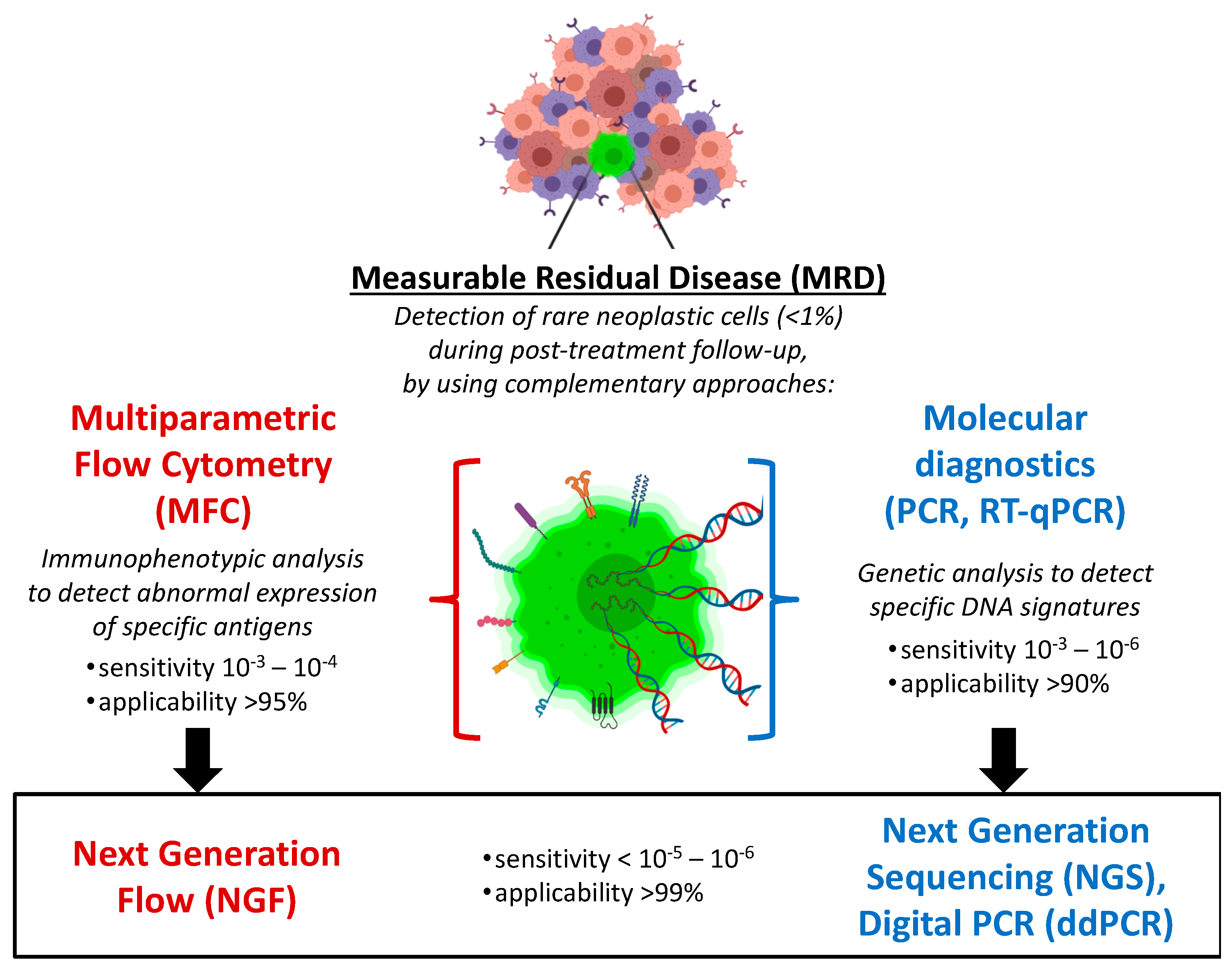{kind=link}
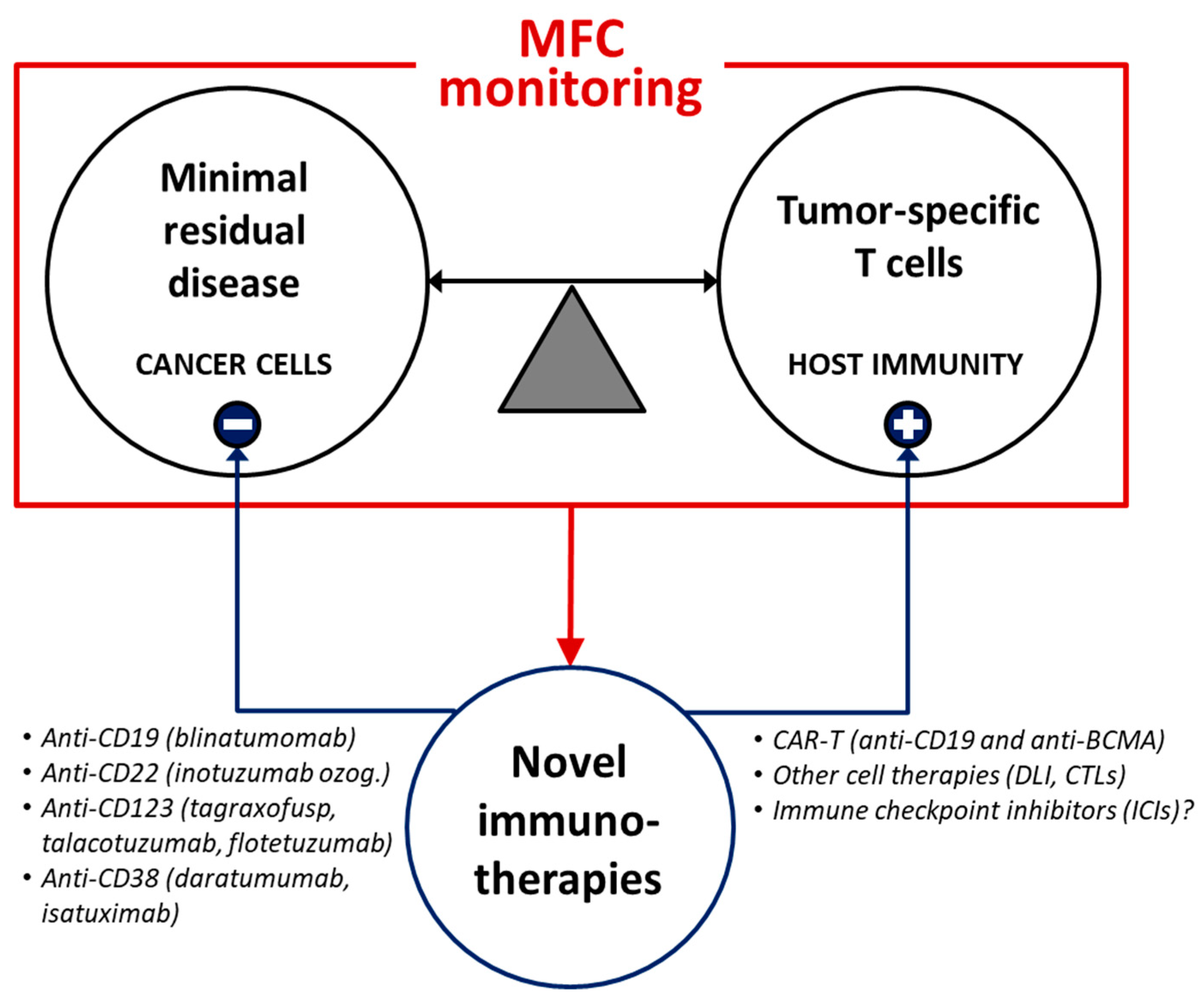{kind=link}
| AML | B-ALL | T-ALL | CLL | MM | |
|---|---|---|---|---|---|
| Sensitivity | 10−3–10−5 | 10−4–10−5 | 10−4–10−5 | 10−4–10−5 | 10−5–10−6 |
| Sample origin | BM | BM | BM, PB | PB, BM | BM |
| N° of cells required | 3 × 106 | 4 × 106 | 4 × 106 | 3 × 106 | 5–20 × 106 |
| Applicability (% of cases) | >97% | >99% | >99% | >95% | >99% |
| MRD “positivity”threshold | ≥10−3 | ≥10−4 | ≥10−4 | ≥10−4 | ≥10−5 |
| Follow-up timepoints | Poorly standardized: usually performed early, after initial therapy (i.e., post-induction/consolidation treatments), then usually guided by clinical protocols (usually, every 3–6 months) | ||||
| Backbone panel | CD34, CD117, CD45, CD13, CD33, CD15, CD7 | CD34, CD19, CD10, CD20, CD38, CD45 | CD2, CD3, CD5, CD7, CD4, CD8, CD34, CD45, CD99, CD1a | CD19, CD20, CD5, CD79b, CD43, CD81 | CD138, CD38, CD45, CD56, CD19, CD27, CD28, CD117, cy k/λ, CD81 |
| Additional markers | CD14, CD64, HLA-DR, CD4, CD11b, CD123, CD133, CD38, CD90 | CD22, CD81, CD66c, CD123, CD73, CD304 | CD10, CD38, CD56, TdT | CD200, CD23, CD160, ROR1 | CD33, CD54, CD200, CD229, CD307, CD319, CD150, VS38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riva, G.; Nasillo, V.; Ottomano, A.M.; Bergonzini, G.; Paolini, A.; Forghieri, F.; Lusenti, B.; Barozzi, P.; Lagreca, I.; Fiorcari, S.; et al. Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges. Cancers 2021, 13, 4582. https://doi.org/10.3390/cancers13184582
Riva G, Nasillo V, Ottomano AM, Bergonzini G, Paolini A, Forghieri F, Lusenti B, Barozzi P, Lagreca I, Fiorcari S, et al. Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges. Cancers. 2021; 13(18):4582. https://doi.org/10.3390/cancers13184582
Chicago/Turabian StyleRiva, Giovanni, Vincenzo Nasillo, Anna Maria Ottomano, Giuliano Bergonzini, Ambra Paolini, Fabio Forghieri, Beatrice Lusenti, Patrizia Barozzi, Ivana Lagreca, Stefania Fiorcari, and et al. 2021. "Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges" Cancers 13, no. 18: 4582. https://doi.org/10.3390/cancers13184582
APA StyleRiva, G., Nasillo, V., Ottomano, A. M., Bergonzini, G., Paolini, A., Forghieri, F., Lusenti, B., Barozzi, P., Lagreca, I., Fiorcari, S., Martinelli, S., Maffei, R., Marasca, R., Potenza, L., Comoli, P., Manfredini, R., Tagliafico, E., Trenti, T., & Luppi, M. (2021). Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges. Cancers, 13(18), 4582. https://doi.org/10.3390/cancers13184582