
Gene Signatures Induced by Ionizing Radiation as Prognostic Tools in an In Vitro Experimental Breast Cancer Model

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Radiation Overview
3. Radiation and Its Biological Effects
4. Radiation Effects and Gene Expression in Other Organs
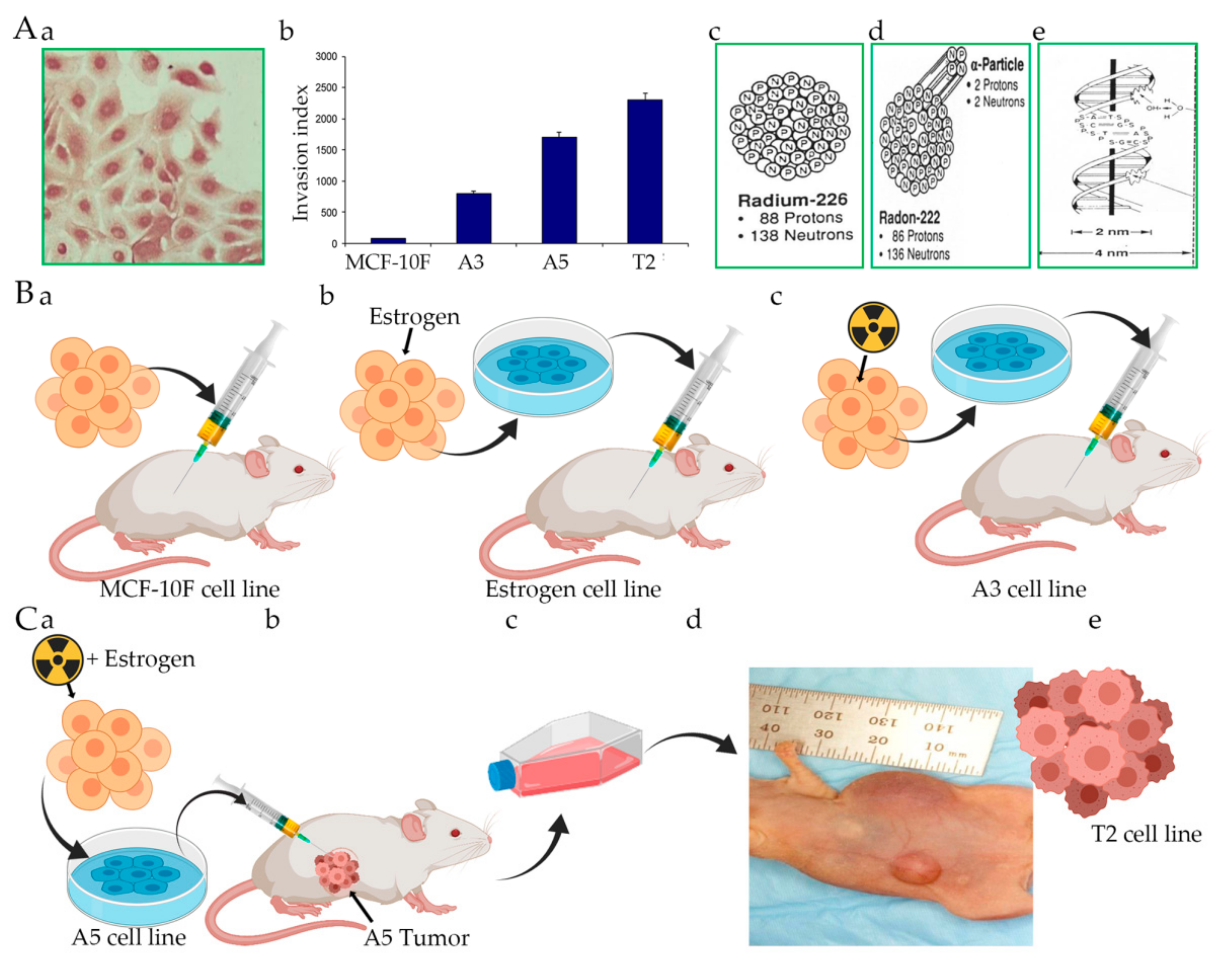
5. Alpha Model, Radiation, and Carcinogenesis
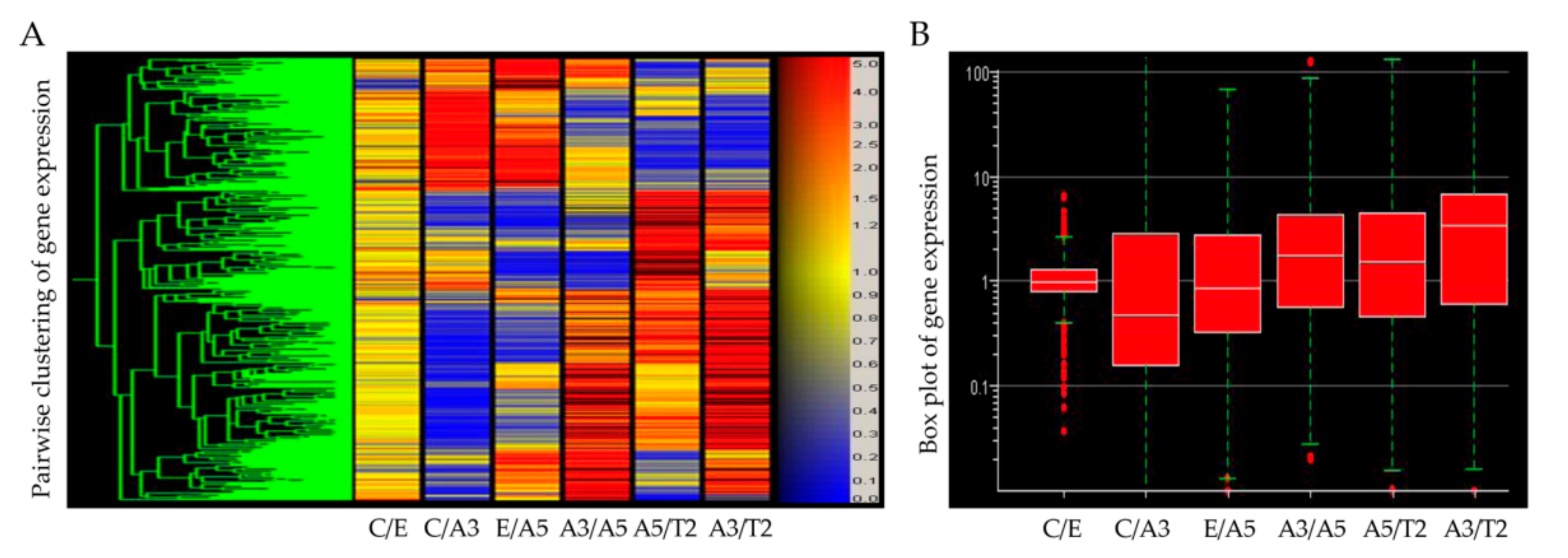
6. Gene Expression Induced by Radiation
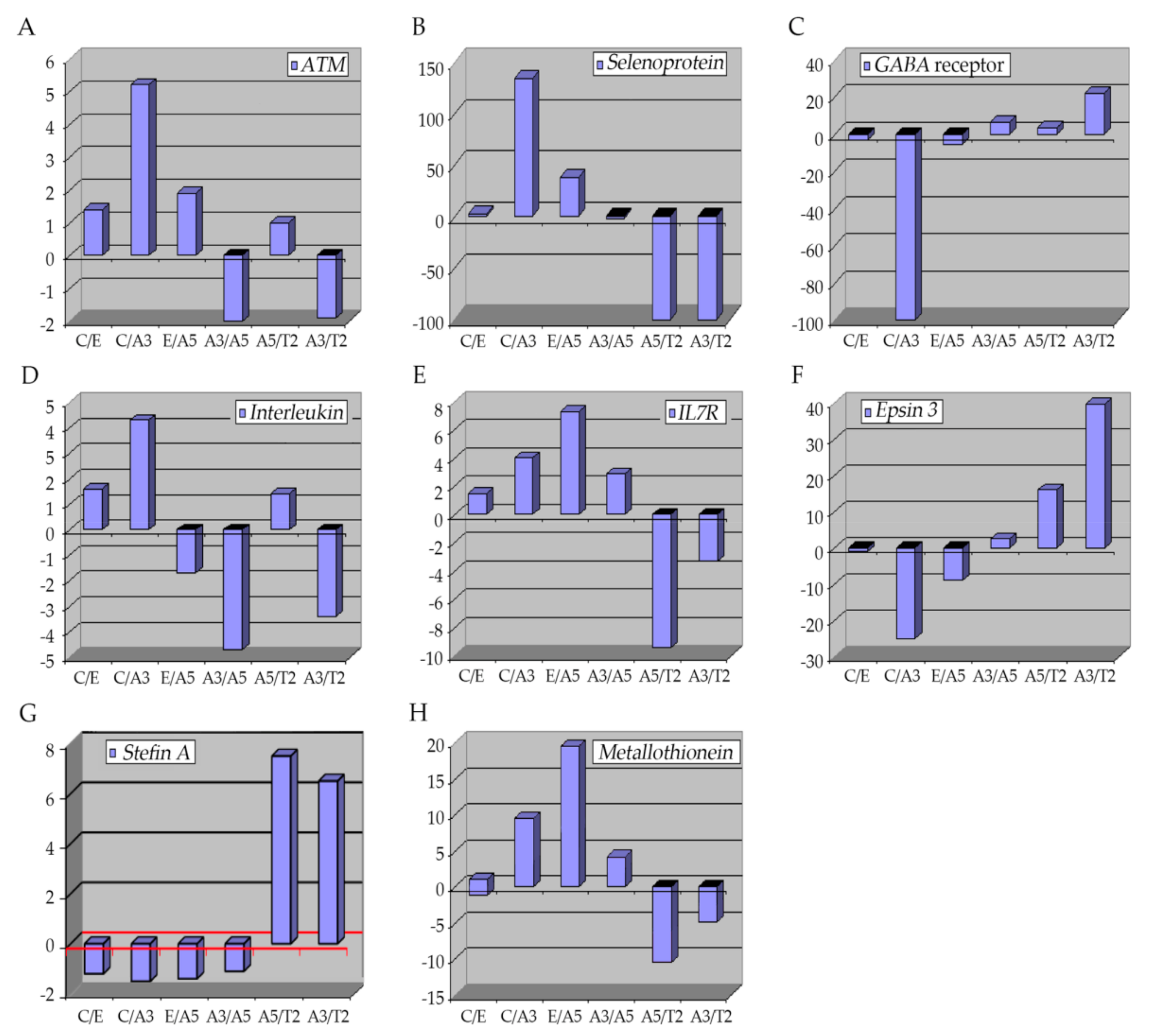
6.1. The Ataxia-Telangiectasia Mutated Gene
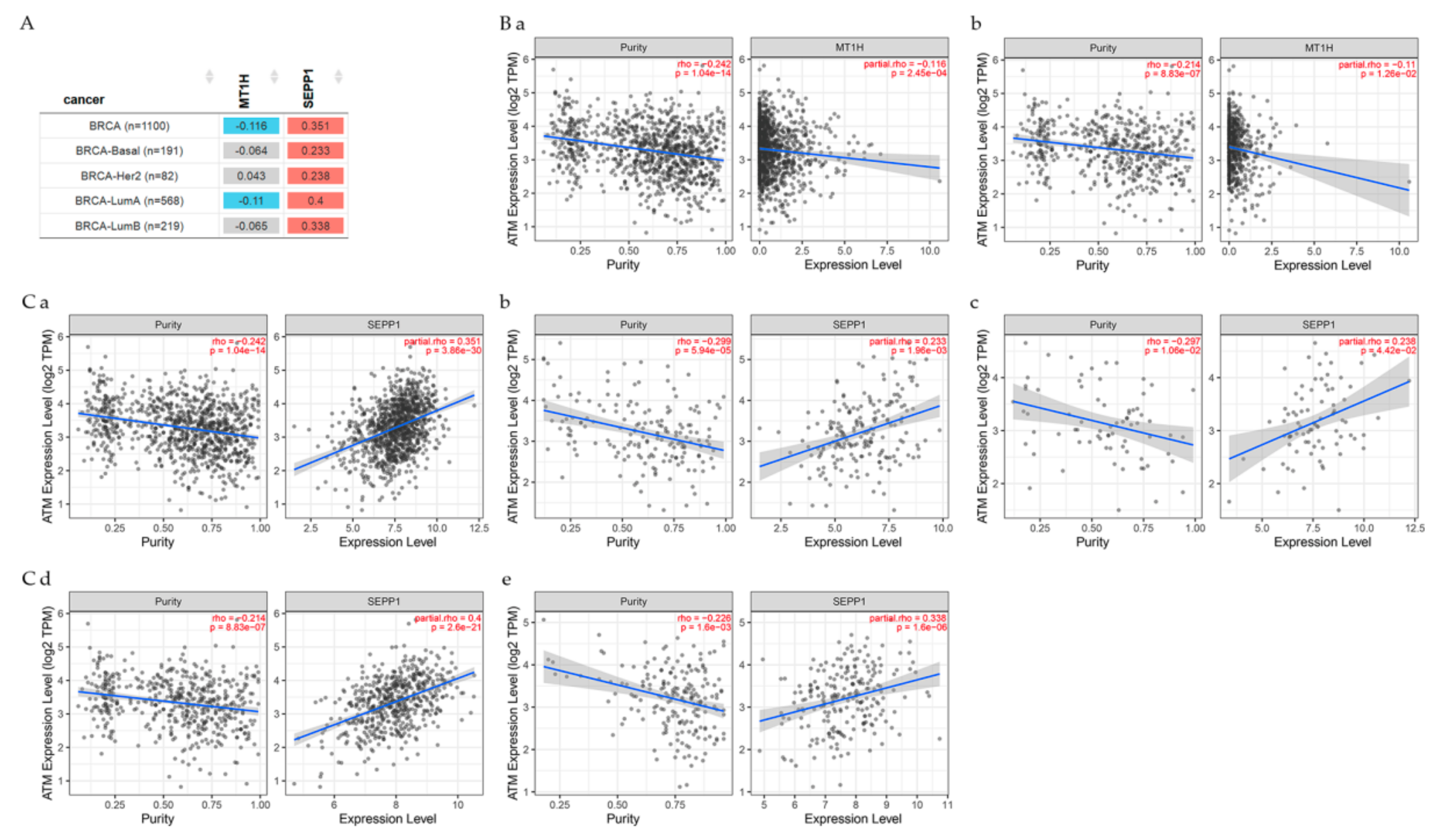
6.2. Selenoproteins (SEPP1)
6.3. GABA Receptor
6.4. Interleukins
6.5. Epsins 3
6.6. Stefin A (Cystatin A)
6.7. Metallothioneins
7. Relationship between Genes and Clinical Aspects
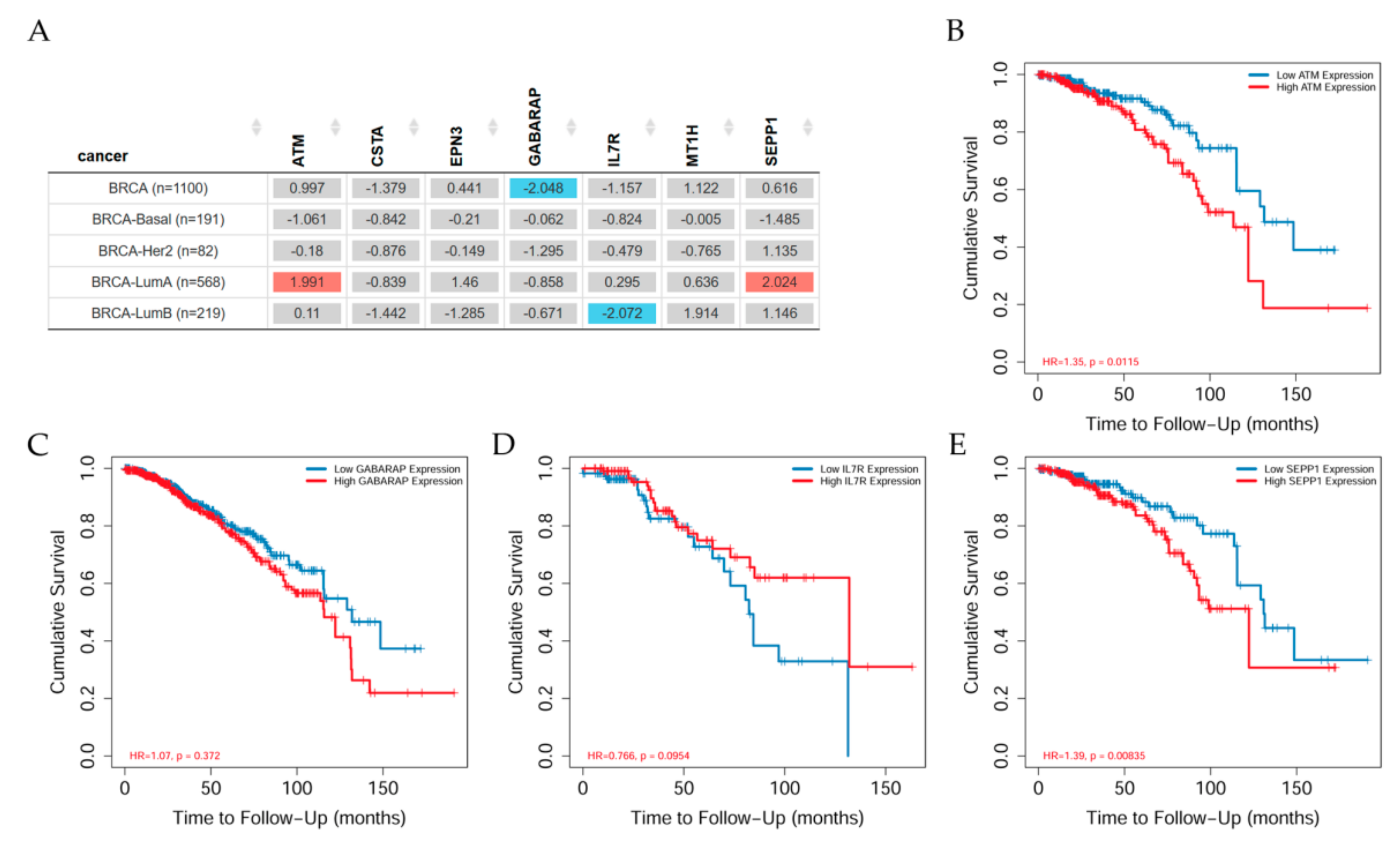
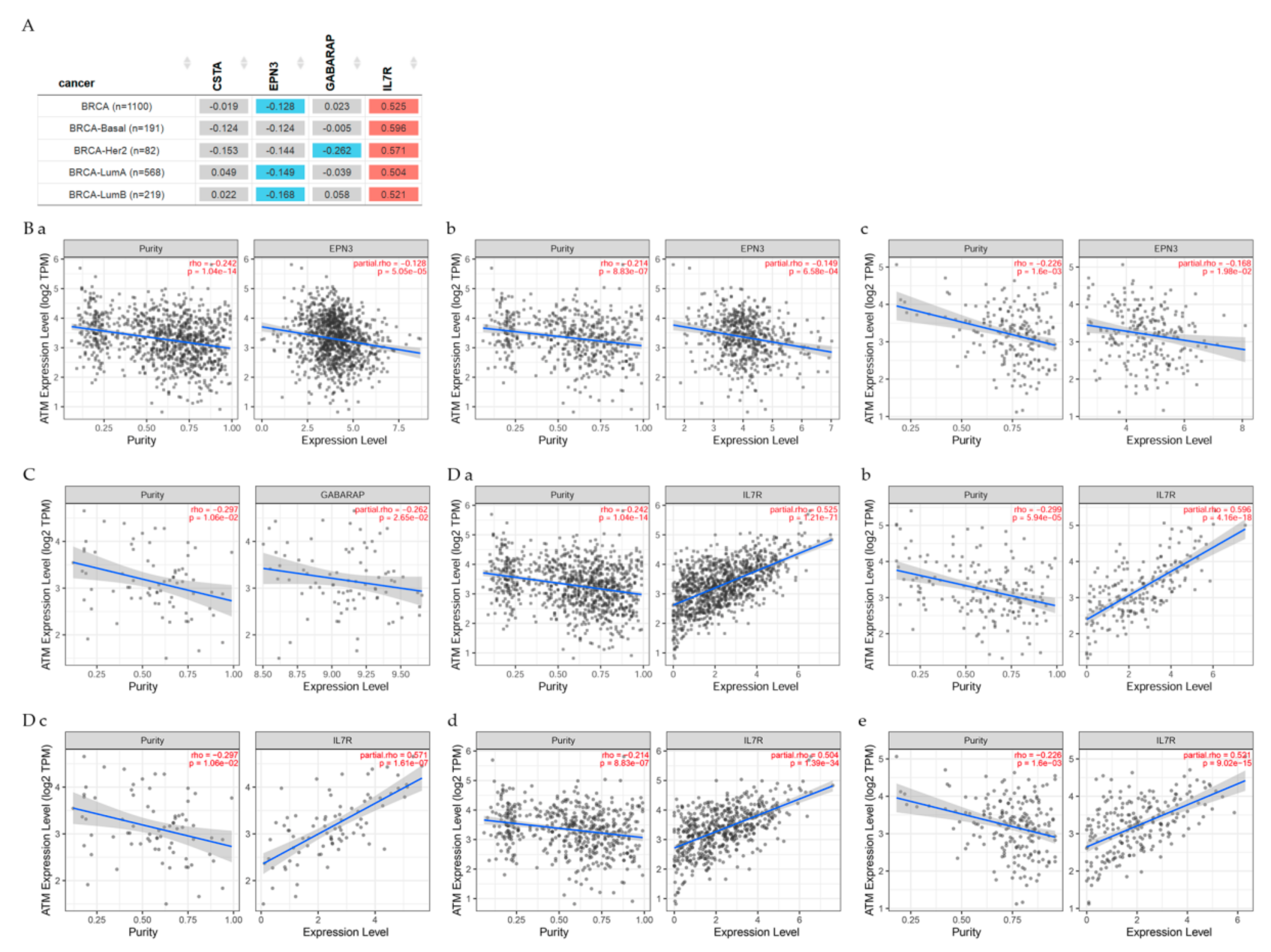
7.1. Genes Related to Clinical Relevance in Breast Cancer Patients
7.2. Gene Correlation between ATM and Other Genes
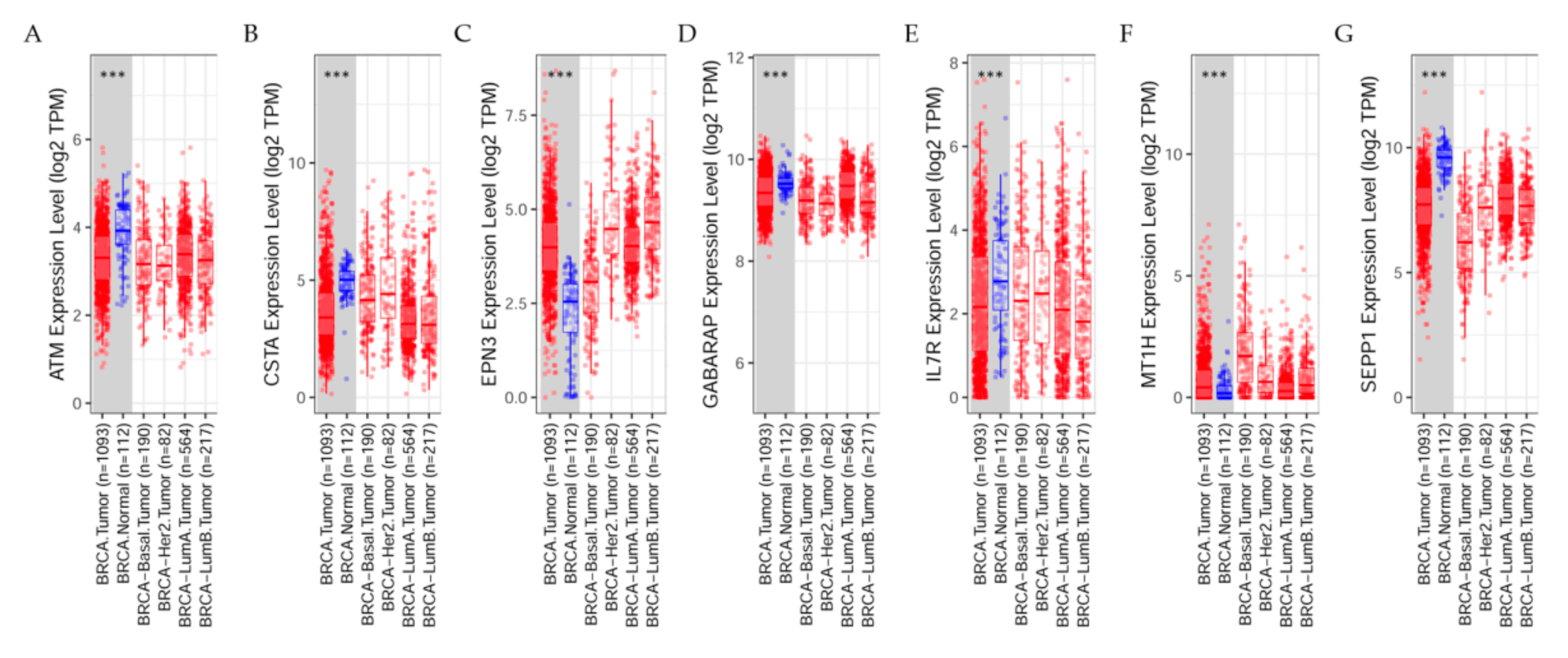
7.3. Differential Gene Expression Levels between Breast Tumors and Adjacent Normal Tissues across All TCGA Tumors
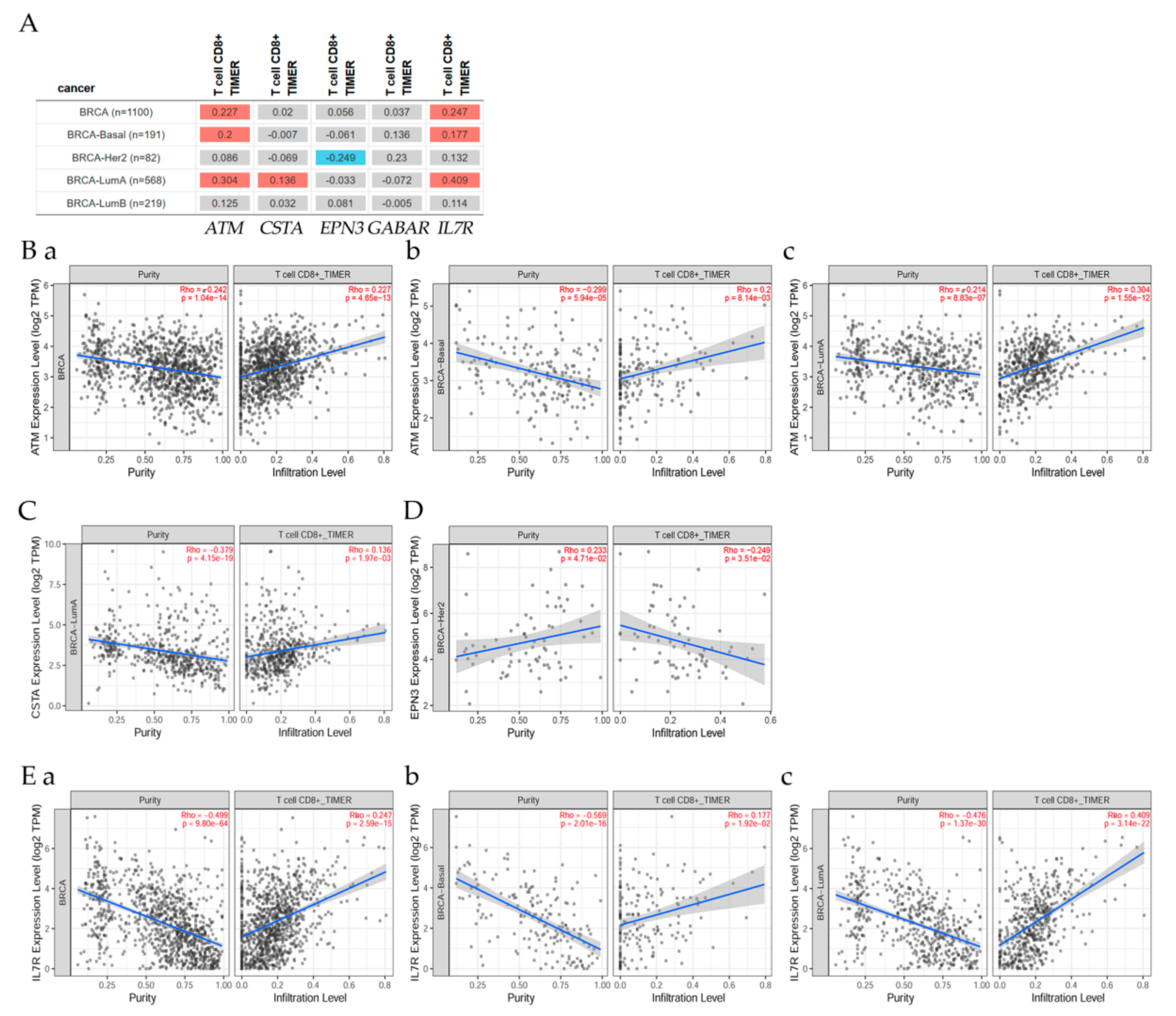
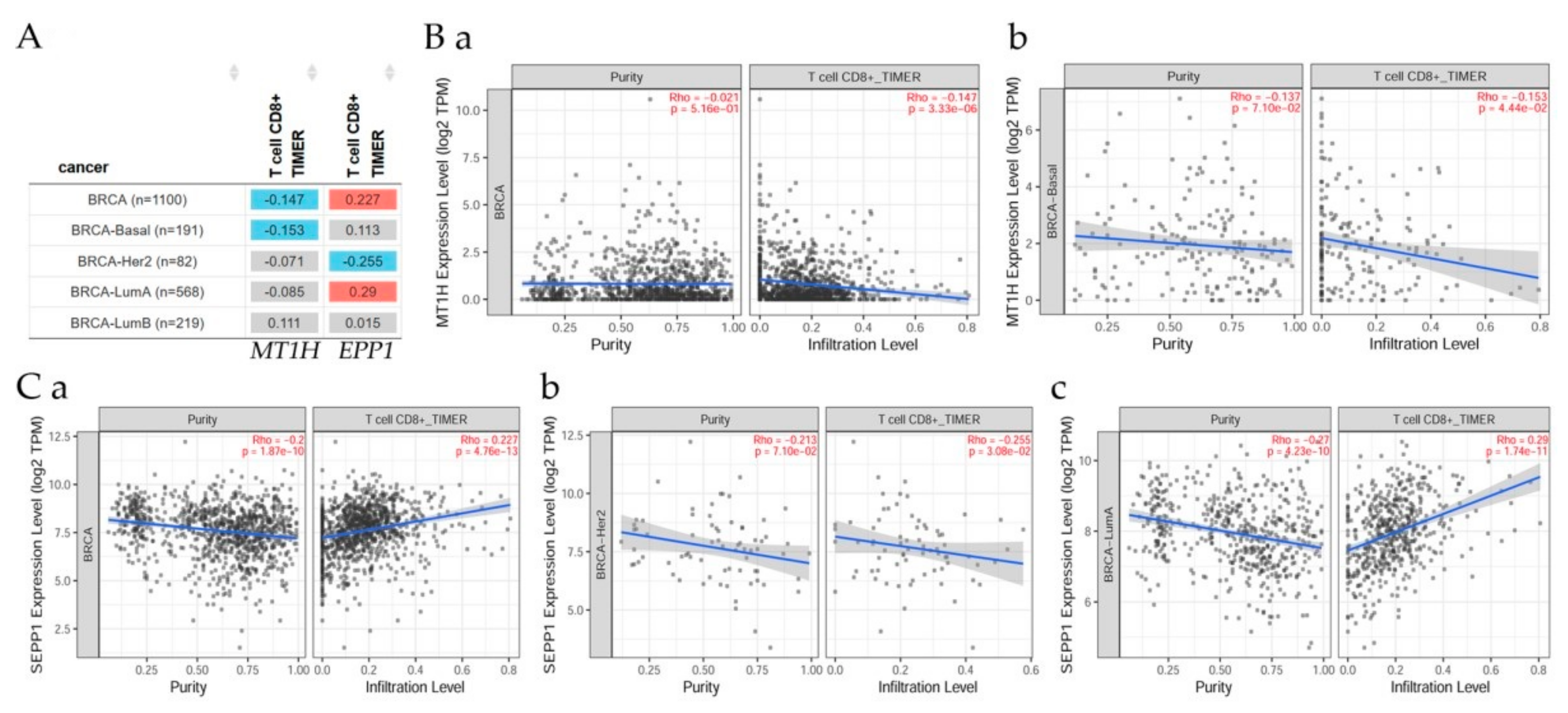
7.4. Correlation of ATM Gene and Its Expression with Immune Infiltration Level in Diverse Breast Cancer Types
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Health Physics Society (HPS): Radiation Fact Sheets. Available online: http://hps.org/hpspublications/radiationfactsheets.html (accessed on 8 April 2021).
- EPA. Radiation Basics. United States Environmental Protection Agency. Available online: https://www.epa.gov/radiation/radiation-basics (accessed on 10 May 2021).
- IARC. Non-ionizing radiation, part 2: Radiofrequency electromagnetic fields. In IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Monograph 102; International Agency for Research on Cancer: Lyon, France, 2012; pp. 1–481. [Google Scholar]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of Ionizing Radiation on Biological Molecules—Mechanisms of Damage and Emerging Methods of Detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Biological Effects of Ionizing Radiation (BEIR V). Genetic Effects of Radiation. In Health Effects of Exposure to Low Levels of Ionizing Radiation: BEIR V; National Research Council (US) Committee on the Biological Effects of Ionizing Radiation; National Academies Press (US): Washington, DC, USA, 1990; pp. 1–16. [Google Scholar]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.-W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- UNSCEAR. Effects of Ionizing Radiation: UNSCEAR 2006 Report; United Nations II Scientific Annexes C, D and E; United Nations Scientific Committee on the Effects of Atomic Radiation: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Mavragani, I.V.; Laskaratou, D.; Gika, V.; Moskvin, V.; Theofilatos, K.; Vougas, K.; Stewart, R.D.; Georgakilas, A.G. Systemic mechanisms and effects of ionizing radiation: A new old paradigm of how the bystanders and distant can become the players. Semin. Cancer Biol. 2016, 37–38, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Georgakilas, A.G.; Ravanat, J.-L. Targeted and Off-Target (Bystander and Abscopal) Effects of Radiation Therapy: Redox Mechanisms and Risk/Benefit Analysis. Antioxid. Redox Signal. 2018, 29, 1447–1487. [Google Scholar] [CrossRef]
- Ng, K.H. Non-Ionizing radiations—sources, biological effects, emissions and exposures. Electromagnetic Field sand Our Health. In Proceedings of the International Conferenceon Non-Ionizing Radiation at UNITEN(ICNIR2003), Kuala Lumpur, Malaysia, 20–22 October 2003. [Google Scholar]
- Baba, A.I.; Catoi, C. Carcinogenesis. In Comparative Oncology; The Publishing House of the Romanian: Bucharest, Romania, 2007. [Google Scholar]
- United Nations Scientific Committee on the Effects of Atomic Radiation. Sources and Effects of Ionizing Radiation: UNSCEAR 2008 Report to the General Assembly Scientific Annexes A and B; United Nations: New York, NY, USA, 2010; p. 683. [Google Scholar]
- Hendry, J.H.; Simon, S.L.; Wojcik, A.; Sohrabi, M.; Burkart, W.; Cardis, E.; Laurier, D.; Tirmarche, M.; Hayata, I. Human exposure to high natural background radiation: What can it teach us about radiation risks? J. Radiol. Prot. 2009, 29, A29–A42. [Google Scholar] [CrossRef]
- Parkin, D.M.; Darby, S.C. 12. Cancers in 2010 attributable to ionising radiation exposure in the UK. Br. J. Cancer 2011, 105, S57–S65. [Google Scholar] [CrossRef] [PubMed]
- Keith, S.; Doyle, J.R.; Harper, C.; Mumtaz, M.; Tarrago, O.; Wohlers, D.W.; Diamond, G.L.; Citra, M.; Barber, L.E. Toxicological Profile for Radon; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2012; p. 283. [Google Scholar]
- USNRC. Radiation Basics. United States Nuclear Regulatory Commission. Available online: https://www.nrc.gov/about-nrc/radiation/health-effects/radiation-basics.html (accessed on 10 May 2021).
- USNRC. Uses of Radiation. United States Nuclear Regulatory Commission. Available online: https://www.nrc.gov/about-nrc/radiation/around-us/uses-radiation.html (accessed on 10 May 2021).
- This Month in Physics History: November 8, 1895: Roentgen’s Discovery of X-rays. Available online: https://www.aps.org/publications/apsnews/200111/history.cfm (accessed on 6 August 2021).
- Bernstein, M.; Gutin, P.H. Interstitial Irradiation of Brain Tumors. Neurosurgery 1981, 9, 741–750. [Google Scholar] [CrossRef]
- Bravatà, V.; Cava, C.; Minafra, L.; Cammarata, F.P.; Russo, G.; Gilardi, M.C.; Castiglioni, I.; Forte, G.I. Radiation-Induced Gene Expression Changes in High and Low Grade Breast Cancer Cell Types. Int. J. Mol. Sci. 2018, 19, 1084. [Google Scholar] [CrossRef]
- Handa, E.; Puspitasari, I.M.; Abdulah, R.; Yamazaki, C.; Kameo, S.; Nakano, T.; Koyama, H. Recent advances in clinical studies of selenium supplementation in radiotherapy. J. Trace Elem. Med. Biol. 2020, 62, 126653. [Google Scholar] [CrossRef] [PubMed]
- Little, J.B.; Loeb, K.R.; Loeb, L.A. Radiation carcinogenesis. Carcinogenesis 2000, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.E.; Citrin, D.E.; Mitchell, J.B.; Mitchell, J.B. Mechanisms of Normal Tissue Injury From Irradiation. Semin. Radiat. Oncol. 2017, 27, 316–324. [Google Scholar] [CrossRef]
- Son, H.; Lee, S.M.; Yoon, R.G.; Lee, H.; Lee, I.; Kim, S.; Chung, W.Y.; Lee, J.W. Effect of selenium supplementation for protection of salivary glands from iodine-131 radiation damage in patients with differentiated thyroid cancer. Hell. J. Nucl. Med. 2017. [Google Scholar] [CrossRef]
- Lowe, D.J.; Herzog, M.; Mosler, T.; Cohen, H.; Felton, S.; Beli, P.; Raj, K.; Galanty, Y.; Jackson, S.P. Chronic irradiation of human cells reduces histone levels and deregulates gene expression. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Autsavapromporn, N.; Suzuki, M.; Funayama, T.; Usami, N.; Plante, I.; Yokota, Y.; Mutou, Y.; Ikeda, H.; Kobayashi, K.; Kobayashi, Y.; et al. Gap junction communication and the propagation of bystander effects induced by microbeam irradiation in human fibroblast cultures: The impact of radiation quality. Radiat. Res. 2013, 180, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Seth, I.; Schwartz, J.L.; Stewart, R.D.; Emery, R.; Joiner, M.C.; Tucker, J.D. Neutron Exposures in Human Cells: Bystander Effect and Relative Biological Effectiveness. PLoS ONE 2014, 9, e98947. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Chrystal, J.A.; Robinson, J.I.; Coates, P.; Wright, E.G. Chromosomal Instability in Unirradiated Hemaopoietic Cells Induced by Macrophages Exposed In vivo to Ionizing Radiation. Cancer Res. 2008, 68, 8122–8126. [Google Scholar] [CrossRef]
- Lyng, F.M.; Maguire, P.; Kilmurray, N.; Mothersill, C.; Shao, C.; Folkard, M.; Prise, K.M. Apoptosis is initiated in human keratinocytes exposed to signalling factors from microbeam irradiated cells. Int. J. Radiat. Biol. 2006, 82, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Rugo, R.; Hendricks, C.; Loree, J.; Thibault, B.; Kutanzi, K.; Pogribny, I.; Yanch, J.C.; Engelward, B.P.; Kovalchuk, O. Irradiation induces DNA damage and modulates epigenetic effectors in distant bystander tissue in vivo. Oncogene 2006, 25, 4267–4275. [Google Scholar] [CrossRef]
- Watson, G.; Lorimore, S.; Macdonald, D.; Wright, E.G. Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Res. 2000, 60, 5608–5611. [Google Scholar] [PubMed]
- Xue, L.Y.; Butler, N.J.; Makrigiorgos, G.M.; Adelstein, S.J.; Kassis, A.I. Bystander effect produced by radiolabeled tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.B.; Mothersill, C. Radiation-induced bystander effects—implications for cancer. Nat. Rev. Cancer 2004, 4, 158–164. [Google Scholar] [CrossRef]
- Eom, H.S.; Park, H.R.; Jo, S.K.; Kim, Y.S.; Moon, C.; Kim, S.-H.; Jung, U. Ionizing Radiation Induces Altered Neuronal Differentiation by mGluR1 through PI3K-STAT3 Signaling in C17.2 Mouse Neural Stem-Like Cells. PLoS ONE 2016, 11, e0147538. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.M.A.C. Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int. J. Radiat. Biol. 1997, 71, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C.B. Cell-Cell Contact during Gamma Irradiation Is Not Required to Induce a Bystander Effect in Normal Human Keratinocytes: Evidence for Release during Irradiation of a Signal Controlling Survival into the Medium. Radiat. Res. 1998, 149, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Marín, A.; Martín, M.; Liñán, O.; Alvarenga, F.; López, M.; Fernández, L.; Büchser, D.; Cerezo, L. Bystander effects and radiotherapy. Rep. Pr. Oncol. Radiother. 2015, 20, 12–21. [Google Scholar] [CrossRef]
- Maes, M.; Anderson, G.; Kubera, M.; Berk, M. Targeting classical IL-6 signalling or IL-6trans-signalling in depression? Expert Opin. Ther. Targets 2014, 18, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Vinken, M.; Rogiers, V.; Bukauskas, F.F.; Bultynck, G.; Leybaert, L. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 2013, 1828, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.; Neumann, R. Changes in gene expression as one of the key mechanisms involved in radiation-induced bystander effect (Review). Biomed. Rep. 2018, 9, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar]
- Azzam, E.; De Toledo, S.M.; Spitz, D.R.; Little, J.B. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from alpha-particle-irradiated normal human fibroblast cultures. Cancer Res. 2002, 62, 5436–5442. [Google Scholar]
- Lyng, F.; Seymour, C.B.; Mothersill, C. Production of a signal by irradiated cells which leads to a response in unirradiated cells characteristic of initiation of apoptosis. Br. J. Cancer 2000, 83, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-J.; Randers-Pehrson, G.; Xu, A.; Waldren, C.A.; Geard, C.R.; Yu, Z.; Hei, T.K. Targeted cytoplasmic irradiation with alpha particles induces mutations in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 4959–4964. [Google Scholar] [CrossRef]
- Zhou, H.; Randers-Pehrson, G.; Waldren, C.A.; Vannais, D.; Hall, E.J.; Hei, T.K. Induction of a bystander mutagenic effect of alpha particles in mammalian cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Sawant, S.G.; Randers-Pehrson, G.; Geard, C.R.; Brenner, D.J.; Hall, E.J. The bystander effect in radiation oncogenesis: I. Transformation in C3H 10T1/2 cells in vitro can be initiated in the unirradiated neighbors of irradiated cells. Radiat Res. 2001, 155, 397–401. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C.B.; Joiner, M.C. Relationship between Radiation-Induced Low-Dose Hypersensitivity and the Bystander Effect. Radiat. Res. 2002, 157, 526–532. [Google Scholar] [CrossRef]
- Mitchell, S.A.; Marino, S.A.; Brenner, D.J.; Hall, E.J. Bystander effect and adaptive response in C3H 10T½ cells. Int. J. Radiat. Biol. 2004, 80, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Ponnaiya, B.; Jenkins-Baker, G.; Brenner, D.J.; Hall, E.J.; Randers-Pehrson, G.; Geard, C.R. Biological responses in known bystander cells relative to known microbeam-irradiated cells. Radiat. Res. 2004, 162, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.V.; Smilenov, L.B.; Hall, E.J.; Panyutin, I.G.; Bonner, W.M.; Sedelnikova, O. Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene 2005, 24, 7257–7265. [Google Scholar] [CrossRef] [PubMed]
- Shuryak, I.; Brenner, D.J.; Ullrich, R.L. Radiation-Induced Carcinogenesis: Mechanistically Based Differences between Gamma-Rays and Neutrons, and Interactions with DMBA. PLoS ONE 2011, 6, e28559. [Google Scholar] [CrossRef] [PubMed]
- Heeran, A.B.; Berrigan, H.P.; O’Sullivan, J.N. The Radiation-Induced Bystander Effect (RIBE) and its Connections with the Hallmarks of Cancer. Radiat. Res. 2019, 192, 668. [Google Scholar] [CrossRef]
- Singh, G.K.; Yadav, V.; Singh, P.; Bhowmik, K.T. Radiation-Induced Malignancies Making Radiotherapy a “Two-Edged Sword”: A Review of Literature. World J. Oncol. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pierce, D.A.; Shimizu, Y.; Preston, D.L.; Vaeth, M.; Mabuchi, K. Studies of the Mortality of Atomic Bomb Survivors. Report 12, Part I. Cancer: 1950–1990. Radiat. Res. 1996, 146, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. Radiation Mutagenesis: The Initial DNA Lesions Responsible. Radiat. Res. 1995, 142, 362. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, K. Epidemiological research on radiation-induced cancer in atomic bomb survivors. J. Radiat. Res. 2016, 57, i112–i117. [Google Scholar] [CrossRef]
- Best, T.; Li, D.; Skol, A.D.; Kirchhoff, T.; Jackson, S.; Yasui, Y.; Bhatia, S.; Strong, L.C.; Domchek, S.M.; Nathanson, K.; et al. Variants at 6q21 implicate PRDM1 in the etiology of therapy-induced second malignancies after Hodgkin’s lymphoma. Nat. Med. 2011, 17, 941–943. [Google Scholar] [CrossRef]
- Cahan, W.G.; Woodard, H.Q.; Higinbotham, N.L.; Stewart, F.W.; Coley, B.L. Sarcoma arising in irradiated bone: Report of eleven cases. Cancer 1948, 1, 3–29. [Google Scholar] [CrossRef]
- IARC. Ionizing radiation, Part I, X- and gamma (y)-radiation, and neutrons. In IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Monograph 75; International Agency for Research on Cancer: Lyon, France, 2000; pp. 1–448. [Google Scholar]
- IARC. Ionizing radiation, part 2: Some internally deposited radionuclides. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; Monograph 78; International Agency for Research on Cancer: Lyon, France, 2001; pp. 1–559. [Google Scholar]
- Fry, R.; Storer, J.B. External Radiation Carcinogenesis1 1This research was sponsored jointly by the Office of Health and Environmental Research, U.S. Department of Energy, under contract DE-AC05–840R21400 with the Martin Marietta Energy Systems, Inc., and National Cancer Institute Contracts Y-l-CM-20112 and Y-l-CM-20113. Adv. Radiat. Biol. 1987, 13, 31–90. [Google Scholar] [CrossRef]
- Chadwick, K.H.; Seymour, C.; Barnhart, B. Cell Transformation and Radiation-induced Cancer; Adam Hikger: Bristol, NY, USA, 1989; pp. 1–414. [Google Scholar]
- Upton, A.C. Biological aspects of radiation carcinogenesis. In Radiation Carcinogenesis: Epidemiology and Biological Significance; Boice, J.D., Fraumeni, J.F., Eds.; Raven Press: New York, NY, USA, 1984; pp. 9–20. [Google Scholar]
- Edimecheva, M.A.K.I.P. The damage to phospholipids caused by free radical attack on glycerol and sphingosine backbone. Int. J. Radiat. Biol. 1997, 71, 555–560. [Google Scholar] [CrossRef]
- Shao, C.; Folkard, M.; Michael, B.D.; Prise, K.M. Targeted cytoplasmic irradiation induces bystander responses. Proc. Natl. Acad. Sci. USA 2004, 101, 13495–13500. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Kosova, F.; Ari, Z.; Sakarya, A.; Kaya, Y. Effect of oncological treatment on serum adipocytokine levels in patients with stage II–III breast cancer. Mol. Clin. Oncol. 2016, 4, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Scott, R.E.; Wille, J.J., Jr.; Wier, M.L. Mechanisms for the initiation and promotion of carcinogenesis: A review and a new concept. Mayo Clin. Proc. 1984, 59, 107–117. [Google Scholar] [CrossRef]
- Berenblum, I. Sequential aspects of chemical carcinogenesis: Skin. In Cancer: A Comprehensive Treatise, 2nd ed.; Becker, F.F., Ed.; Plenum Press: New York, NY, USA, 1982; Volume I, pp. 451–484. [Google Scholar]
- Anderson, M.W.; Reynolds, S.H.; You, M.; Maronpot, R.M. Role of proto-oncogene activation in carcinogenesis. Environ. Health Perspect. 1992, 98, 13–24. [Google Scholar] [CrossRef]
- Bishop, J. Molecular themes in oncogenesis. Cell 1991, 64, 235–248. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Msaouel, P.; Pissimissis, N.; Halapas, A.; Koutsilieris, M. Mechanisms of bone metastasis in prostate cancer: Clinical implications. Best Pr. Res. Clin. Endocrinol. Metab. 2008, 22, 341–355. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef]
- Souhami, R.L.; Tobias, J.S. Cancer and Its Management, 4th ed.; Blackwell Science: Malden, MA, USA, 2003; p. 624. [Google Scholar]
- Tang, Z.; Zeng, Q.; Li, Y.; Zhang, X.; Ma, J.; Suto, M.J.; Xu, B.; Yi, N. Development of a radiosensitivity gene signature for patients with soft tissue sarcoma. Oncotarget 2017, 8, 27428–27439. [Google Scholar] [CrossRef] [PubMed]
- Gerszten, P.C.; Mendel, E.; Yamada, Y. Radiotherapy and Radiosurgery for Metastatic Spine Disease. Spine 2009, 34, S78–S92. [Google Scholar] [CrossRef]
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Nehs, M.A.; Lin, C.-I.; Kozono, D.E.; Whang, E.E.; Cho, N.L.; Zhu, K.; Moalem, J.; Moore, F.D.; Ruan, D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011, 150, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, M.; Bhatt, A.N.; Das, A.; Dwarakanath, B.S.; Sharma, K. Radiation-induced autophagy: Mechanisms and consequences. Free Radic. Res. 2016, 50, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Walden, T.L.; Hughes, H.N. Prostaglandin and Lipid Metabolism in Radiation Injury, 1st ed.; Springer US: Boston, MA, USA, 1987; pp. 1–420. [Google Scholar]
- Dixon, S.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.F.; Chaudhary, K.R.; Zandkarimi, F.; Harken, A.D.; Kinslow, C.J.; Upadhyayula, P.S.; Dovas, A.; Higgins, D.M.; Tan, H.; Zhang, Y.; et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem. Biol. 2020, 15, 469–484. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Freitas, F.P.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2017, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Fujii, J. Cysteine preservation confers resistance to glutathione-depleted cells against ferroptosis via CDGSH iron sulphur domain-containing proteins (CISDs). Free Radic. Res. 2020, 54, 397–407. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; De Toledo, S.M.; Gooding, T.; Little, J.B. Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat. Res. 1998, 150, 497. [Google Scholar] [CrossRef]
- Azzam, E.I.; de Toledo, S.M.; Little, J.B. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha -particle irradiated to nonirradiated cells. Proc. Natl. Acad. Sci. USA 2001, 98, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.; De Toledo, S.M.; Little, J.B. Expression of CONNEXIN43 is highly sensitive to ionizing radiation and other environmental stresses. Cancer Res. 2003, 63, 7128–7135. [Google Scholar]
- Olsson, M.G.; Nilsson, E.J.C.; Rutardóttir, S.; Paczesny, J.; Pallon, J.; Åkerström, B. Bystander Cell Death and Stress Response is Inhibited by the Radical Scavenger α1-Microglobulin in Irradiated Cell Cultures. Radiat. Res. 2010, 174, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Herok, R.; Konopacka, M.; Polanska, J.; Swierniak, A.; Rogolinski, J.; Jaksik, R.; Hancock, R.; Rzeszowska-Wolny, J. Bystander Effects Induced by Medium From Irradiated Cells: Similar Transcriptome Responses in Irradiated and Bystander K562 Cells. Int. J. Radiat. Oncol. 2010, 77, 244–252. [Google Scholar] [CrossRef]
- Facoetti, A.; Ballarini, F.; Cherubini, R.; Gerardi, S.; Nano, R.; Prise, K.M.; Trott, K.R.; Ottolenghi, A.; Zilio, C. Gamma ray-induced bystander effect in tumour glioblastoma cells: A specific study on cell survival, cytokine release and cytokine receptors. Radiat. Prot. Dosim. 2006, 122, 271–274. [Google Scholar] [CrossRef]
- Tian, W.; Yin, X.; Wang, L.; Wang, J.; Zhu, W.; Cao, J.; Yang, H. The key role of miR-21-regulated SOD2 in the medium-mediated bystander responses in human fibroblasts induced by α-irradiated keratinocytes. Mutat. Res. Mol. Mech. Mutagen. 2015, 780, 77–85. [Google Scholar] [CrossRef]
- Chaudhry, M.A.; Omaruddin, R.A. Differential regulation of MicroRNA expression in irradiated and bystander cells. Mol. Biol. 2012, 46, 569–578. [Google Scholar] [CrossRef]
- Hu, W.; Xu, S.; Yao, B.; Hong, M.; Wu, X.; Pei, H.; Chang, L.; Ding, N.; Gao, X.; Ye, C.; et al. MiR-663 inhibits radiation-induced bystander effects by targetingTGFB1in a feedback mode. RNA Biol. 2014, 11, 1189–1198. [Google Scholar] [CrossRef]
- Xu, S.; Ding, N.; Pei, H.; Hu, W.; Wei, W.; Zhang, X.; Zhou, G.; Wang, J. MiR-21 is involved in radiation-induced bystander effects. RNA Biol. 2014, 11, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Ghandhi, S.; Sinha, A.; Markatou, M.; Amundson, S. Time-series clustering of gene expression in irradiated and bystander fibroblasts: An application of FBPA clustering. BMC Genom. 2011, 12, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, A.; Mychasiuk, R.; Muhammad, A.; Hossain, S.; Ilnytskyy, S.; Ghose, A.; Kirkby, C.; Ghasroddashti, E.; Kovalchuk, O.; Kolb, B. Liver irradiation causes distal bystander effects in the rat brain and affects animal behaviour. Oncotarget 2015, 7, 4385–4398. [Google Scholar] [CrossRef] [PubMed]
- Aravindan, N.; Natarajan, M.; Ramraj, S.K.; Pandian, V.; Khan, F.H.; Herman, T.S. Abscopal effect of low-LET γ-radiation mediated through Rel protein signal transduction in a mouse model of nontargeted radiation response. Cancer Gene Ther. 2013, 21, 54–59. [Google Scholar] [CrossRef]
- Krolewski, B.; Little, J. Alterations of MDM2 gene in X-ray transformed mouse 10t1/2 cell clones. Int. J. Oncol. 1995, 6, 1123–1127. [Google Scholar] [CrossRef]
- Murnane, J.P. Cell cycle regulation in response to DNA damage in mammalian cells: A historical perspective. Cancer Metastasis Rev. 1995, 14, 17–29. [Google Scholar] [CrossRef]
- Li, C.Y.; Nagasawa, H.; Dahlberg, W.K.; Little, J.B. Diminished capacity for p53 in mediating a radiation-induced G1 arrest in established human tumor cell lines. Oncogene 1995, 11, 1885–1892. [Google Scholar]
- Syljuåsen, R.G.; Krolewski, B.; Little, J.B. Loss of normal G1 checkpoint control is an early step in carcinogenesis, independent of p53 status. Cancer Res. 1999, 59, 1008–1014. [Google Scholar]
- Suetens, A.; Moreels, M.; Quintens, R.; Chiriotti, S.; Tabury, K.; Michaux, A.; Grégoire, V.; Baatout, S. Carbon ion irradiation of the human prostate cancer cell line PC3: A whole genome microarray study. Int. J. Oncol. 2014, 44, 1056–1072. [Google Scholar] [CrossRef]
- Iwadate, Y.; Mizoe, J.-E.; Osaka, Y.; Yamaura, A.; Tsujii, H. High linear energy transfer carbon radiation effectively kills cultured glioma cells with either mutant or wild-type p53. Int. J. Radiat. Oncol. 2001, 50, 803–808. [Google Scholar] [CrossRef]
- Wada, S.; Kobayashi, Y.; Funayama, T.; Natsuhori, M.; Ito, N.; Yamamoto, K. Detection of DNA damage in individual cells induced by heavy-ion irradiation with an non-denaturing comet assay. J. Radiat. Res. 2002, 43, S153–S156. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N. Recent Insights into the Biological Action of Heavy-Ion Radiation. J. Radiat. Res. 2009, 50, 1–9. [Google Scholar] [CrossRef]
- Ishikawa, H.; Tsuji, H.; Kamada, T.; Akakura, K.; Suzuki, H.; Shimazaki, J.; Tsujii, H.; The Working Group for Genitourinary Tumors. Carbon-ion radiation therapy for prostate cancer. Int. J. Urol. 2012, 19, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, R.; Fossati, P.; Rossi, S. The National Center for Oncological Hadron Therapy: Status of the Project and Future Clinical use of the Facility. Tumori J. 2009, 95, 169–176. [Google Scholar] [CrossRef]
- Sedlmayer, F.; Reitsamer, R.; Wenz, F.; Sperk, E.; Fussl, C.; Kaiser, J.; Ziegler, I.; Zehentmayr, F.; Deutschmann, H.; Kopp, P.; et al. Intraoperative radiotherapy (IORT) as boost in breast cancer. Radiat. Oncol. 2017, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.E.R.; Small, W.J. Intraoperative Radiotherapy for Breast Cancer. Front. Oncol. 2017, 7, 317. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Cairns, J.; Little, J.B. Timing of the steps in transformation of C3H 10T½ cells by X-irradiation. Nature 1984, 307, 85–86. [Google Scholar] [CrossRef]
- Port, M.; Boltze, C.; Wang, Y.; Röper, B.; Meineke, V.; Abend, M. A Radiation-Induced Gene Signature Distinguishes Post-Chernobyl from Sporadic Papillary Thyroid Cancers. Radiat. Res. 2007, 168, 639–649. [Google Scholar] [CrossRef]
- Collins, B.J.; Schneider, A.B.; Prinz, R.A.; Xu, X. Low Frequency of BRAF Mutations in Adult Patients with Papillary Thyroid Cancers Following Childhood Radiation Exposure. Thyroid 2006, 16, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, D.; Michel, L.; Rosiere, A.; Trigaux, J.-P.; Donckier, J. Occurrence of Thyroid Papillary Carcinoma in Young Patients. A Chernobyl Connection? J. Pediatr. Endocrinol. Metab. 2001, 14, 503–506. [Google Scholar] [CrossRef]
- Pacini, F.; Vorontsova, T.; Molinaro, E.; Shavrova, E.; Agate, L.; Kuchinskaya, E.; Elisei, R.; Demidchik, E.P.; Pinchera, A. Thyroid consequences of the Chernobyl nuclear accident. Acta Paediatr. 1999, 88, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, L.; Grosclaude, P.; Chérié-Challine, L. Increased Incidence of Thyroid Carcinoma in France: A True Epidemic or Thyroid Nodule Management Effects? Report from the French Thyroid Cancer Committee. Thyroid 2004, 14, 1056–1060. [Google Scholar] [CrossRef]
- Chernobyl, P. Chernobyl Accident: Regional and Global Impacts. Environ. Int. 1988, 14, 69–73. [Google Scholar] [CrossRef]
- Scarpino, S.; D’Alena, F.C.; Di Napoli, A.; Ballarini, F.; Prat, M.; Ruco, L.P. Papillary carcinoma of the thyroid: Evidence for a role for hepatocyte growth factor (HGF) in promoting tumour angiogenesis. J. Pathol. 2003, 199, 243–250. [Google Scholar] [CrossRef]
- Pasieka, Z.; Stepień, H.; Komorowski, J.; Kołomecki, K.; Kuzdak, K. Evaluation of the levels of bFGF, VEGF, sICAM-1, and sVCAM-1 in serum of patients with thyroid cancer. Recent Results Cancer Res. 2003, 162, 189–194. [Google Scholar] [CrossRef]
- Yu, X.-M.; Lo, C.-Y.; Chan, W.-F.; Lam, A.; Leung, P.; Luk, J. Increased Expression of Vascular Endothelial Growth Factor C in Papillary Thyroid Carcinoma Correlates with Cervical Lymph Node Metastases. Clin. Cancer Res. 2005, 11, 8063–8069. [Google Scholar] [CrossRef]
- Siironen, P.; Louhimo, J.; Nordling, S.; Ristimäki, A.; Mäenpää, H.; Haapiainen, R.; Haglund, C. Prognostic Factors in Papillary Thyroid Cancer: An Evaluation of 601 Consecutive Patients. Tumor Biol. 2005, 26, 57–64. [Google Scholar] [CrossRef]
- Kameyama, K. Expression of MMP-1 in the Capsule of Thyroid Cancer—Relationship with Invasiveness. Pathol. Res. Pr. 1996, 192, 20–26. [Google Scholar] [CrossRef]
- Cao, L.-L.; Shen, C.; Zhu, W.-G. Histone modifications in DNA damage response. Sci. China Life Sci. 2016, 59, 257–270. [Google Scholar] [CrossRef]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Miller, K.M.; Jackson, S.P. Histone marks: Repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans. 2012, 40, 370–376. [Google Scholar] [CrossRef]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2011, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Hei, T.K. Establishment of a radiation- and estrogen-induced breast cancer model. Carcinogenesis 2000, 21, 769–776. [Google Scholar] [CrossRef]
- Calaf, G.; Russo, J. Transformation of human breast epithelial cells by chemical carcinogens. Carcinogenesis 1993, 14, 483–492. [Google Scholar] [CrossRef]
- Calaf, G.M.; Hei, T.K. Ionizing radiation induces alterations in cellular proliferation and c-myc, c-jun and c-fos protein expression in breast epithelial cells. Int. J. Oncol. 2004, 25, 1859–1866. [Google Scholar] [CrossRef]
- Roy, D.; Calaf, G.; Hei, T.K. Profiling of differentially expressed genes induced by high linear energy transfer radiation in breast epithelial cells. Mol. Carcinog. 2001, 31, 192–203. [Google Scholar] [CrossRef]
- Calaf, G.; Zhang, P.; Alvarado, M.; Estrada, S.; Russo, J. C-HA-RAS enhances the neoplastic transformation of human breast epithelial-cells treated with chemical carcinogens. Int. J. Oncol. 1995, 6, 5–11. [Google Scholar] [CrossRef]
- Russo, J.; Calaf, G.; Russo, I.H. A critical approach to the malignant transformation of human breast epithelial cells with chemical carcinogens. Crit. Rev. Oncog. 1993, 4, 403–417. [Google Scholar] [PubMed]
- Calaf, G.; Russo, J.; Tait, L.; Estrad, S.; Alvarado, M. Morphological phenotypes in neoplastic progression of human breast epithelial cells. J. Submicrosc. Cytol. Pathol. 2000, 32, 83–96. [Google Scholar] [PubMed]
- Calaf, G.; Russo, J.; Alvarado, M.E. Morphological phenotypes in neoplastic progression of benz(alpha)pyrene-treated breast epithelial cells. J. Submicrosc. Cytol. Pathol. 2000, 32, 535–545. [Google Scholar] [PubMed]
- Stampfer, M.; Hallowes, R.C.; Hackett, A.J. Growth of normal human mammary cells in culture. Vitr. Cell. Dev. Biol. Anim. 1980, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kakunaga, T. Neoplastic transformation of human diploid fibroblast cells by chemical carcinogens. Proc. Natl. Acad. Sci. USA 1978, 75, 1334–1338. [Google Scholar] [CrossRef]
- Sharungbam, G.D.; Schwager, C.; Chiblak, S.; Brons, S.; Hlatky, L.; Haberer, T.; Debus, J.; Abdollahi, A. Identification of stable endogenous control genes for transcriptional profiling of photon, proton and carbon-ion irradiated cells. Radiat. Oncol. 2012, 7, 70. [Google Scholar] [CrossRef]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar]
- Zhou, Y.-C.; Liu, J.-Y.; Li, J.; Zhang, J.; Xu, Y.-Q.; Zhang, H.-W.; Qiu, L.-B.; Ding, G.-R.; Su, X.-M.; Shi, M.; et al. Ionizing Radiation Promotes Migration and Invasion of Cancer Cells Through Transforming Growth Factor-Beta–Mediated Epithelial–Mesenchymal Transition. Int. J. Radiat. Oncol. 2011, 81, 1530–1537. [Google Scholar] [CrossRef]
- Fujita, M.; Otsuka, Y.; Yamada, S.; Iwakawa, M.; Imai, T. X-ray irradiation and Rho-kinase inhibitor additively induce invasiveness of the cells of the pancreatic cancer line, MIAPaCa-2, which exhibits mesenchymal and amoeboid motility. Cancer Sci. 2011, 102, 792–798. [Google Scholar] [CrossRef]
- Sofia Vala, I.; Martins, L.R.; Imaizumi, N.; Nunes, R.J.; Rino, J.; Kuonen, F.; Carvalho, L.M.; Ruegg, C.; Grillo, I.M.; Barata, J.T.; et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS ONE 2010, 5, e11222. [Google Scholar] [CrossRef] [PubMed]
- Pickhard, A.C.; Margraf, J.; Knopf, A.; Stark, T.; Piontek, G.; Beck, C.; Boulesteix, A.-L.; Scherer, E.Q.; Pigorsch, S.; Schlegel, J.; et al. Inhibition of radiation induced migration of human head and neck squamous cell carcinoma cells by blocking of EGF receptor pathways. BMC Cancer 2011, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Roy, D.; Narayan, G.; Balajee, A.S. Differential expression of cell adhesion molecules in an ionizing radiation-induced breast cancer model system. Oncol. Rep. 2013, 30, 285–291. [Google Scholar] [CrossRef]
- Angèle, S.; Treilleux, I.; Tanière, P.; Martel-Planche, G.; Vuillaume, M.; Bailly, C.; Brémond, A.; Montesano, R.; Hall, J. Abnormal expression of the ATM and TP53 genes in sporadic breast carcinomas. Clin. Cancer Res. 2000, 6, 3536–3544. [Google Scholar]
- Prokopcova, J.; Kleibl, Z.; Banwell, C.M.; Pohlreich, P. The role of ATM in breast cancer development. Breast Cancer Res. Treat. 2006, 104, 121–128. [Google Scholar] [CrossRef]
- Cuatrecasas, M.; Santamaria, G.; Velasco, M.; Camacho, E.; Hernandez, L.; Sanchez, M.; Orrit, C.; Murcia, C.; Cardesa, A.; Campo, E.; et al. ATM gene expression is associated with differentiation and angiogenesis in infiltrating breast carcinomas. Histol. Histopathol. 2005, 21, 149–156. [Google Scholar] [CrossRef]
- Kitagawa, R.; Kastan, M. The ATM-dependent DNA Damage Signaling Pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 99–109. [Google Scholar] [CrossRef]
- Teive, H.A.; Moro, A.; Moscovich, M.; Arruda, W.O.; Munhoz, R.P.; Raskin, S.; Ashizawa, T. Ataxia-telangiectasia—A historical review and a proposal for a new designation: ATM syndrome. J. Neurol. Sci. 2015, 355, 3–6. [Google Scholar] [CrossRef]
- Thompson, D.; Duedal, S.; Kirner, J.; McGuffog, L.; Last, J.; Reiman, A.; Byrd, P.; Taylor, M.; Easton, D.F. Cancer Risks and Mortality in Heterozygous ATM Mutation Carriers. J. Natl. Cancer Inst. 2005, 97, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Furtado, S.; Das, S.; Suchowersky, O. A review of the inherited ataxias: Recent advances in genetic, clinical and neuropathologic aspects. Park. Relat. Disord. 1998, 4, 161–169. [Google Scholar] [CrossRef]
- Morrell, D.; Cromartie, E.; Swift, M. Mortality and Cancer Incidence in 263 Patients With Ataxia-Telangiectasia2. J. Natl. Cancer Inst. 1986, 77, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Lang, A.E. Ataxia-telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Mov. Disord. 2018, 33, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Swift, M. Mortality rates among carriers of ataxia-telangiectasia mutant alleles. Ann. Intern. Med. 2000, 133, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Dombernowsky, S.L.; Weischer, M.; Allin, K.H.; Bojesen, S.E.; Tybjjrg-Hansen, A.; Nordestgaard, B.G. Risk of Cancer by ATM Missense Mutations in the General Population. J. Clin. Oncol. 2008, 26, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.; Reitnauer, P.J.; Morrell, D.; Chase, C.L. Breast and Other Cancers in Families with Ataxia-Telangiectasia. N. Engl. J. Med. 1987, 316, 1289–1294. [Google Scholar] [CrossRef]
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583. [Google Scholar]
- Swift, M.; Morrell, D.; Massey, R.B.; Chase, C.L. Incidence of Cancer in 161 Families Affected by Ataxia–Telangiectasia. N. Engl. J. Med. 1991, 325, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Chessa, L.; Lisa, A.; Fiorani, O.; Zei, G. Ataxia-telangiectasia in Italy: Genetic analysis. Int. J. Radiat. Biol. 1994, 66, S31–S33. [Google Scholar] [CrossRef]
- Renwick, A.; The Breast Cancer Susceptibility Collaboration (UK); Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef]
- Van Os, N.; Roeleveld, N.; Weemaes, C.; Jongmans, M.; Janssens, G.O.R.J.; Taylor, A.; Hoogerbrugge, N.; Willemsen, M. Health risks for ataxia-telangiectasia mutated heterozygotes: A systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Marabelli, M.; Cheng, S.-C.; Parmigiani, G. Penetrance ofATMGene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet. Epidemiol. 2016, 40, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Foroughizadeh, M.; Mozdarani, H.; Majidzadeh-A, K.; Kaviani, A. Variation of ATM Gene Expression in Peripheral Blood Cells of Sporadic Breast Carcinomas in Iranian Patients. Avicenna J. Med. Biotechnol. 2012, 4, 95–101. [Google Scholar] [PubMed]
- Vo, Q.N.; Kim, W.-J.; Cvitanovic, L.; Boudreau, D.A.; Ginzinger, D.G.; Brown, K.D. The ATM gene is a target for epigenetic silencing in locally advanced breast cancer. Oncogene 2004, 23, 9432–9437. [Google Scholar] [CrossRef]
- Weissberg, J.B.; Huang, D.-D.; Swift, M. Radiosensitivity of normal tissues in ataxia-telangiectasia heterozygotes. Int. J. Radiat. Oncol. 1998, 42, 1133–1136. [Google Scholar] [CrossRef]
- Kryukov, G.; Castellano, S.; Novoselov, S.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Marciel, M.P.; Hoffmann, P.R. Selenoproteins and Metastasis. Adv. Cancer Res. 2017, 136, 85–108. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Gundimeda, U. Protein Kinase C as a Molecular Target for Cancer Prevention by Selenocompounds. Nutr. Cancer 2001, 40, 55–63. [Google Scholar] [CrossRef]
- Hosseinimehr, S.J. The protective effects of trace elements against side effects induced by ionizing radiation. Radiat. Oncol. J. 2015, 33, 66–74. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Peter, S. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef]
- Wu, W.-S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Muecke, R.; Schomburg, L.; Buentzel, J.; Kisters, K.; Micke, O.; German Working Group Trace Elements and Electrolytes in Oncology-AKTE. Selenium or No Selenium- That Is the Question in Tumor Patients: A New Controversy. Integr. Cancer Ther. 2010, 9, 136–141. [Google Scholar] [CrossRef]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346 Pt 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Capitanio, A.; Selenius, M.; Brodin, O.; Rundlöf, A.-K.; Björnstedt, M. Expression profiles of thioredoxin family proteins in human lung cancer tissue: Correlation with proliferation and differentiation. Histopathology 2009, 55, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Kahlos, K.; Näpänkangas, U.; Kaarteenaho-Wiik, R.; Säily, M.; Koistinen, P.; Pääakkö, P.; Holmgren, A.; Kinnula, V.L. Widespread expression of thioredoxin and thioredoxin reductase in non-small cell lung carcinoma. Clin. Cancer Res. 2001, 7, 1750–1757. [Google Scholar] [PubMed]
- Cañas, A.; López-Sánchez, L.M.; Valverde-Estepa, A.; Hernández, V.; Fuentes, E.; Muñoz-Castañeda, J.R.; López-Pedrera, C.; Rodríguez, J.R.D.L.H.; Aranda, E.; Rodríguez-Ariza, A. Maintenance of S-nitrosothiol homeostasis plays an important role in growth suppression of estrogen receptor-positive breast tumors. Breast Cancer Res. 2012, 14, R153. [Google Scholar] [CrossRef]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44. [Google Scholar] [CrossRef]
- Esen, H.; Erdi, F.; Kaya, B.; Feyzioglu, B.; Keskin, F.; Demir, L.S.; Feyzioglu, B. Tissue thioredoxin reductase-1 expression in astrocytomas of different grades. J. Neuro-Oncol. 2014, 121, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2015, 8, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer. Ther. 2016, 17, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Tsomides, A.; Kim, A.J.; Saunders, D.; Hwang, K.L.; Evason, K.J.; Heidel, J.; Brown, K.K.; Yuan, M.; Lien, E.C.; et al. Selenoprotein H is an essential regulator of redox homeostasis that cooperates with p53 in development and tumorigenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E5562–E5571. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, A.; Burdan, F.; Duma, D.; Solski, J.; Mazurkiewicz, M. γ-amino butyric acid (GABA) level as an overall survival risk factor in breast cancer. Ann. Agric. Environ. Med. 2017, 24, 435–439. [Google Scholar] [CrossRef]
- Serrano-Regal, M.P.; Bayón-Cordero, L.; Ordaz, R.P.; Garay, E.; Limon, A.; Arellano, R.O.; Matute, C.; Sánchez-Gómez, M.V. Expression and Function of GABA Receptors in Myelinating Cells. Front. Cell. Neurosci. 2020, 14, 256. [Google Scholar] [CrossRef]
- Wu, P.H.; Coultrap, S.; Pinnix, C.; Davies, K.D.; Tailor, R.; Ang, K.K.; Browning, M.D.; Grosshans, D.R. Radiation Induces Acute Alterations in Neuronal Function. PLoS ONE 2012, 7, e37677. [Google Scholar] [CrossRef]
- Lang, M.; Moradi-Chameh, H.; Zahid, T.; Gane, J.; Wu, C.; Valiante, T.A.; Zhang, L. Regulating hippocampal hyperexcitability through GABAB Receptors. Physiol. Rep. 2014, 2, e00278. [Google Scholar] [CrossRef]
- Takehara, A.; Hosokawa, M.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Nakamura, Y.; Nakagawa, H. γ-Aminobutyric Acid (GABA) Stimulates Pancreatic Cancer Growth through Overexpressing GABAA Receptor π Subunit. Cancer Res. 2007, 67, 9704–9712. [Google Scholar] [CrossRef] [PubMed]
- Matuszek, M.; Jesipowicz, M.; Kleinrok, Z. GABA content and GAD activity in gastric cancer. Med. Sci. Monit. 2001, 7, 377–381. [Google Scholar]
- Papadopoulos, V.; Kapsis, A.; Li, H.; Amri, H.; Hardwick, M.; Culty, M.; Kasprzyk, P.G.; Carlson, M.; Moreau, J.P.; Drieu, K. Drug-induced inhibition of the peripheral-type benzodiazepine receptor expression and cell proliferation in human breast cancer cells. Anticancer Res. 2000, 20, 2835–2847. [Google Scholar] [PubMed]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 2008, 29, 1979–1985. [Google Scholar] [CrossRef]
- Wang, T.; Huang, W.; Chen, F. Baclofen, a GABAB receptor agonist, inhibits human hepatocellular carcinoma cell growth in vitro and in vivo. Life Sci. 2008, 82, 536–541. [Google Scholar] [CrossRef]
- Opolski, A.; Mazurkiewicz, M.; Wietrzyk, J.; Kleinrok, Z.; Radzikowski, C. The role of GABA-ergic system in human mammary gland pathology and in growth of transplantable murine mammary cancer. J. Exp. Clin. Cancer Res. 2000, 19, 383–390. [Google Scholar]
- Watanabe, M.; Maemura, K.; Oki, K.; Shiraishi, N.; Shibayama, Y.; Katsu, K. Gamma-aminobutyric acid (GABA) and cell proliferation: Focus on cancer cells. Histol. Histopathol. 2006, 21, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Fasoulakis, Z.; Kolios, G.; Papamanolis, V.; Kontomanolis, E.N. Interleukins Associated with Breast Cancer. Cureus 2018, 10, e3549. [Google Scholar] [CrossRef]
- Dmitrieva, O.S.; Shilovskiy, I.; Khaitov, M.; Grivennikov, S.I. Interleukins 1 and 6 as main mediators of inflammation and cancer. Biochemistry 2016, 81, 80–90. [Google Scholar] [CrossRef]
- Anestakis, D.; Petanidis, S.; Kalyvas, S.; Nday, C.M.; Tsave, O.; Kioseoglou, E.; Salifoglou, A. Mechanisms and Applications of Interleukins in Cancer Immunotherapy. Int. J. Mol. Sci. 2015, 16, 1691–1710. [Google Scholar] [CrossRef]
- Vitiello, G.A.F.; Guembarovski, R.L.; Amarante, M.K.; Ceribelli, J.R.; Carmelo, E.C.B.; Watanabe, M.A.E. Interleukin 7 receptor alpha Thr244Ile genetic polymorphism is associated with susceptibility and prognostic markers in breast cancer subgroups. Cytokine 2018, 103, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Marconi, R.; Serafini, A.; Giovanetti, A.; Bartoleschi, C.; Pardini, M.C.; Bossi, G.; Strigari, L. Cytokine Modulation in Breast Cancer Patients Undergoing Radiotherapy: A Revision of the Most Recent Studies. Int. J. Mol. Sci. 2019, 20, 382. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-Y.; Jung, J.-S.; Kim, T.H.; Lim, S.-J.; Oh, E.-S.; Kim, J.-Y.; Ji, K.-A.; Joe, E.-H.; Cho, K.-H.; Han, I.-O. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol. Dis. 2006, 21, 457–467. [Google Scholar] [CrossRef]
- Lee, W.H.; Sonntag, W.; Mitschelen, M.; Yan, H.; Lee, Y.W. Irradiation induces regionally specific alterations in pro-inflammatory environments in rat brain. Int. J. Radiat. Biol. 2010, 86, 132–144. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Coates, P.J.; Scobie, G.E.; Milne, G.; Wright, E.G. Inflammatory-type responses after exposure to ionizing radiation in vivo: A mechanism for radiation-induced bystander effects? Oncogene 2001, 20, 7085–7095. [Google Scholar] [CrossRef]
- Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Pilones, K.A.; García-Martínez, E.; Rudqvist, N.; Formenti, S.C.; DeMaria, S. Barriers to Radiation-Induced In Situ Tumor Vaccination. Front. Immunol. 2017, 8, 229. [Google Scholar] [CrossRef]
- Song, K.; Cai, X.; Dong, Y.; Wu, H.; Wei, Y.; Shankavaram, U.T.; Cui, K.; Lee, Y.; Zhu, B.; Bhattacharjee, S.; et al. Epsins 1 and 2 promote NEMO linear ubiquitination via LUBAC to drive breast cancer development. J. Clin. Investig. 2021, 131, e129374. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fre, S.; Slepnev, V.I.; Capua, M.R.; Takei, K.; Butler, M.H.; Di Fiore, P.P.; De Camilli, P. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature 1998, 394, 793–797. [Google Scholar] [CrossRef]
- Ko, G.; Paradise, S.; Chen, H.; Graham, M.; Vecchi, M.; Bianchi, F.; Cremona, O.; Di Fiore, P.P.; De Camilli, P. Selective high-level expression of epsin 3 in gastric parietal cells, where it is localized at endocytic sites of apical canaliculi. Proc. Natl. Acad. Sci. USA 2010, 107, 21511–21516. [Google Scholar] [CrossRef] [PubMed]
- Spradling, K.D.; McDaniel, A.E.; Lohi, J.; Pilcher, B.K. Epsin 3 Is a Novel Extracellular Matrix-induced Transcript Specific to Wounded Epithelia. J. Biol. Chem. 2001, 276, 29257–29267. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Lee, Y.; Zhu, B.; Wu, H.; Chen, Y.; Chen, H. Epsins in vascular development, function and disease. Cell. Mol. Life Sci. 2020, 78, 833–842. [Google Scholar] [CrossRef]
- Windler, S.L.; Bilder, D. Endocytic Internalization Routes Required for Delta/Notch Signaling. Curr. Biol. 2010, 20, 538–543. [Google Scholar] [CrossRef]
- Aguilar, R.C.; Longhi, S.A.; Shaw, J.D.; Yeh, L.-Y.; Kim, S.; Schon, A.; Freire, E.; Hsu, A.; McCormick, W.K.; Watson, H.A.; et al. Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4116–4121. [Google Scholar] [CrossRef]
- Rahman, H.A.; Wu, H.; Dong, Y.; Pasula, S.; Wen, A.; Sun, Y.; Brophy, M.L.; Tessneer, K.L.; Cai, X.; McManus, J.; et al. Selective Targeting of a Novel Epsin–VEGFR2 Interaction Promotes VEGF-Mediated Angiogenesis. Circ. Res. 2016, 118, 957–969. [Google Scholar] [CrossRef]
- Liu, X.; Pasula, S.; Song, H.; Tessneer, K.L.; Dong, Y.; Hahn, S.; Yago, T.; Brophy, M.L.; Chang, B.; Cai, X.; et al. Temporal and spatial regulation of epsin abundance and VEGFR3 signaling are required for lymphatic valve formation and function. Sci. Signal. 2014, 7, ra97. [Google Scholar] [CrossRef]
- Coon, B.G.; Burgner, J.; Camonis, J.H.; Aguilar, R.C. The Epsin Family of Endocytic Adaptors Promotes Fibrosarcoma Migration and Invasion. J. Biol. Chem. 2010, 285, 33073–33081. [Google Scholar] [CrossRef] [PubMed]
- Coon, B.G.; DiRenzo, D.M.; Konieczny, S.F.; Aguilar, R.C. Epsins’ novel role in cancer cell invasion. Commun. Integr. Biol. 2011, 4, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, W.; Kan, P.; Huang, C.; Ma, Z.; Wu, Q.; Yao, X.; Zhang, B. Overexpression of Epsin 3 enhances migration and invasion of glioma cells by inducing epithelialmesenchymal transition. Oncol. Rep. 2018, 40, 3049–3059. [Google Scholar] [CrossRef] [PubMed]
- Sloane, B.F.; Rozhin, J.; Johnson, K.; Taylor, H.; Crissman, J.D.; Honn, K.V. Cathepsin B: Association with plasma membrane in metastatic tumors. Proc. Natl. Acad. Sci. USA 1986, 83, 2483–2487. [Google Scholar] [CrossRef] [PubMed]
- Brix, K. Lysosomal Proteases: Revival of the Sleeping Beauty. In Madame Curie Bioscience Database (Formerly, Eurekah Bioscience Database); Saftig, P., Ed.; Landes Bioscience: Austin, TX, USA, 2005. [Google Scholar]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2011, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Guardia, C.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef]
- Joyce, J.A.; Baruch, A.; Chehade, K.; Meyer-Morse, N.; Giraudo, E.; Tsai, F.-Y.; Greenbaum, D.C.; Hager, J.H.; Bogyo, M.; Hanahan, D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 2004, 5, 443–453. [Google Scholar] [CrossRef]
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.-P. Cathepsin S Controls Angiogenesis and Tumor Growth via Matrix-derived Angiogenic Factors. J. Biol. Chem. 2006, 281, 6020–6029. [Google Scholar] [CrossRef]
- Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Small, D.; Quinn, D.J.; Hegarty, S.M.; Ward, C.; Walker, B.; Johnston, J.A.; Olwill, S.A.; et al. Antibody-Mediated Inhibition of Cathepsin S Blocks Colorectal Tumor Invasion and Angiogenesis. Clin. Cancer Res. 2009, 15, 6042–6051. [Google Scholar] [CrossRef]
- Urbich, C.; Heeschen, C.; Aicher, A.; Sasaki, K.-I.; Bruhl, T.; Farhadi, M.R.; Vajkoczy, P.; Hofmann, W.K.; Peters, C.; Pennacchio, L.; et al. Cathepsin L is required for endothelial progenitor cell–induced neovascularization. Nat. Med. 2005, 11, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Cox, J.L. Cathepsin L increases invasion and migration of B16 melanoma. Cancer Cell Int. 2007, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Rousselet, N.; Mills, L.; Jean, D.; Tellez, C.; Bar-Eli, M.; Frade, R. Inhibition of Tumorigenicity and Metastasis of Human Melanoma Cells by Anti-Cathepsin L Single Chain Variable Fragment. Cancer Res. 2004, 64, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Joyce, J.A. Cysteine Cathepsins and the Cutting Edge of Cancer Invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef]
- S Sudhan, D.R.; Siemann, D.W. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin. Exp. Metastasis 2013, 30, 891–902. [Google Scholar] [CrossRef]
- Bratovš, A.; Kramer, L.; Mikhaylov, G.; Vasiljeva, O.; Turk, B. Stefin A-functionalized liposomes as a system for cathepsins S and L-targeted drug delivery. Biochimie 2019, 166, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Lecaille, F.; Kaleta, J.; Brömme, D. Human and Parasitic Papain-Like Cysteine Proteases: Their Role in Physiology and Pathology and Recent Developments in Inhibitor Design. Chem. Rev. 2002, 102, 4459–4488. [Google Scholar] [CrossRef]
- Turk, V.; Turk, B.; Turk, D. NEW EMBO MEMBERS’ REVIEW: Lysosomal cysteine proteases: Facts and opportunities. EMBO J. 2001, 20, 4629–4633. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A. The cystatins: A diverse superfamily of cysteine peptidase inhibitors. Biomed. Biochim. Acta 1986, 45, 1363–1374. [Google Scholar]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.; Daglioglu, C.; Tutar, Y.; Zamyatnin, J.A.A. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Lah, T.T.; Kokalj-Kunovar, M.; Strukelj, B.; Pungerčar, J.; Barlič-Maganja, D.; Drobnic-Kosorok, M.; Kastelic, L.; Babnik, J.; Golouh, R.; Turk, V. Stefins and lysosomal cathepsins B, L and D in human breast carcinoma. Int. J. Cancer 1992, 50, 36–44. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, Y.; Li, Y.; Grün, K.; Berndt, A.; Zhou, Z.; Petersen, I. Cystatin A suppresses tumor cell growth through inhibiting epithelial to mesenchymal transition in human lung cancer. Oncotarget 2017, 9, 14084–14098. [Google Scholar] [CrossRef] [PubMed]
- Mirtti, T.; Alanen, K.; Kallajoki, M.; Rinne, A.; Söderström, K.-O. Expression of cystatins, high molecular weight cytokeratin, and proliferation markers in prostatic adenocarcinoma and hyperplasia. Prostate 2003, 54, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Liyong, Z.; Ding, F.; Zhang, L.; Liu, Z.; Wu, Y.; Luo, A.; Wu, M.; Wang, M.; Zhan, Q.; Liu, Z. Overexpression of Stefin A in Human Esophageal Squamous Cell Carcinoma Cells Inhibits Tumor Cell Growth, Angiogenesis, Invasion, and Metastasis. Clin. Cancer Res. 2005, 11, 8753–8762. [Google Scholar] [CrossRef]
- Kuopio, T.; Kankaanranta, A.; Jalava, P.; Kronqvist, P.; Kotkansalo, T.; Weber, E.; Collan, Y. Cysteine proteinase inhibitor cystatin A in breast cancer. Cancer Res. 1998, 58, 432–436. [Google Scholar]
- Duivenvoorden, H.M.; Rautela, J.; Edgington-Mitchell, L.E.; Spurling, A.; Greening, D.W.; Nowell, C.J.; Molloy, T.J.; Robbins, E.; Brockwell, N.K.; Lee, C.S.; et al. Myoepithelial cell-specific expression of stefin A as a suppressor of early breast cancer invasion. J. Pathol. 2017, 243, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Larsen, A.; Stoltenberg, M.; Penkowa, M. The role of metallothionein in oncogenesis and cancer prognosis. Prog. Histochem. Cytochem. 2009, 44, 29–64. [Google Scholar] [CrossRef]
- Thirumoorthy, N. Metallothionein: An overview. World J. Gastroenterol. 2007, 13, 993–996. [Google Scholar] [CrossRef]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Experientia 2002, 59, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Maret, W. The Functions of Metamorphic Metallothioneins in Zinc and Copper Metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef]
- Kumari, M.R.; Hiramatsu, M.; Ebadi, M. Free radical scavenging actions of metallothionein isoforms I and II. Free Radic. Res. 1998, 29, 93–101. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The Role of Metallothionein in Oxidative Stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Liu, J.; Diwan, B.A. Metallothionein protection of cadmium toxicity. Toxicol. Appl. Pharmacol. 2009, 238, 215–220. [Google Scholar] [CrossRef]
- Arriaga, J.M.; Levy, E.M.; Bravo, A.I.; Bayo, S.M.; Amat, M.; Aris, M.; Hannois, A.; Bruno, L.; Roberti, M.P.; Loria, F.S.; et al. Metallothionein expression in colorectal cancer: Relevance of different isoforms for tumor progression and patient survival. Hum. Pathol. 2012, 43, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, R.; Daniunaite, K.; Bakavicius, A.; Sabaliauskaite, R.; Skeberdyte, A.; Petroska, D.; Laurinavicius, A.; Jankevicius, F.; Lazutka, J.R.; Jarmalaite, S. Decreased expression of MT1E is a potential biomarker of prostate cancer progression. Oncotarget 2017, 8, 61709–61718. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiang, L.; Hu, Y.; Xiao, C.; Xu, N.; Zhou, J.; Zhou, X. Metallothionein 1H (MT1H) functions as a tumor suppressor in hepatocellular carcinoma through regulating Wnt/β-catenin signaling pathway. BMC Cancer 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gomulkiewicz, A.; Podhorska-Okolow, M.; Szulc, R.; Smorag, Z.; Wojnar, A.; Zabel, M.; Dzięgiel, P. Correlation between metallothionein (MT) expression and selected prognostic factors in ductal breast cancers. Folia Histochem. Cytobiol. 2010, 48, 242–248. [Google Scholar] [CrossRef]
- Hengstler, J.; Pilch, H.; Schmidt, M.; Dahlenburg, H.; Schiffer, I.; Oesch, F.; Knapstein, P.; Kaina, B.; Tanner, B. Metallothionein expression in ovarian cancer in relation to histopathological parameters and molecular markers of prognosis. Int. J. Cancer 2001, 95, 121–127. [Google Scholar] [CrossRef]
- Wülfing, C.; Van Ahlen, H.; Eltze, E.; Piechota, H.; Hertle, L.; Schmid, K.-W. Metallothionein in bladder cancer: Correlation of overexpression with poor outcome after chemotherapy. World J. Urol. 2007, 25, 199–205. [Google Scholar] [CrossRef]
- Jayasurya, A.; Bay, B.; Yap, W.; Tan, N.; Tan, B. Proliferative potential in nasopharyngeal carcinoma: Correlations with metallothionein expression and tissue zinc levels. Carcinogenesis 2000, 21, 1809–1812. [Google Scholar] [CrossRef][Green Version]
- Weinlich, G.; Eisendle, K.; Hassler, E.; Baltaci, M.; O Fritsch, P.; Zelger, B. Metallothionein – overexpression as a highly significant prognostic factor in melanoma: A prospective study on 1270 patients. Br. J. Cancer 2006, 94, 835–841. [Google Scholar] [CrossRef]
- Werynska, B.; Pula, B.; Muszczynska-Bernhard, B.; Piotrowska, A.; Jethon, A.; Podhorska-Okolow, M.; Dziegiel, P.; Jankowska, R. Correlation between expression of metallothionein and expression of Ki-67 and MCM-2 proliferation markers in non-small cell lung cancer. Anticancer. Res. 2011, 31, 2833–2839. [Google Scholar]
- Jin, R.; Chow, V.T.-K.; Tan, P.-H.; Dheen, S.T.; Duan, W.; Bay, B.-H. Metallothionein 2A expression is associated with cell proliferation in breast cancer. Carcinogenesis 2002, 23, 81–86. [Google Scholar] [CrossRef]
- Jin, R.; Bay, B.-H.; Chow, V.T.-K.; Tan, P.-H. Metallothionein 1F mRNA expression correlates with histological grade in breast carcinoma. Breast Cancer Res. Treat. 2001, 66, 265–272. [Google Scholar] [CrossRef]
- Wierzowiecka, B.; Gomulkiewicz, A.; Cwynar-Zając, L.; Olbromski, M.; Grzegrzolka, J.; Kobierzycki, C.; Podhorska-Okolow, M.; Dziegiel, P. Expression of Metallothionein and Vascular Endothelial Growth Factor Isoforms in Breast Cancer Cells. In Vivo 2016, 30, 271–278. [Google Scholar]
- Haerslev, T.; Jacobsen, K.; Nedergaard, L.; Zedeler, K. Immunohistochemical detection of metallothionein in primary breast carcinomas and their axillary lymph node metastases. Pathol. Res. Pr. 1994, 190, 675–681. [Google Scholar] [CrossRef]
- Hishikawa, Y.; Kohno, H.; Ueda, S.; Kimoto, T.; Dhar, D.K.; Kubota, H.; Tachibana, M.; Koji, T.; Nagasue, N. Expression of Metallothionein in Colorectal Cancers and Synchronous Liver Metastases. Oncology 2001, 61, 162–167. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, J.Y.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Park, B.H.; Jeong, H.G. Metallothionein-2A overexpression increases the expression of matrix metalloproteinase-9 and invasion of breast cancer cells. FEBS Lett. 2010, 585, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, A.; Pula, B.; Suchański, J.; Olbromski, M.; Gomułkiewicz, A.; Owczarek, T.; Kruczak, A.; Ambicka, A.; Rys, J.; Ugorski, M.; et al. Metallothionein-3 Increases Triple-Negative Breast Cancer Cell Invasiveness via Induction of Metalloproteinase Expression. PLoS ONE 2015, 10, e0124865. [Google Scholar] [CrossRef]
- Gomułkiewicz, A.; Jabłońska, K.; Pula, B.; Grzegrzółka, J.; Borska, S.; Podhorska-Okolow, M.; Wojnar, A.; Rys, J.; Ambicka, A.; Ugorski, M.; et al. Expression of metallothionein 3 in ductal breast cancer. Int. J. Oncol. 2016, 49, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Goulding, H.; Jasani, B.; Pereira, H.; Reid, A.; Galea, M.; Bell, J.A.; Elston, C.W.; Robertson, J.F.; Blamey, R.W.; Nicholson, R.A.; et al. Metallothionein expression in human breast cancer. Br. J. Cancer 1995, 72, 968–972. [Google Scholar] [CrossRef]
- Jin, R.; Huang, J.; Tan, P.-H.; Bay, B.-H. Clinicopathological significance of metallothioneins in breast cancer. Pathol. Oncol. Res. 2004, 10, 74–79. [Google Scholar] [CrossRef]
- Sens, M.A.; Somji, S.; Garrett, S.H.; Beall, C.L.; Sens, D.A. Metallothionein Isoform 3 Overexpression Is Associated with Breast Cancers Having a Poor Prognosis. Am. J. Pathol. 2001, 159, 21–26. [Google Scholar] [CrossRef]
- Surowiak, P.; Materna, V.; Kaplenko, I.; Spaczyński, M.; Dietel, M.; Lage, H.; Zabel, M. Augmented expression of metallothionein and glutathione S-transferase pi as unfavourable prognostic factors in cisplatin-treated ovarian cancer patients. Virchows Arch. 2005, 447, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Dziegiel, P.; Jeleń, M.; Muszczyńska, B.; Maciejczyk, A.; Szulc, A.; Podhorska-OkoŁów, M.; Cegielski, M.; Zabel, M. Role of metallothionein expression in non-small cell lung carcinomas. Rocz. Akad. Med. Bialymst. (1995) 2004, 49, 43–45. (In Polish) [Google Scholar]
- Dutsch-Wicherek, M.; Lazar, A.; Tomaszewska, R. The Potential Role of MT and Vimentin Immunoreactivity in the Remodeling of the Microenvironment of Parotid Adenocarcinoma. Cancer Microenviron. 2010, 4, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Dutsch-Wicherek, M.; Lazar, A.; Tomaszewska, R.; Kazmierczak, W.; Wicherek, L. Analysis of metallothionein and vimentin immunoreactivity in pharyngeal squamous cell carcinoma and its microenvironment. Cell Tissue Res. 2013, 352, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Dutsch-Wicherek, M. REVIEW ARTICLE: RCAS1, MT, and Vimentin as Potential Markers of Tumor Microenvironment Remodeling. Am. J. Reprod. Immunol. 2010, 63, 181–188. [Google Scholar] [CrossRef]
- Nagel, W.W.; Vallee, B.L. Cell cycle regulation of metallothionein in human colonic cancer cells. Proc. Natl. Acad. Sci. USA 1995, 92, 579–583. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Liu, X.S. TIMER2.0. Dana Farber Cancer Institute. Available online: http://timer.cistrome.org/ (accessed on 6 August 2021).









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calaf, G.M.; Crispin, L.A.; Roy, D.; Aguayo, F.; Muñoz, J.P.; Bleak, T.C. Gene Signatures Induced by Ionizing Radiation as Prognostic Tools in an In Vitro Experimental Breast Cancer Model. Cancers 2021, 13, 4571. https://doi.org/10.3390/cancers13184571
Calaf GM, Crispin LA, Roy D, Aguayo F, Muñoz JP, Bleak TC. Gene Signatures Induced by Ionizing Radiation as Prognostic Tools in an In Vitro Experimental Breast Cancer Model. Cancers. 2021; 13(18):4571. https://doi.org/10.3390/cancers13184571
Chicago/Turabian StyleCalaf, Gloria M., Leodan A. Crispin, Debasish Roy, Francisco Aguayo, Juan P. Muñoz, and Tammy C. Bleak. 2021. "Gene Signatures Induced by Ionizing Radiation as Prognostic Tools in an In Vitro Experimental Breast Cancer Model" Cancers 13, no. 18: 4571. https://doi.org/10.3390/cancers13184571
APA StyleCalaf, G. M., Crispin, L. A., Roy, D., Aguayo, F., Muñoz, J. P., & Bleak, T. C. (2021). Gene Signatures Induced by Ionizing Radiation as Prognostic Tools in an In Vitro Experimental Breast Cancer Model. Cancers, 13(18), 4571. https://doi.org/10.3390/cancers13184571